
Innate Immunity and Immune Evasion by Enterovirus 71


Abstract
:1. Introduction

{kind=link}
{kind=link}
| Pattern-Recognition Receptor (PRR) | Pathogen-Associated Molecular Patterns (PAMPs) | |
|---|---|---|
| ||
| Viral Glycoproteins (RSV, MMTV) | |
| Viral Glycoproteins (Measles virus, HCMV, HSV-1, Epstein-Barr virus (EBV), RSV, LCMV) | |
| dsRNA (Reovirus, LCMV, MCMV, VSV, West Nile Virus, Punta Toro Virus) | |
| ssRNA (Influenza virus, VSV, Sendai virus, HIV, Coxsackievirus B) | |
| Viral DNA (HSV-1/-2, MCMV, EBV) | |
| ||
| Uncapped 5′ppp ssRNA, short dsRNA (VSV, Rabies virus, Sendai virus, NDV, RSV, Influenza A&B, HCV, Japanese encephalitis virus, and Ebola virus) | |
| dsRNA (EV71, CVB, EMCV, theiler’s virus, and mengo virus) | |
| ||
|  | Viral DNA (HSV) |
| ||
| ||
| ||
| ||
| ||
| dsRNA | |
1.1. Clinical Features of EV71 Infection
1.2. Virus Infection and Innate Immune Response
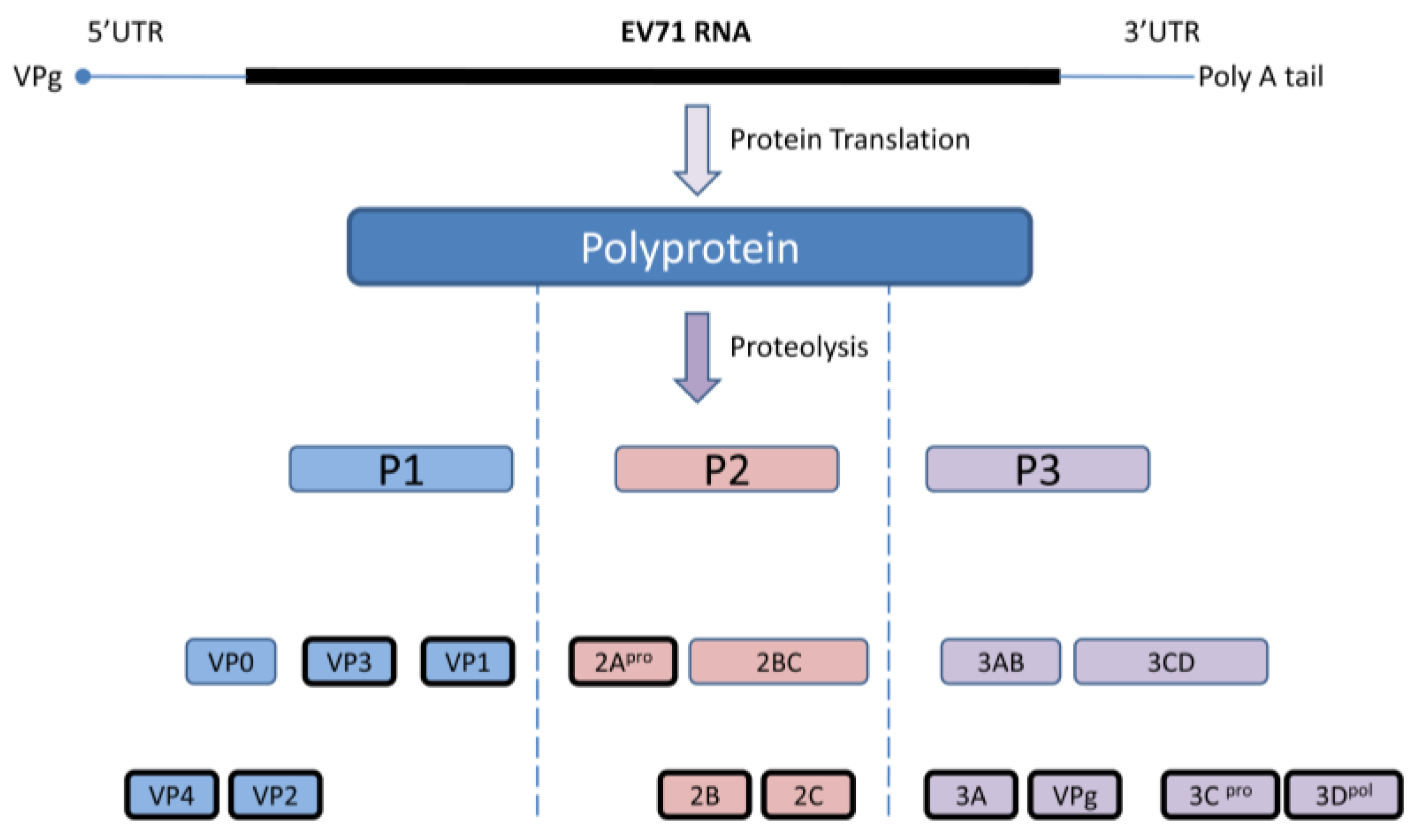
2. EV 71 Genome and Replication
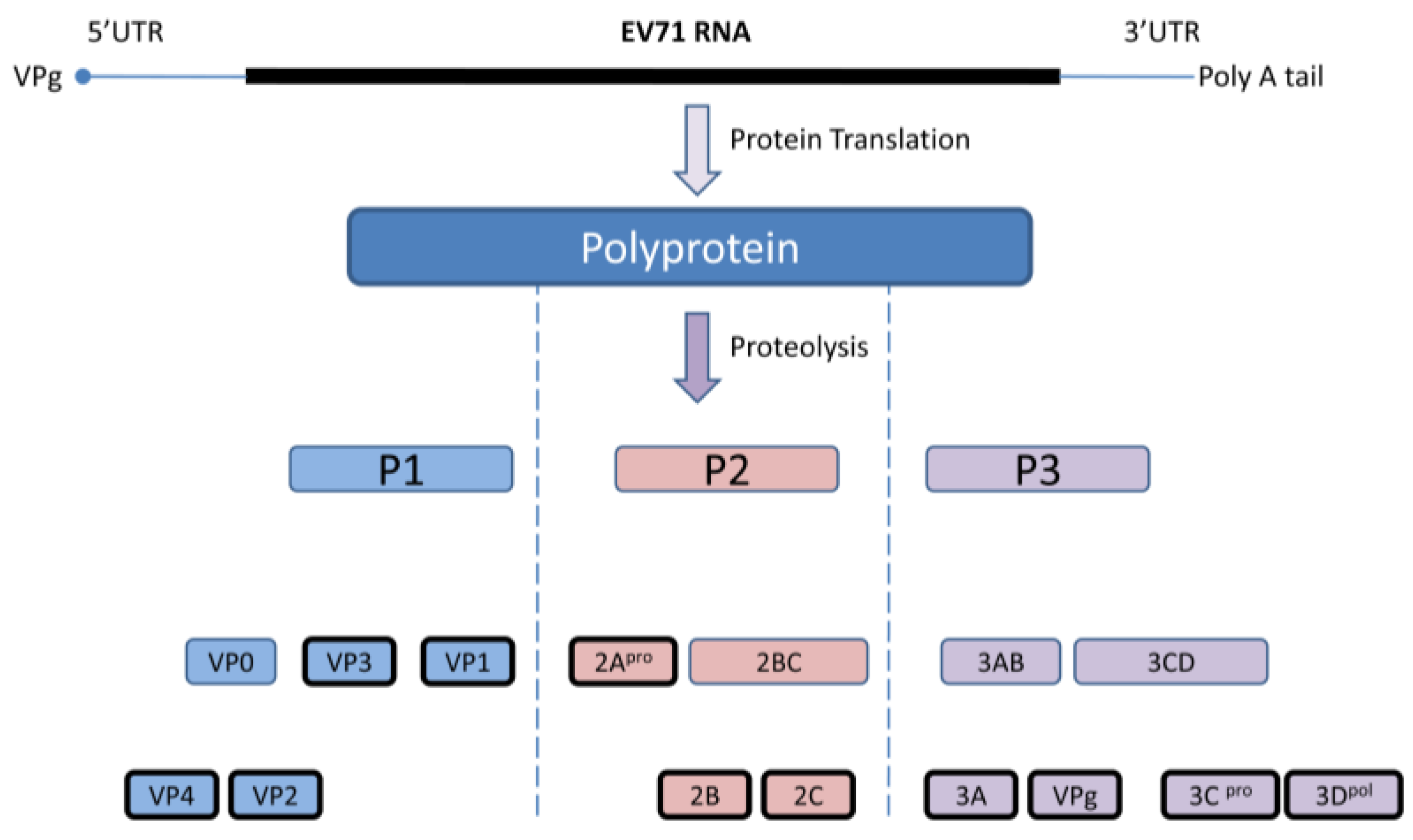
3. Role of Type I IFNs on EV71 Infections
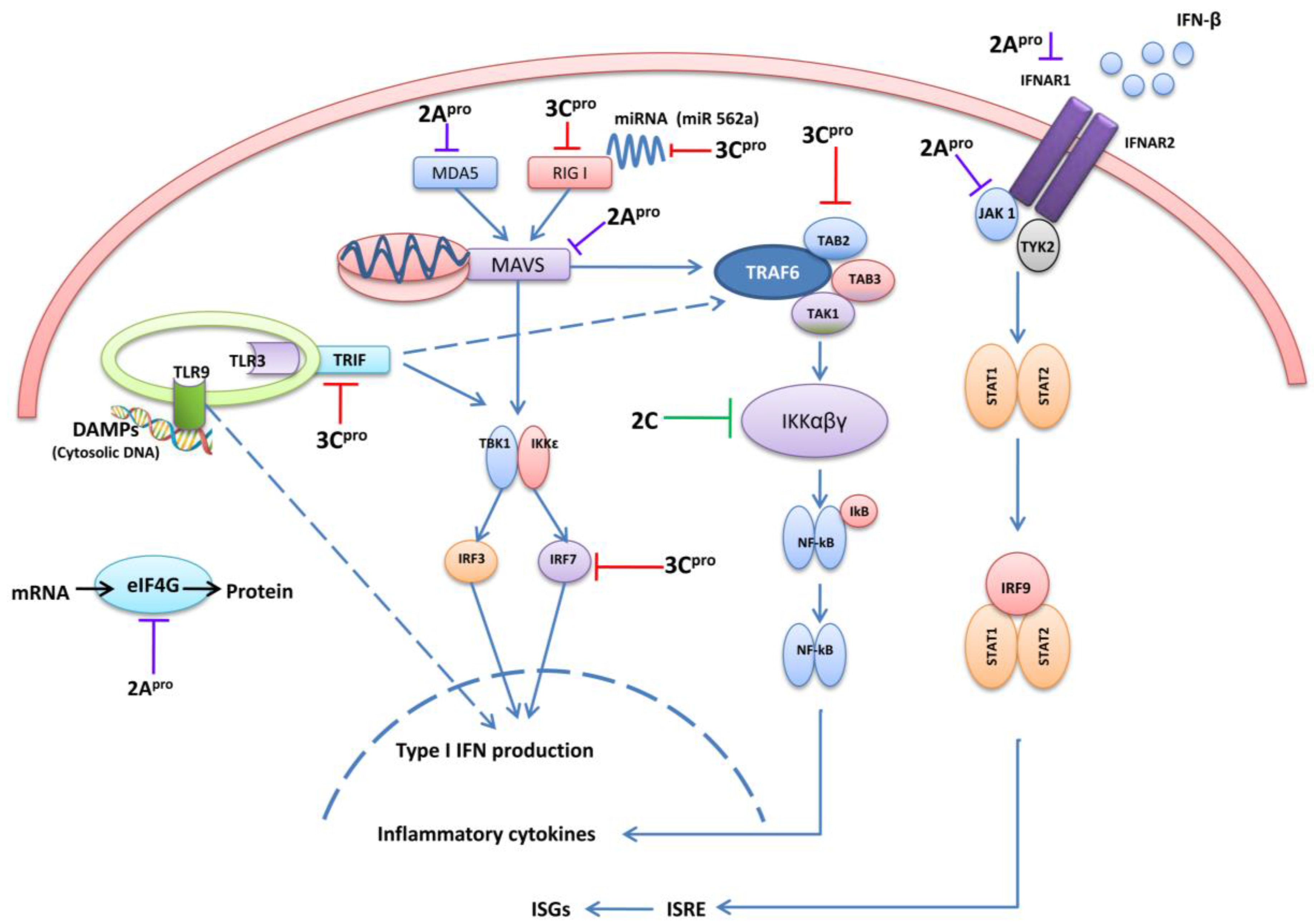
4. 3C and 2A Proteases of EV71 Are the Main Antagonists of Type I IFN
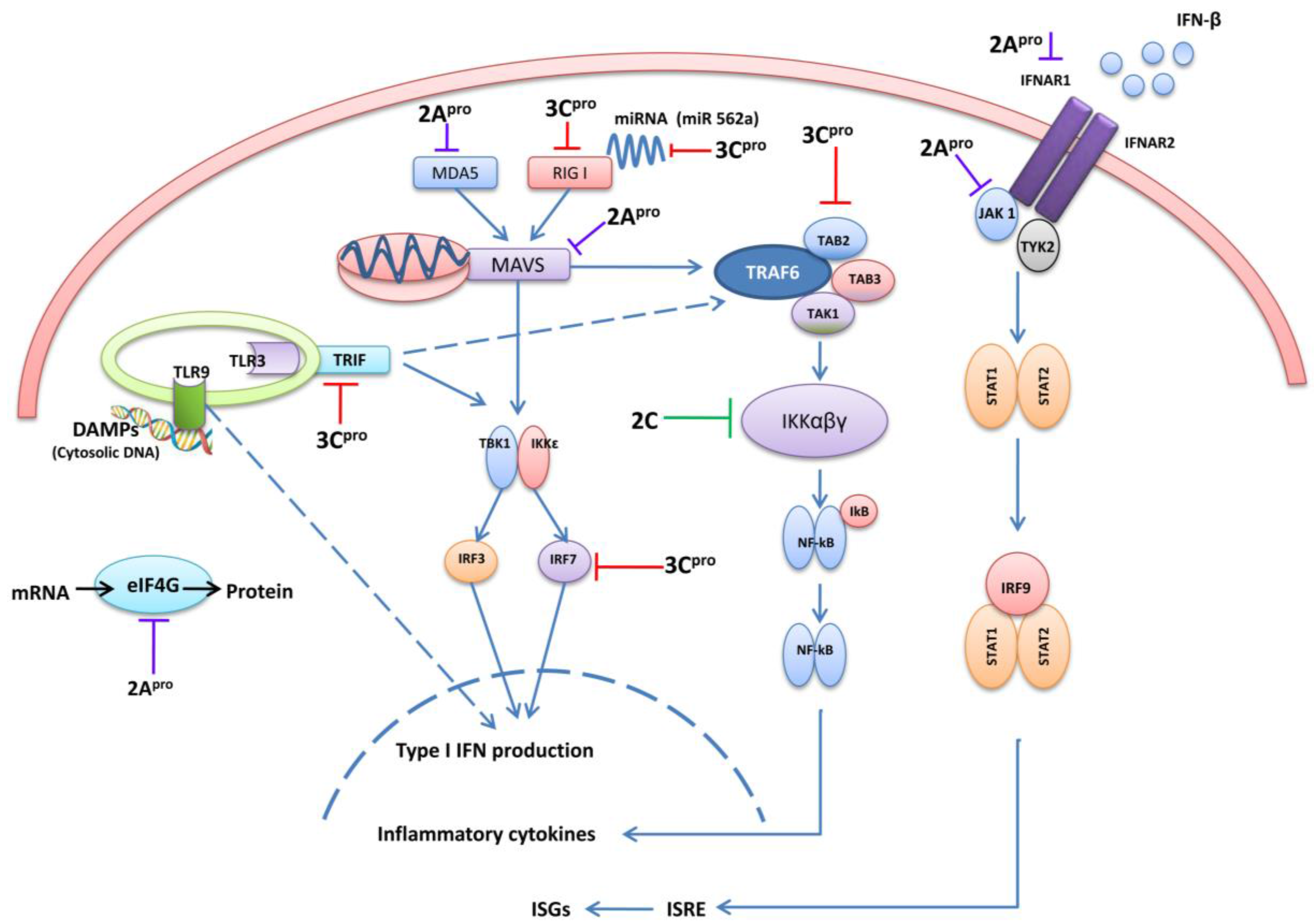
4.1. 3C pro Interacts with RIG-I
4.2. The Role of TRIF and Cleavage by 3Cpro
4.3. 3C pro Cleaves Interferon Regulatory Factor 7 (IRF7) and IRF9
4.4. 3Cpro Cleaves the TAK1/TAB1/TAB2/TAB3 Complex
4.5. 3Cpro Cleaves CSTF-64
4.6. 3Cpro Manipulates Cellular Micro RNAs
5. EV71 2Apro Disrupts IFN Signaling
5.1. 2Apro Cleaves MAVS
5.2. 2Apro Cleaves MDA5
5.3. 2Apro Reduces Interferon Receptor I (IFNAR I) Expression
5.4. 2Apro Induces eIF4G Cleavage
6. EV71 2Cpro Inhibits NF-κB Activation
7. Potential Antiviral Therapeutics
8. Discussion
Conflicts of Interest
References
- Brown, B.A.; Pallansch, M.A. Complete nucleotide sequence of enterovirus 71 is distinct from poliovirus. Virus Res. 1995, 39, 195–205. [Google Scholar] [CrossRef]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Schenten, D.; Medzhitov, R. The control of adaptive immune responses by the innate immune system. Adv. Immunol. 2011, 109, 87–124. [Google Scholar] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Akira, S. Pathogen recognition by innate receptors. J. Infect. Chemother. 2008, 14, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.H.; Wong, S.C.; Lewthwaite, P.; Cardosa, M.J.; Solomon, T. Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010, 9, 1097–1105. [Google Scholar] [CrossRef]
- Liu, C.C.; Tseng, H.W.; Wang, S.M.; Wang, J.R.; Su, I.J. An outbreak of enterovirus 71 infection in Taiwan, 1998: Epidemiologic and clinical manifestations. J. Clin. Virol. 2000, 17, 23–30. [Google Scholar] [CrossRef]
- Merovitz, L.; Demers, A.M.; Newby, D.; McDonald, J. Enterovirus 71 infections at a Canadian center. Pediatr. Infect. Dis. J. 2000, 19, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Yao, P.P.; Xia, Y.; Qian, L.; Yang, Z.N.; Xie, R.H.; Sun, Y.S.; Lu, H.J.; Miao, Z.P.; Li, C.; et al. Enterovirus 71 infection causes severe pulmonary lesions in gerbils, merionesunguiculatus, which can be prevented by passive immunization with specific antisera. PLoS ONE 2015, 10, e0119173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, W.; Liu, L.; Wang, J.; Zhao, H.; Liao, Y.; Na, R.; Dong, C.; Wang, L.; Xie, Z.; et al. Pathogenesis study of enterovirus 71 infection in rhesus monkeys. Lab. Investig. 2011, 91, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Lee, Y.P.; Hung, Y.T.; Lin, C.H.; Chuang, J.I.; Lei, H.Y.; Su, I.J.; Yu, C.K. Exogenous interleukin-6, interleukin-13, and interferon-gamma provoke pulmonary abnormality with mild edema in enterovirus 71-infected mice. Respir. Res. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.A.; Randall, R.E.; Taylor, G.L. Crystal structure of human IPS-1/MAVS/VISA/Cardifcaspase activation recruitment domain. BMC Struct. Biol. 2008, 8. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 2008, 20, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Nishiyama, Y. Mitochondria and viruses. Mitochondrion 2011, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhou, Y.; Shen, P. NF-kappaB and its regulation on the immune system. Cell. Mol. Immunol. 2004, 1, 343–350. [Google Scholar] [PubMed]
- Oshiumi, H.; Okamoto, M.; Fujii, K.; Kawanishi, T.; Matsumoto, M.; Koike, S.; Seya, T. The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J. Immunol. 2011, 187, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Fujii, K.; Nagata, N.; Takeuchi, O.; Akira, S.; Oshiumi, H.; Matsumoto, M.; Seya, T.; Koike, S. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J. Virol. 2012, 86, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Richer, M.J.; Lavallee, D.J.; Shanina, I.; Horwitz, M.S. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS ONE 2009, 4, e4127. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Osawa, T.; Ogami, K.; Ouyang, X.; Sakaguchi, S.; Koshiba, R.; Yanai, H.; Seko, Y.; Shitara, H.; Bishop, K.; et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20446–20451. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Hacker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Yamashita, Y.; Li, J.; Hanagata, N.; Minowa, T.; Takemura, T.; Koike, S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 2009, 15, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chuang, H.; Yang, K.D. Sialylatedglycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol. J. 2009, 6, 141. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Chen, T.C.; Weng, K.F.; Chang, S.C.; Chen, L.L.; Shih, S.R. Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 2009, 16. [Google Scholar] [CrossRef] [PubMed]
- Kontsek, P. Human type I interferons: Structure and function. ActaVirol. 1994, 38, 345–360. [Google Scholar]
- Parmar, S.; Platanias, L.C. Interferons: Mechanisms of action and clinical applications. Curr. Opin. Oncol. 2003, 15, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Yanai, H. Interferon signalling network in innate defence. Cell. Microbiol. 2006, 8, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; He, Y.; Chen, Y.; Kung, H.F.; He, M.L. Potent inhibition of human enterovirus 71 replication by type I interferon subtypes. Antivir. Ther. 2011, 16, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Lee, Y.P.; Wang, Y.F.; Lei, H.Y.; Liu, C.C.; Wang, S.M.; Su, I.J.; Wang, J.R.; Yeh, T.M.; Chen, S.H.; et al. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 2005, 86, 3263–3269. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Wang, H.C.; Shih, S.R.; Teng, I.F.; Tseng, C.P.; Hsu, J.T. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 2011, 203, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yi, L.; Zhao, J.; Yu, J.; Chen, Y.; Lin, M.C.; Kung, H.F.; He, M.L. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 2012, 86, 3767–3776. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; West, H.; Paludan, S.R. IFN-lambda: Novel antiviral cytokines. J. Interferon Cytokine Res. 2006, 26, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.; Svedin, E.; Utorova, R.; Stone, V.M.; Flodstrom-Tullberg, M. Type III interferons are expressed by Coxsackievirus-infected human primary hepatocytes and regulate hepatocyte permissiveness to infection. Clin. Exp. Immunol. 2014, 177, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.; Richardson, S.J.; Leete, P.; Morgan, N.G.; Korsgren, O.; Flodstrom-Tullberg, M. Induction of an antiviral state and attenuated coxsackievirus replication in type III interferon-treated primary human pancreatic islets. J. Virol. 2013, 87, 7646–7654. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Wang, J.; Fan, T.; Qin, B.; Guo, L.; Lei, X.; Wang, J.; Wang, M.; Jin, Q. Crystal structure of human enterovirus 71 3C protease. J. Mol. Biol. 2011, 408, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.R.; Chiang, C.; Chen, T.C.; Wu, C.N.; Hsu, J.T.; Lee, J.C.; Hwang, M.J.; Li, M.L.; Chen, G.W.; Ho, M.S. Mutations at KFRDI and VGK domains of enterovirus 71 3C protease affect its RNA binding and proteolytic activities. J. Biomed. Sci. 2004, 11, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Sim, A.C.; Luhur, A.; Tan, T.M.; Chow, V.T.; Poh, C.L. RNA interference against enterovirus 71 infection. Virology 2005, 341, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yameen, M.; Liu, W.; Gao, Z.; Li, Y.; Peng, X.; Cai, Y.; Wu, C.; Zheng, Q.; Li, J.; et al. Conformational plasticity of the 2A proteinase from enterovirus 71. J. Virol. 2013, 87, 7348–7356. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.L.; Kao, L.T.; Lin, S.J.; Wang, R.Y.; Shih, S.R. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS ONE 2013, 8, e63431. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Huhn, M.H.; McCartney, S.A.; Lind, K.; Svedin, E.; Colonna, M.; Flodstrom-Tullberg, M. Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after Coxsackievirus infection. Virology 2010, 401, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. RIG-I is cleaved during picornavirus infection. Virology 2009, 391, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoSPathog. 2010, 6, e1001178. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Lamothe, B.; Campos, A.D.; Webster, W.K.; Maddineni, U.; Lin, S.C.; Wu, H.; Darnay, B.G. TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 2007, 282, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Han, N.; Xiao, X.; Jin, Q.; He, B.; Wang, J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J. Virol. 2014, 88, 9830–9841. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, Y.; Manley, J.L.; MacDonald, C.C.; Wilusz, J.; Shenk, T. A multisubunit factor, CstF, is required for polyadenylation of mammalian pre-mRNAs. Genes Dev. 1990, 4, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Weng, K.F.; Li, M.L.; Hung, C.T.; Shih, S.R. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoSPathog. 2009, 5, e1000593. [Google Scholar] [CrossRef] [PubMed]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef]
- Tsitsiou, E.; Lindsay, M.A. microRNAs and the immune response. Curr. Opin. Pharmacol. 2009, 9, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Lui, Y.L.; Tan, T.L.; Woo, W.H.; Timms, P.; Hafner, L.M.; Tan, K.H.; Tan, E.L. Enterovirus71 (EV71) utilise host microRNAs to mediate host immune system enhancing survival during infection. PLoS ONE 2014, 9, e102997. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Yu, S.L.; Chen, J.J.; Chang, S.Y.; Yan, B.S.; Hong, Q.S.; Singh, S.; Kao, C.L.; Chen, H.Y.; Su, K.Y.; et al. Enterovirus-induced miR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor eIF4E. Cell Host Microbe 2011, 9, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Sukhatme, V.P.; Cao, X.M.; Chang, L.C.; Tsai-Morris, C.H.; Stamenkovich, D.; Ferreira, P.C.; Cohen, D.R.; Edwards, S.A.; Shows, T.B.; Curran, T.; et al. A zinc finger-encoding gene coregulated with c-fos during growth and differentiation, and after cellular depolarization. Cell 1988, 53, 37–43. [Google Scholar] [CrossRef]
- Ho, B.C.; Yu, I.S.; Lu, L.F.; Rudensky, A.; Chen, H.Y.; Tsai, C.W.; Chang, Y.L.; Wu, C.T.; Chang, L.Y.; Shih, S.R.; et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, X.; Zheng, Z.; Zhang, Z.; Wei, C.; Guan, K.; Hou, L.; Zhang, B.; Zhu, L.; Cao, Y.; et al. Downregulation of microRNA miR-526a by enterovirus inhibits RIG-I-dependent innate immune response. J. Virol. 2014, 88, 11356–11368. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Penaranda, S.; Maher, K.; Pallansch, M.A. Complete genome sequences of all members of the species Human enterovirus A. J. Gen. Virol. 2004, 85, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.L.; Kung, S.H.; Hsu, Y.Y.; Liu, W.T. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 2002, 83, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Lacoste, J.; Nakhaei, P.; Sun, Q.; Yang, L.; Paz, S.; Wilkinson, P.; Julkunen, I.; Vitour, D.; Meurs, E.; et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 2006, 80, 6072–6083. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef] [PubMed]
- Drahos, J.; Racaniello, V.R. Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 2009, 83, 11581–11587. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoSPathog. 2011, 7, e1001311. [Google Scholar]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoSPathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Vakakis, E.; Kar, S.; Richer, E.; Evans, G.L.; Triantafilou, M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 2012, 125, 4761–4769. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.M.; Racaniello, V.R. Proteinase 2Apro is essential for enterovirus replication in type I interferon-treated cells. J. Virol. 2009, 83, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.C.; Parsons, K.; Barr, I.; Lowther, S.; Middleton, D.; Hansbro, P.M.; Wark, P.A. Critical role of constitutive type I interferon response in bronchial epithelial cell to influenza infection. PLoS ONE 2012, 7, e32947. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Zhao, X.; Yu, R.; Zhang, X.; Wu, S.; Liu, J.; Chi, X.; Song, X.; Fu, L.; et al. Enterovirus 71 inhibits cellular type I interferon signaling by downregulating JAK1 protein expression. Viral Immunol. 2014, 27, 267–276. [Google Scholar] [CrossRef]
- Bovee, M.L.; Marissen, W.E.; Zamora, M.; Lloyd, R.E. The predominant elF4G-specific cleavage activity in poliovirus-infected HeLa cells is distinct from 2A protease. Virology 1998, 245, 229–240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sommergruber, W.; Ahorn, H.; Klump, H.; Seipelt, J.; Zoephel, A.; Fessl, F.; Krystek, E.; Blaas, D.; Kuechler, E.; Liebig, H.D.; et al. 2A proteinases of coxsackie- and rhinovirus cleave peptides derived from eIF-4 gamma via a common recognition motif. Virology 1994, 198, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Haghighat, A.; Svitkin, Y.; Novoa, I.; Kuechler, E.; Skern, T.; Sonenberg, N. The eIF4G-eIF4E complex is the target for direct cleavage by the rhinovirus 2A proteinase. J. Virol. 1996, 70, 8444–8450. [Google Scholar] [PubMed]
- Baeuerle, P.A.; Baltimore, D. NF-κB: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Nguyen, T.L.; Arguello, M.; Nakhaei, P.; Paz, S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene 2006, 25, 6844–6867. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, H.; Zhang, Z.; Meng, J.; Mao, D.; Bai, B.; Lu, B.; Mao, P.; Hu, Q.; Wang, H. Enterovirus 71 2C protein inhibits TNF-α-mediated activation of NF-κB by suppressing IκB kinase β phosphorylation. J. Immunol. 2011, 187, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, Z.; Qin, B.; Zhang, X.; Chen, L.; Hu, Y.; Yuan, Z. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: Evaluation of antiviral efficacy in a murine infection model. Antivir. Res. 2013, 97, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Xu, M.; Yin, Z. Antiviral drug discovery for the treatment of enterovirus 71 infections. Antivir. Res. 2013, 97, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Laxton, C.; Rodman, J.; Myangar, N.; Horscroft, N.; Parkinson, T. Investigating Toll-like receptor agonists for potential to treat hepatitis C virus infection. Antimicrob. Agents Chemother. 2007, 51, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Horsmans, Y.; Berg, T.; Desager, J.P.; Mueller, T.; Schott, E.; Fletcher, S.P.; Steffy, K.R.; Bauman, L.A.; Kerr, B.M.; Averett, D.R. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology 2005, 42, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wu, C.C.; Lee, K.J.; Chuang, T.H.; Katakura, K.; Liu, Y.T.; Chan, M.; Tawatao, R.; Chung, M.; Shen, C.; et al. Activation of anti-hepatitis C virus responses via Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2006, 103, 1828–1833. [Google Scholar] [CrossRef] [PubMed]
- Hammerbeck, D.M.; Burleson, G.R.; Schuller, C.J.; Vasilakos, J.P.; Tomai, M.; Egging, E.; Cochran, F.R.; Woulfe, S.; Miller, R.L. Administration of a dual toll-like receptor 7 and toll-like receptor 8 agonist protects against influenza in rats. Antivir. Res. 2007, 73, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cluff, C.W.; Baldridge, J.R.; Stover, A.G.; Evans, J.T.; Johnson, D.A.; Lacy, M.J.; Clawson, V.G.; Yorgensen, V.M.; Johnson, C.L.; Livesay, M.T.; et al. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect. Immun. 2005, 73, 3044–3052. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Watanabe, I.; Ito, S.; Fujii, H.; Moriyama, M.; Tamura, S.; Takahashi, H.; Sawa, H.; Chiba, J.; Kurata, T.; et al. Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J. Virol. 2005, 79, 2910–2919. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.P.; Christopher, M.E.; Salazar, A.M.; Dale, R.M.; Sun, L.Q.; Wang, M. Nucleic acid-based antiviral drugs against seasonal and avian influenza viruses. Vaccine 2007, 25, 3175–3178. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Suter, E.E.; Miller, R.L.; Wessels, M.R. Unique efficacy of Toll-like receptor 8 agonists in activating human neonatal antigen-presenting cells. Blood 2006, 108, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.B.; Chou, A.H.; Lin, S.I.; Chen, I.H.; Lien, S.P.; Liu, C.C.; Chong, P.; Liu, S.J. Toll-like receptor 9-mediated protection of enterovirus 71 infection in mice is due to the release of danger-associated molecular patterns. J. Virol. 2014, 88, 11658–11670. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.; Lear, Z.; Herod, M.R.; Ryan, M.; Rowlands, D.J.; Stonehouse, N.J. Inhibition of the foot-and-mouth disease virus subgenomic replicon by RNA aptamers. J. Gen. Virol. 2014, 95, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.M.; Kim, K.S.; Lee, J.M.; Shim, H.S.; Cho, S.J.; Lee, W.K.; Ko, H.W.; Keum, Y.S.; Kim, S.Y.; Pathinayake, P.; et al. Single-stranded DNA aptamer that specifically binds to the influenza virus NS1 protein suppresses interferon antagonism. Antivir. Res. 2013, 100, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Ida-Hosonuma, M.; Iwasaki, T.; Yoshikawa, T.; Nagata, N.; Sato, Y.; Sata, T.; Yoneyama, M.; Fujita, T.; Taya, C.; Yonekawa, H.; et al. The α/β interferon response controls tissue tropism and pathogenicity of poliovirus. J. Virol. 2005, 79, 4460–4469. [Google Scholar] [CrossRef] [PubMed]
- Wessely, R.; Klingel, K.; Knowlton, K.U.; Kandolf, R. Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: Implications for mortality and early viral replication. Circulation 2001, 103, 756–761. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathinayake, P.S.; Hsu, A.C.-Y.; Wark, P.A.B. Innate Immunity and Immune Evasion by Enterovirus 71. Viruses 2015, 7, 6613-6630. https://doi.org/10.3390/v7122961
Pathinayake PS, Hsu AC-Y, Wark PAB. Innate Immunity and Immune Evasion by Enterovirus 71. Viruses. 2015; 7(12):6613-6630. https://doi.org/10.3390/v7122961
Chicago/Turabian StylePathinayake, Prabuddha S., Alan C-Y. Hsu, and Peter A.B. Wark. 2015. "Innate Immunity and Immune Evasion by Enterovirus 71" Viruses 7, no. 12: 6613-6630. https://doi.org/10.3390/v7122961
APA StylePathinayake, P. S., Hsu, A. C.-Y., & Wark, P. A. B. (2015). Innate Immunity and Immune Evasion by Enterovirus 71. Viruses, 7(12), 6613-6630. https://doi.org/10.3390/v7122961