
CDC42 Use in Viral Cell Entry Processes by RNA Viruses

{kind=link}
{kind=link}
Abstract
:1. Introduction
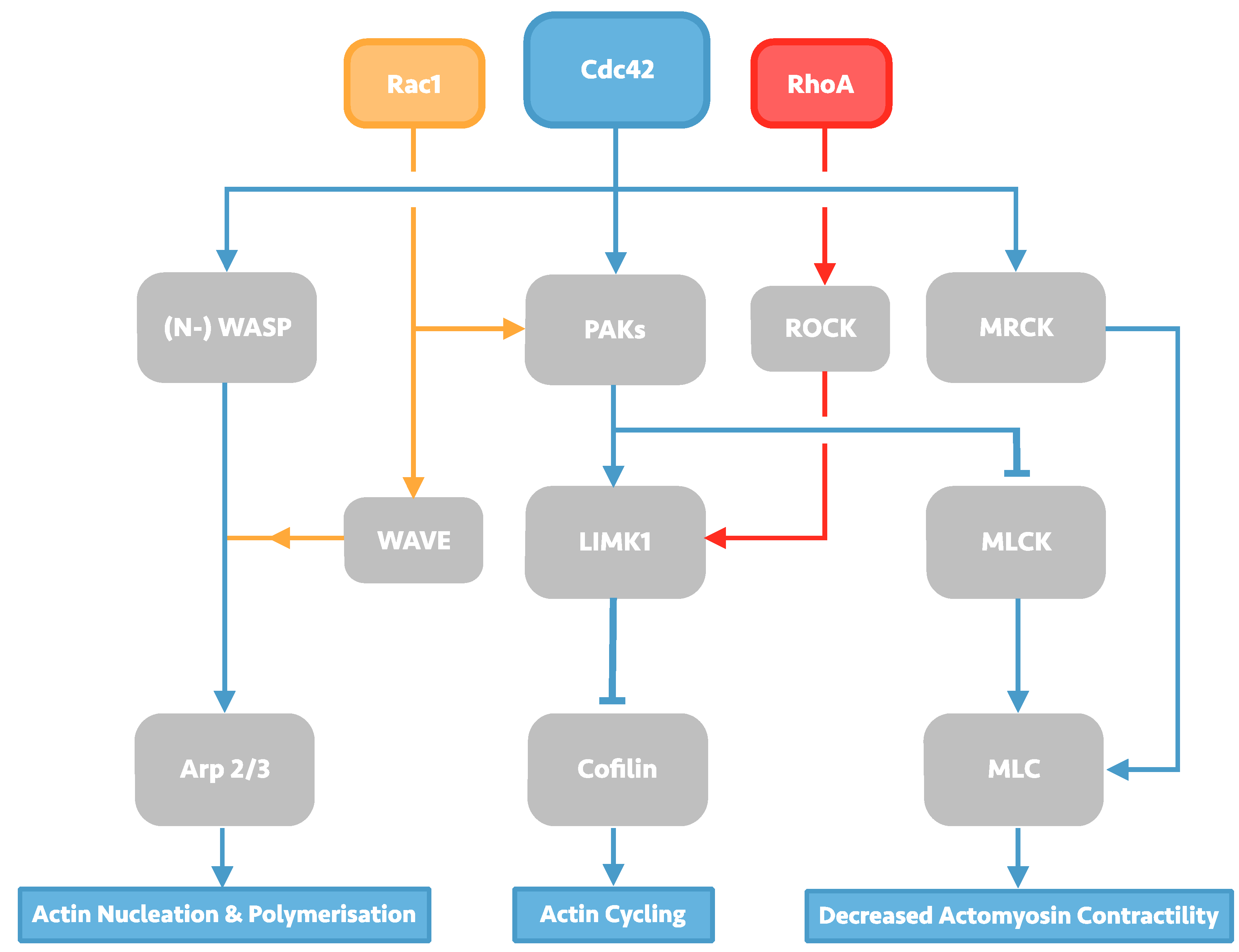
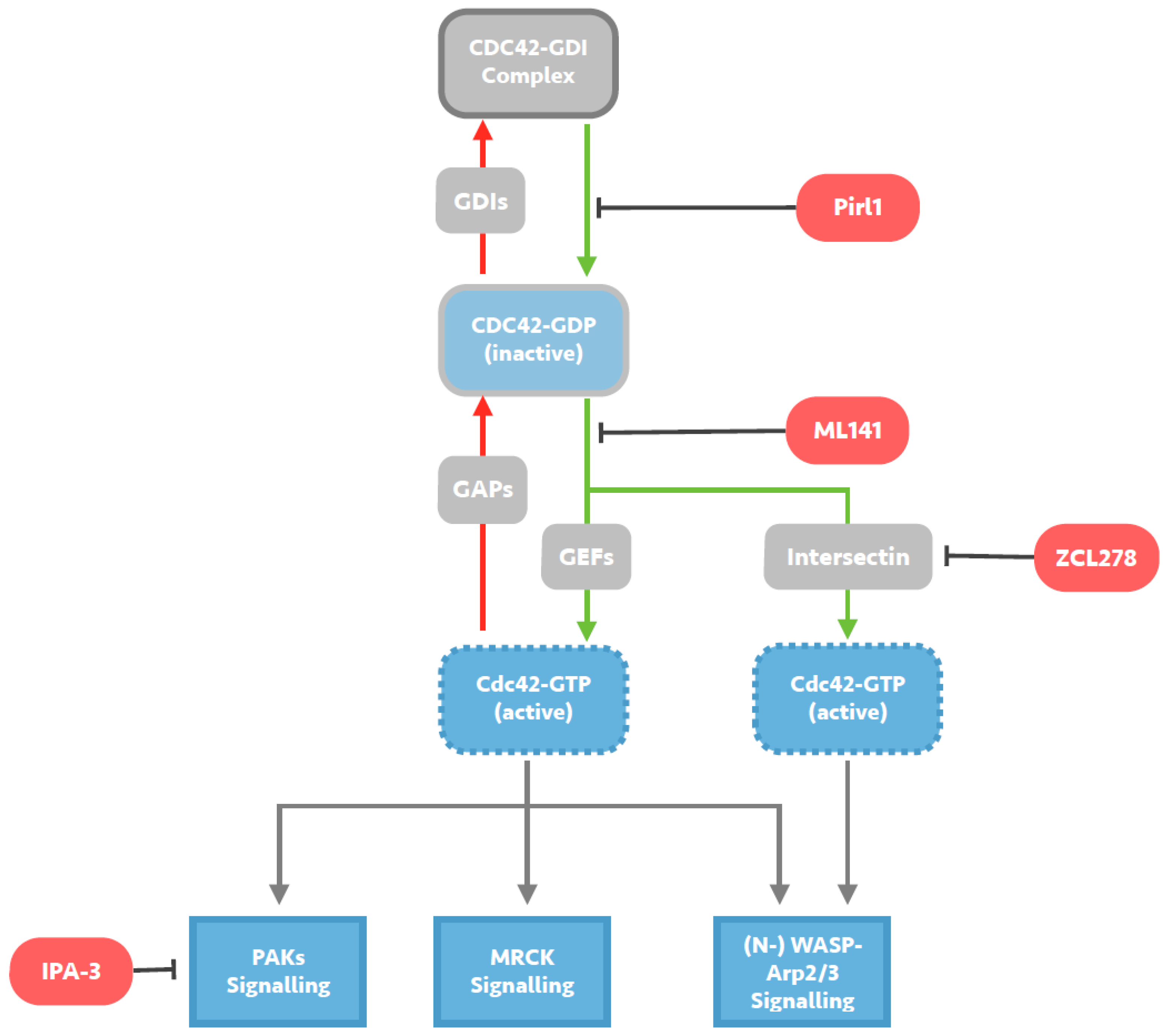
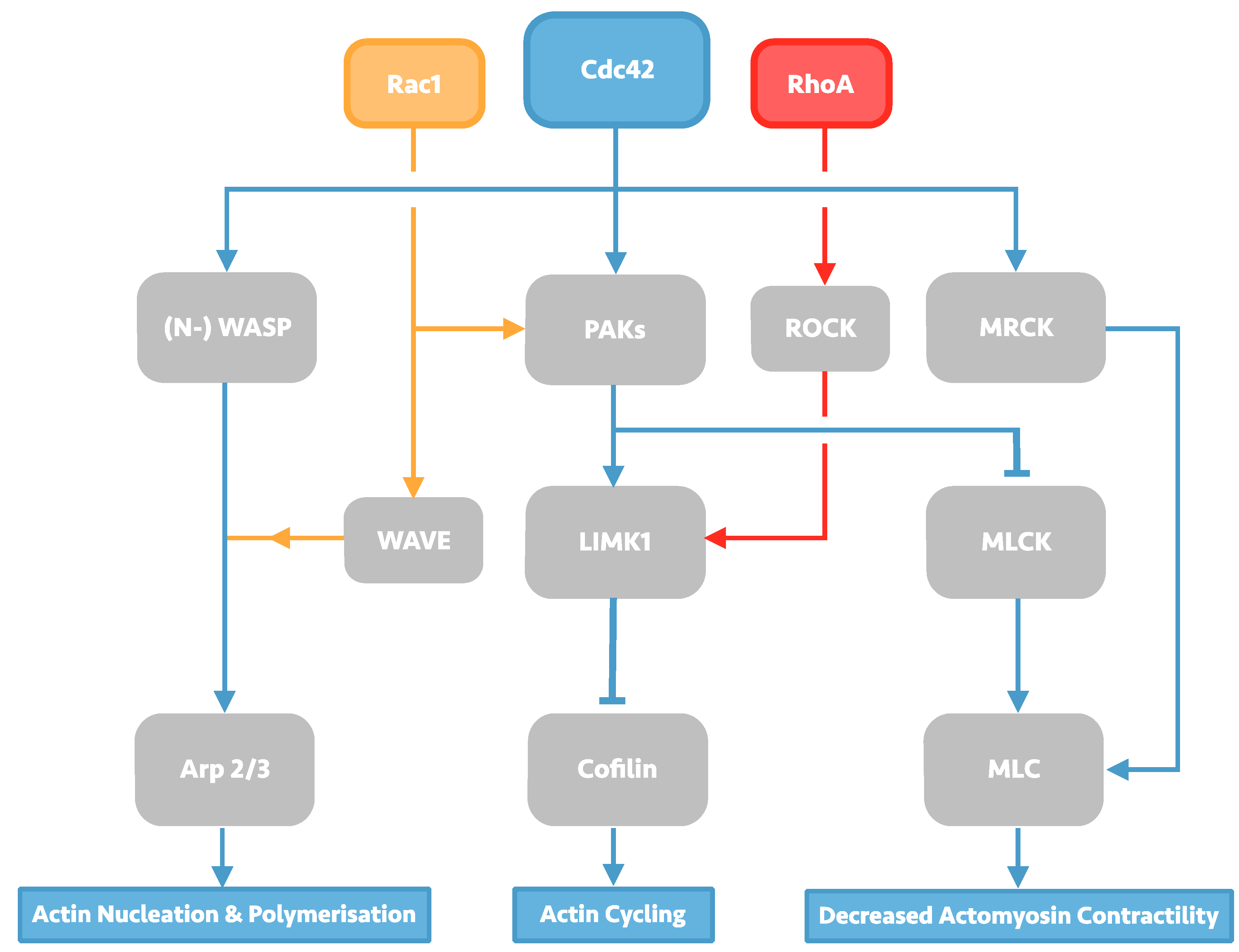
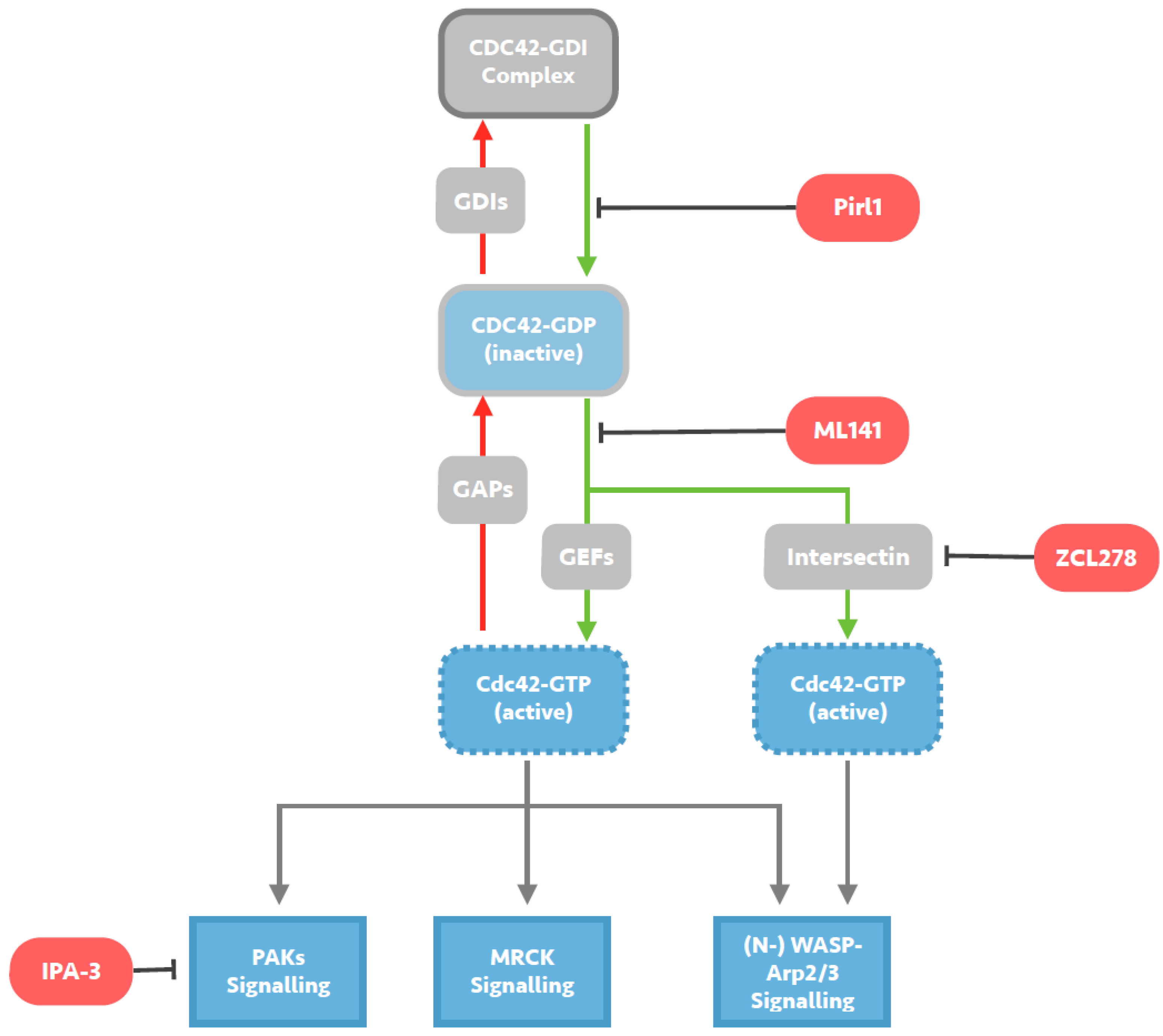
2. Investigation of the HIV-1 Entry Process
Investigating a Specific Role for CDC42 in HIV entry
3. Cdc42 Involvement in other RNA Virus Cell Entry
3.1. Respiratory Syncytial Virus
3.2. Rotaviruses
3.3. Coronaviruses
3.4. Ebola Virus
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Pantaloni, D.; Le Clainche, C.; Carlier, M.F. Mechanism of actin-based motility. Science 2001, 292, 1502–1506. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Coyne, C.B. The Actin Cytoskeleton as a Barrier to Virus Infection of Polarized Epithelial Cells. Viruses 2011, 3, 2462–2477. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Itoh, R.E.; Yoshizaki, H.; Nakamura, Y.O.T.; Matsuda, M. Coactivation of Rac1 and Cdc42 at Lamellipodia and Membrane Ruffles Induced by Epidermal Growth Factor. Mol. Biol. Cell 2004, 15, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Diebold, B.A.; Fowler, B.; Lu, J.; Dinauer, M.C.; Bokoch, G.M. Antagonistic cross-talk between Rac and Cdc42 GTPases regulates generation of reactive oxygen species. J. Biol. Chem. 2004, 279, 28136–28142. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Garcia-Mata, R.; Burridge, K. Rho protein crosstalk: Another social network? Trends Cell Biol. 2011, 21, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A. Jasplakinolide: An actin-specific reagent that promotes actin polymerization. Methods Mol. Biol. 2001, 161, 109–120. [Google Scholar] [PubMed]
- Voth, D.E.; Ballard, J.D. Clostridium difficile Toxins: Mechanism of Action and Role in Disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Orgaz, J.L.; Herraiz, C.; Sanz-Moreno, V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5, e29019. [Google Scholar] [CrossRef] [PubMed]
- Arias-Romero, L.E.; Chernoff, J. Targeting Cdc42 in cancer. Expert Opin. Ther. Targets 2013, 17, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.; Martin, T.; Weeks, H.P.; Jiang, W.G. Structure and role of WASP and WAVE in Rho GTPase signalling in cancer. Cancer Genom. Proteom. 2014, 11, 155–165. [Google Scholar]
- Wilson, K.F.; Erickson, J.W.; Antonyak, M.A.; Cerione, R.A. Rho GTPases and their roles in cancer metabolism. Trends Mol. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Samstag, Y.; Eibert, S.M.; Klemke, M.; Wabnitz, G.H. Actin cytoskeletal dynamics in T lymphocyte activation and migration. J. Leukoc. Biol. 2003, 73, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 2012, 18. [Google Scholar] [CrossRef] [PubMed]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S.Y. HIV-1 entry into quiescent primary lymphocytes: Molecular analysis reveals a labile, latent viral structure. Cell 2015, 61, 213–222. [Google Scholar] [CrossRef]
- Vatakis, D.N.; Nixon, C.C.; Zack, J.A. Quiescent T cells and HIV: An unresolved relationship. Immunol. Res. 2010, 48. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, L.; Burstein, E.; Guha-Niyogi, A.; Louder, M.K.; Mascola, J.R.; Klomp, L.W.J.; Wijmenga, C.; Duckett, C.S.; Nabel, G.J. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature 2003, 426, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Baldauf, H.-M.; Keppler, O.T.; Fackler, O.T. Restrictions to HIV-1 replication in resting CD4(+) T lymphocytes. Cell Res. 2013, 23, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.; Han, Y.; Zhou, Y.; Siliciano, J.; Siliciano, R.F. The multifactorial nature of HIV-1 latency. Trends Mol. Med. 2004, 10, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wind-Rotolo, M.; Yang, H.-C.; Siliciano, J.D.; Siliciano, R.F. Experimental approaches to the study of HIV-1 latency. Nat. Rev. Microbiol. 2007, 5, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, W.; Yoder, A.; Spear, M.; Wu, Y. The HIV envelope but not VSV glycoprotein is capable of mediating HIV latent infection of resting CD4 T cells. PLoS Pathog. 2009, 5, e1000633. [Google Scholar] [CrossRef] [PubMed]
- Cicala, C.; Arthos, J.; Selig, S.M.; Dennis, G.J.; Hosack, D.A.; Van Ryk, D.; Spangler, M.L.; Steenbeke, T.D.; Khazanie, P.; Gupta, N.; et al. HIV envelope induces a cascade of cell signals in non-proliferating target cells that favor virus replication. Proc. Natl. Acad. Sci. USA 2002, 99, 9380–9385. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Baranda, S.; Gomez-Mouton, C.; Rojas, A.; Martinez-Prats, L.; Mira, E.; Ana Lacalle, R.; Valencia, A.; Dimitrov, D.S.; Viola, A.; Delgado, R.; et al. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007, 9, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yoder, A.; Yu, D.; Wang, W.; Liu, J.; Barrett, T.; Wheeler, D.; Schlauch, K. Cofilin activation in peripheral CD4 T cells of HIV-1 infected patients: A pilot study. Retrovirology 2008, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.; Yu, D.; Dong, L.; Iyer, S.R.; Xu, X.; Kelly, J.; Liu, J.; Wang, W.; Vorster, P.J.; Agulto, L.; et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 2008, 134, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Barrero-Villar, M.; Cabrero, J.R.; Gordon-Alonso, M.; Barroso-Gonzalez, J.; Alvarez-Losada, S.; Munoz-Fernandez, M.A.; Sanchez-Madrid, F.; Valenzuela-Fernandez, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Vorster, P.J.; Guo, J.; Yoder, A.; Wang, W.; Zheng, Y.; Xu, X.; Yu, D.; Spear, M.; Wu, Y. LIM kinase 1 modulates cortical actin and CXCR4 cycling and is activated by HIV-1 to initiate viral infection. J. Biol. Chem. 2011, 286, 12554–12564. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Guo, J.; Wu, Y. The trinity of the cortical actin in the initiation of HIV-1 infection. Retrovirology 2012, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Guo, J.; Turner, A.; Yu, D.; Wang, W.; Meltzer, B.; He, S.; Hu, X.; Shang, H.; Kuhn, J.; et al. HIV-1 triggers WAVE2 phosphorylation in primary CD4 T cells and macrophages, mediating Arp2/3-dependent nuclear migration. J. Biol. Chem. 2014, 289, 6949–6959. [Google Scholar] [CrossRef] [PubMed]
- Pontow, S.; Harmon, B.; Campbell, N.; Ratner, L. Antiviral activity of a Rac GEF inhibitor characterized with a sensitive HIV/SIV fusion assay. Virology 2007, 368, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Denton, P.W.; Othieno, F.; Martinez-Torres, F.; Zou, W.; Krisko, J.F.; Fleming, E.; Zein, S.; Powell, D.A.; Wahl, A.; Kwak, Y.T.; et al. One percent tenofovir applied topically to humanized BLT mice and used according to the CAPRISA 004 experimental design demonstrates partial protection from vaginal HIV infection, validating the BLT model for evaluation of new microbicide candidates. J. Virol. 2011, 85, 7582–7593. [Google Scholar] [CrossRef] [PubMed]
- Amy, C.; Dittmar, M.T. The Role of GTPases and Their Regulators within Early Steps of the HIV-1 Lifecycle, and Their Potential as a Target against HIV-1 Infection; Queen Mary University: London, UK, 2013. [Google Scholar]
- Swaine, T.; Dittmar, M.T. The Effect of Pharmacological Inhibition of RhoA and Cdc42 on the Early Steps of the HIV-1 Infection Lifecycle; Queen Mary University: London, UK, 2014. [Google Scholar]
- Chen, C.; Song, X.; Ma, S.; Wang, X.; Xu, J.; Zhang, H.; Wu, Q.; Zhao, K.; Cao, J.; Qiao, J.; et al. Cdc42 inhibitor ML141 enhances G-CSF-induced hematopoietic stem and progenitor cell mobilization. Int. J. Hematol. 2015, 101, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Waller, A.; Strouse, J.J.; Bologa, C.; Ursu, O.; Salas, V.; Parkinson, J.F.; Phillips, G.K.; Romero, E.; Wandinger-Ness, A.; et al. A Potent and Selective Inhibitor of Cdc42 GTPase. In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Chen, H.-Y.; Yang, Y.M.; Stevens, B.M.; Noble, M. Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol. Med. 2013, 5, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Friesland, A.; Zhao, Y.; Chen, Y.-H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Malinowsky, K.; Luksza, J.; Dittmar, M.T. Susceptibility to virus-cell fusion at the plasma membrane is reduced through expression of HIV gp41 cytoplasmic domains. Virology 2008, 376, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Lohrengel, S.; Hermann, F.; Hagmann, I.; Oberwinkler, H.; Scrivano, L.; Hoffmann, C.; von Laer, D.; Dittmar, M.T. Determinants of human immunodeficiency virus type 1 resistance to membrane-anchored gp41-derived peptides. J. Virol. 2005, 79, 10237–10246. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Noth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N.K.; Jenna, S.; Glogauer, M.; Quinn, C.C.; Wasiak, S.; Guipponi, M.; Antonarakis, S.E.; Kay, B.K.; Stossel, T.P.; Lamarche-Vane, N.; et al. Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nat. Cell Biol. 2001, 3, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Jenna, S.; Hussain, N.K.; Danek, E.I.; Triki, I.; Wasiak, S.; McPherson, P.S.; Lamarche-Vane, N. The activity of the GTPase-activating protein CdGAP is regulated by the endocytic protein intersectin. J. Biol. Chem. 2002, 277, 6366–6373. [Google Scholar] [CrossRef] [PubMed]
- Klein, I.K.; Predescu, D.N.; Sharma, T.; Knezevic, I.; Malik, A.B.; Predescu, S. Intersectin-2L regulates caveola endocytosis secondary to Cdc42-mediated actin polymerization. J. Biol. Chem. 2009, 284, 25953–25961. [Google Scholar] [CrossRef] [PubMed]
- McGavin, M.K.; Badour, K.; Hardy, L.A.; Kubiseski, T.J.; Zhang, J.; Siminovitch, K.A. The intersectin 2 adaptor links Wiskott Aldrich Syndrome protein (WASp)-mediated actin polymerization to T cell antigen receptor endocytosis. J. Exp. Med. 2001, 194, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Srinivasakumar, N.; Ogra, P.L.; Flanagan, T.D. Characteristics of fusion of respiratory syncytial virus with HEp-2 cells as measured by R18 fluorescence dequenching assay. J. Virol. 1991, 65, 4063–4069. [Google Scholar] [PubMed]
- Ohki, S.; Liu, J.-Z.; Schaller, J.; Welliver, R.C. The compound DATEM inhibits respiratory syncytial virus fusion activity with epithelial cells. Antivir. Res. 2003, 58, 115–124. [Google Scholar] [CrossRef]
- Razinkov, V.; Huntley, C.; Ellestad, G.; Krishnamurthy, G. RSV entry inhibitors block F-protein mediated fusion with model membranes. Antivir. Res. 2002, 55, 189–200. [Google Scholar] [CrossRef]
- Huang, K.; Incognito, L.; Cheng, X.; Ulbrandt, N.D.; Wu, H. Respiratory Syncytial Virus-Neutralizing Monoclonal Antibodies Motavizumab and Palivizumab Inhibit Fusion. J. Virol. 2010, 84, 8132–8140. [Google Scholar] [CrossRef] [PubMed]
- Kolokoltsov, A.A.; Deniger, D.; Fleming, E.H.; Roberts, N.J.; Karpilow, J.M.; Davey, R.A. Small Interfering RNA Profiling Reveals Key Role of Clathrin-Mediated Endocytosis and Early Endosome Formation for Infection by Respiratory Syncytial Virus. J. Virol. 2007, 81, 7786–7800. [Google Scholar] [CrossRef] [PubMed]
- San-Juan-Vergara, H.; Sampayo-Escobar, V.; Reyes, N.; Cha, B.; Pacheco-Lugo, L.; Wong, T.; Peeples, M.E.; Collins, P.L.; Castano, M.E.; Mohapatra, S.S. Cholesterol-rich microdomains as docking platforms for respiratory syncytial virus in normal human bronchial epithelial cells. J. Virol. 2012, 86, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Krzyzaniak, M.A.; Zumstein, M.T.; Gerez, J.A.; Picotti, P.; Helenius, A. Host Cell Entry of Respiratory Syncytial Virus Involves Macropinocytosis Followed by Proteolytic Activation of the F Protein. PLoS Pathog. 2013, 9, e1003309. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.R.; Lebensohn, A.M.; Pelish, H.E.; Kirschner, M.W. Biochemical suppression of small molecule inhibitors: A new strategy to identify inhibitor targets and signaling pathway components. Chem. Biol. 2006, 13, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Cuadras, M.A.; Arias, C.F.; Lopez, S. Rotaviruses induce an early membrane permeabilization of MA104 cells and do not require a low intracellular Ca2+ concentration to initiate their replication cycle. J. Virol. 1997, 71, 9065–9074. [Google Scholar] [PubMed]
- Bass, D.M.; Baylor, M.; Chen, C.; Upadhyayula, U. Dansylcadaverine and cytochalasin D enhance rotavirus infection of murine L cells. Virology 1995, 212, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kaljot, K.T.; Shaw, R.D.; Rubin, D.H.; Greenberg, H.B. Infectious rotavirus enters cells by direct cell membrane penetration, not by endocytosis. J. Virol. 1988, 62, 1136–1144. [Google Scholar] [PubMed]
- Sánchez-San Martín, C.; López, T.; Arias, C.F.; López, S. Characterization of Rotavirus Cell Entry. J. Virol. 2004, 78, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Salinas, M.A.; Romero, P.; Espinosa, R.; Hoshino, Y.; López, S.; Arias, C.F. The Spike Protein VP4 Defines the Endocytic Pathway Used by Rotavirus To Enter MA104 Cells. J. Virol. 2013, 87, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, M.; Isa, P.; Sánchez-San Martín, C.; Pérez-Vargas, J.; Espinosa, R.; Arias, C.F.; López, S. Different Rotavirus Strains Enter MA104 Cells through Different Endocytic Pathways: The Role of Clathrin-Mediated Endocytosis. J. Virol. 2010, 84, 9161–9169. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Salinas, M.A.; Silva-Ayala, D.; López, S.; Arias, C.F. Rotaviruses Reach Late Endosomes and Require the Cation-Dependent Mannose-6-Phosphate Receptor and the Activity of Cathepsin Proteases To Enter the Cell. J. Virol. 2014, 88, 4389–4402. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Nomura, R.; Kiyota, A.; Suzaki, E.; Kataoka, K.; Ohe, Y.; Miyamoto, K.; Senda, T.; Fujimoto, T. Human Coronavirus 229E Binds to CD13 in Rafts and Enters the Cell through Caveolae. J. Virol. 2004, 78, 8701–8708. [Google Scholar] [CrossRef] [PubMed]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Hahn, K.M.; Grinstein, S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrowicz, P.; Marzi, A.; Biedenkopf, N.; Beimforde, N.; Becker, S.; Hoenen, T.; Feldmann, H.; Schnittler, H.-J. Ebola Virus Enters Host Cells by Macropinocytosis and Clathrin-Mediated Endocytosis. J. Infect. Dis. 2011, 204, S957–S967. [Google Scholar] [CrossRef] [PubMed]
- Quinn, K.; Brindley, M.A.; Weller, M.L.; Kaludov, N.; Kondratowicz, A.; Hunt, C.L.; Sinn, P.L.; McCray, P.B.; Stein, C.S.; Davidson, B.L.; et al. Rho GTPases Modulate Entry of Ebola Virus and Vesicular Stomatitis Virus Pseudotyped Vectors. J. Virol. 2009, 83, 10176–10186. [Google Scholar] [CrossRef] [PubMed]
- Mulherkar, N.; Raaben, M.; de la Torre, J.C.; Whelan, S.P.; Chandran, K. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 2011, 419, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Guo, J.; Wu, Y. Novel anti-HIV therapeutics targeting chemokine receptors and actin regulatory pathways. Immunol. Rev. 2013, 256, 300–312. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swaine, T.; Dittmar, M.T. CDC42 Use in Viral Cell Entry Processes by RNA Viruses. Viruses 2015, 7, 6526-6536. https://doi.org/10.3390/v7122955
Swaine T, Dittmar MT. CDC42 Use in Viral Cell Entry Processes by RNA Viruses. Viruses. 2015; 7(12):6526-6536. https://doi.org/10.3390/v7122955
Chicago/Turabian StyleSwaine, Thomas, and Matthias T. Dittmar. 2015. "CDC42 Use in Viral Cell Entry Processes by RNA Viruses" Viruses 7, no. 12: 6526-6536. https://doi.org/10.3390/v7122955
APA StyleSwaine, T., & Dittmar, M. T. (2015). CDC42 Use in Viral Cell Entry Processes by RNA Viruses. Viruses, 7(12), 6526-6536. https://doi.org/10.3390/v7122955