
Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells


Abstract
:1. Virology and Electron Microscopy (EM)
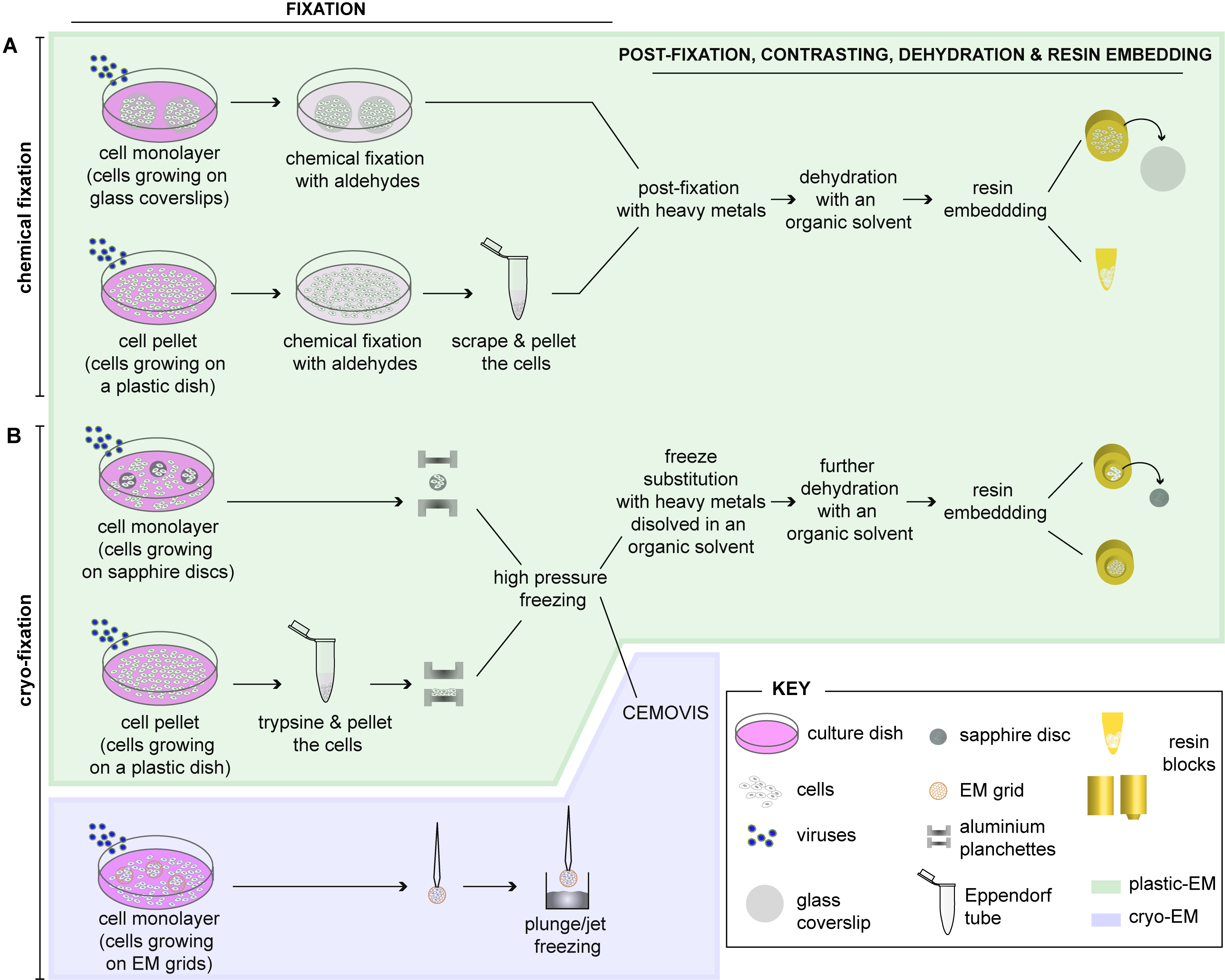
2. Preparation of Cells for EM
2.1. Fixation of Cells
2.1.1. Chemical Fixation
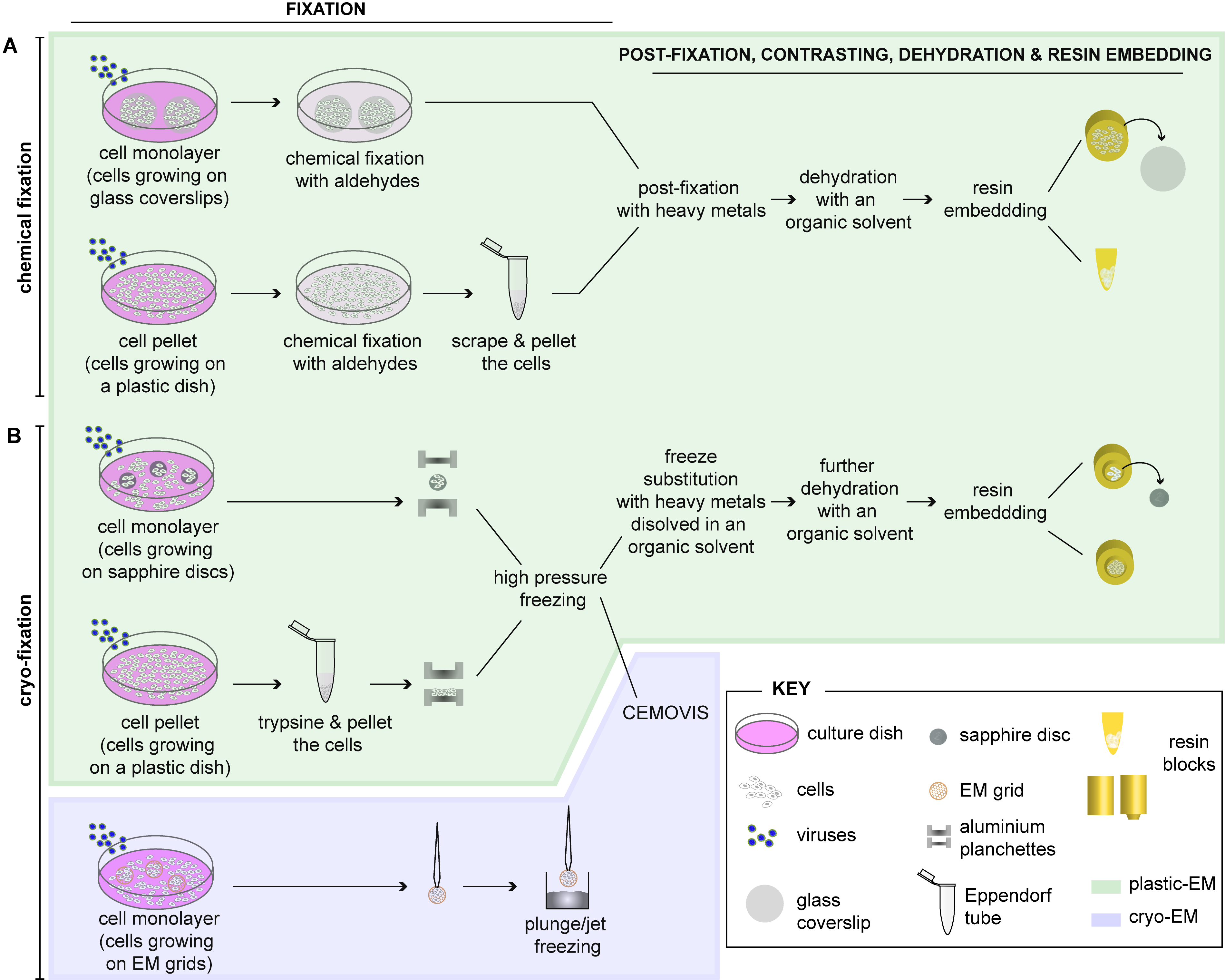
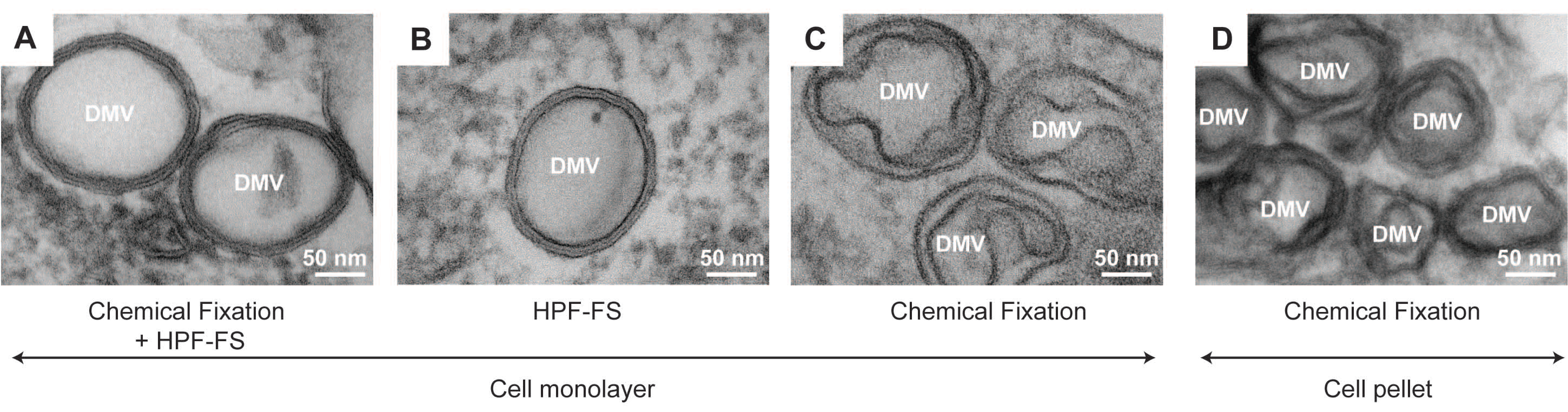
2.1.2. Cryo-Fixation
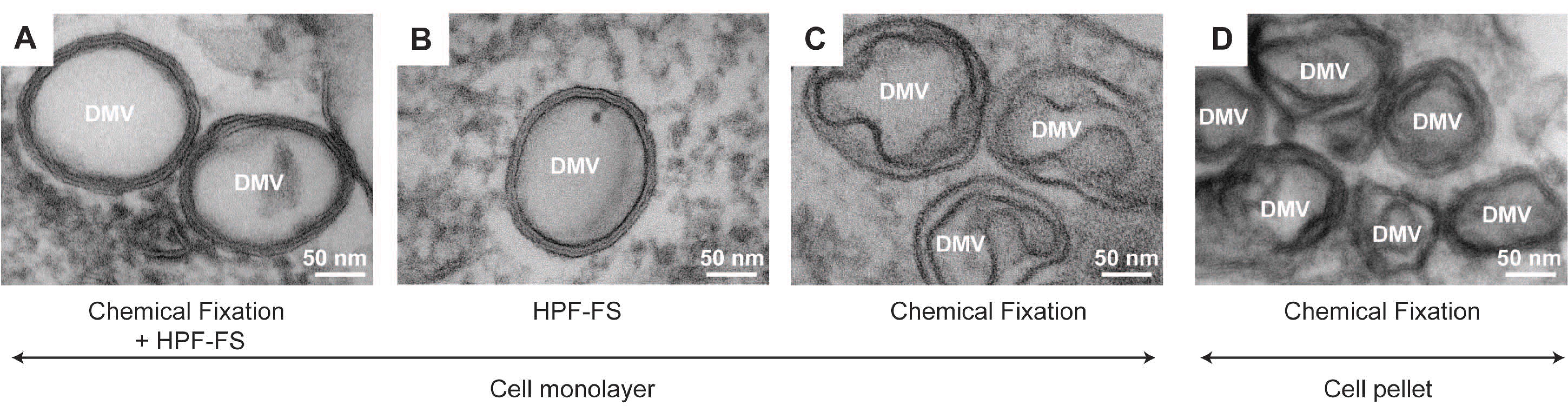
2.1.3. Combination of Chemical and Cryo-Fixation
2.2. Embedding of Cells
2.2.1. Embedding of Chemically Fixed Cells
2.2.2. Embedding of Cryo-Fixed Cells
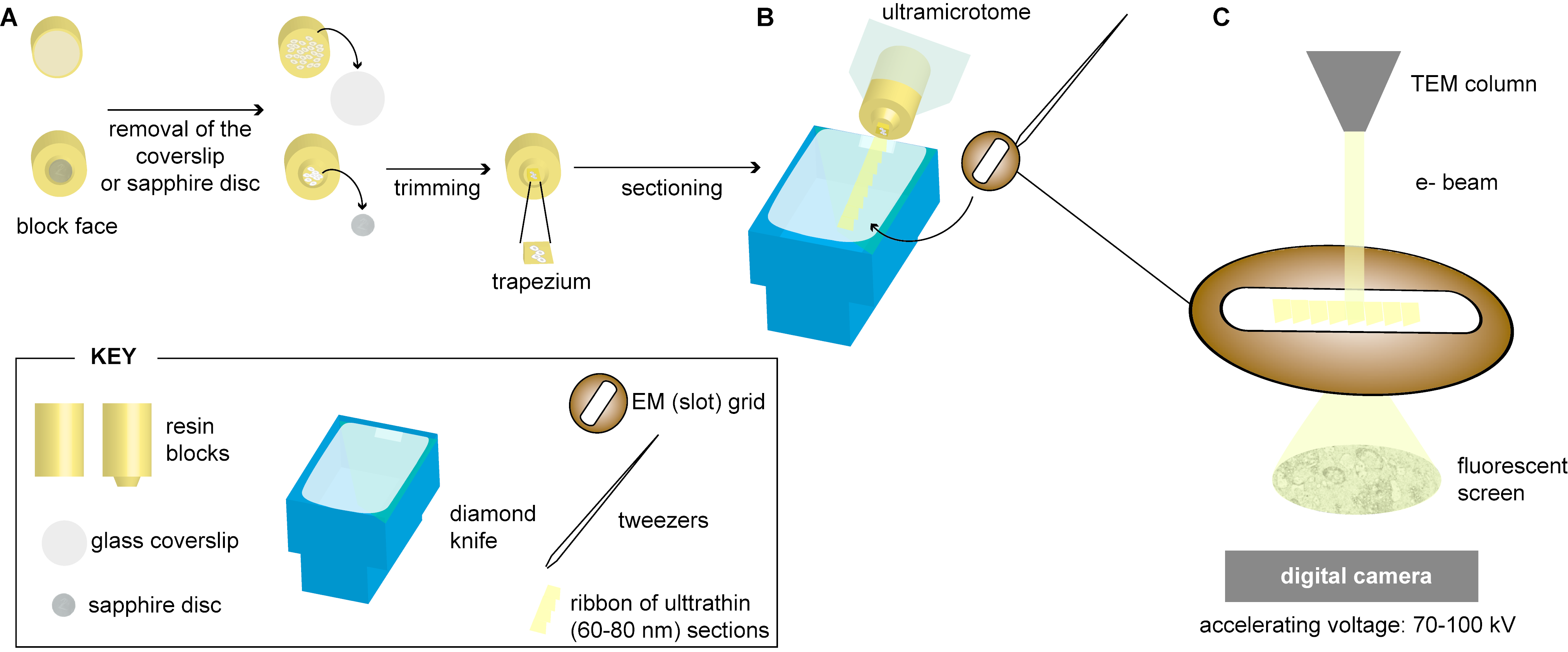
2.3. Sectioning of Resin-Embedded Cells
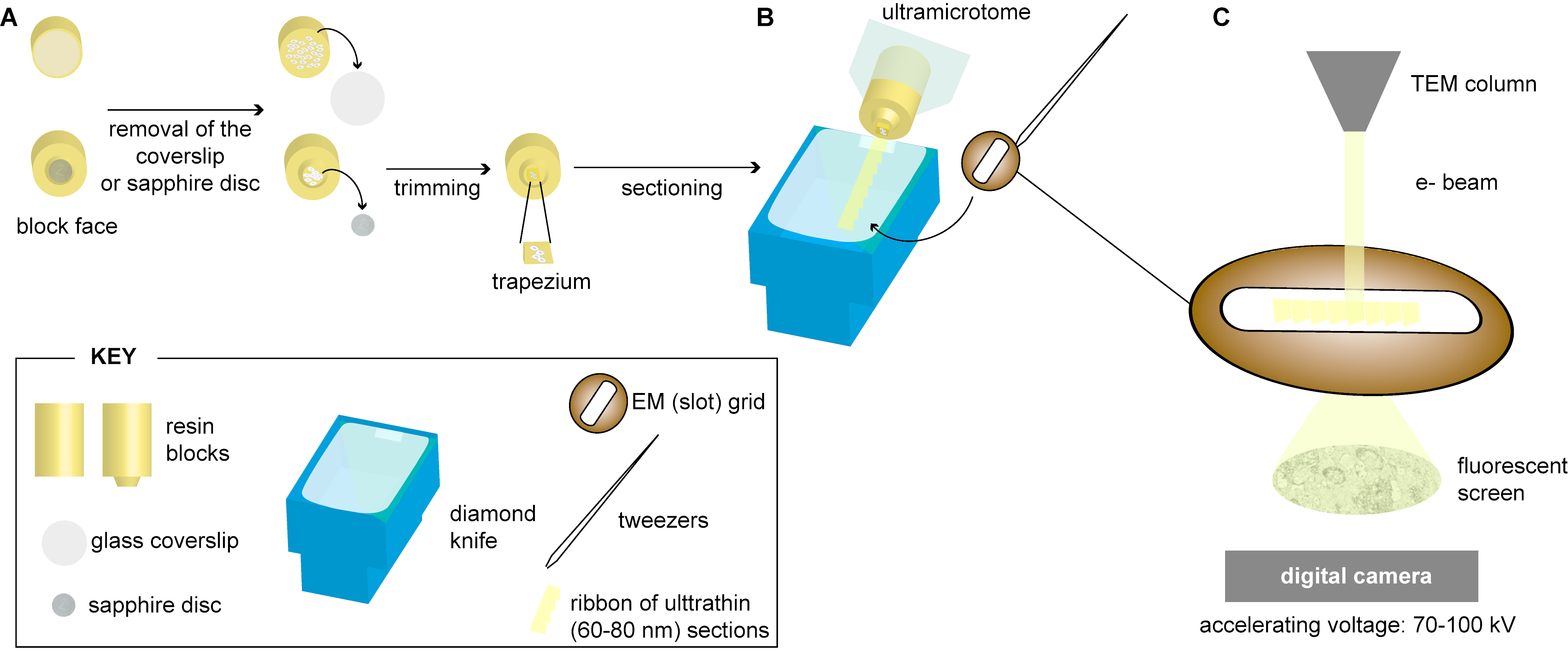
2.4. Resin-Free Processing of Cells
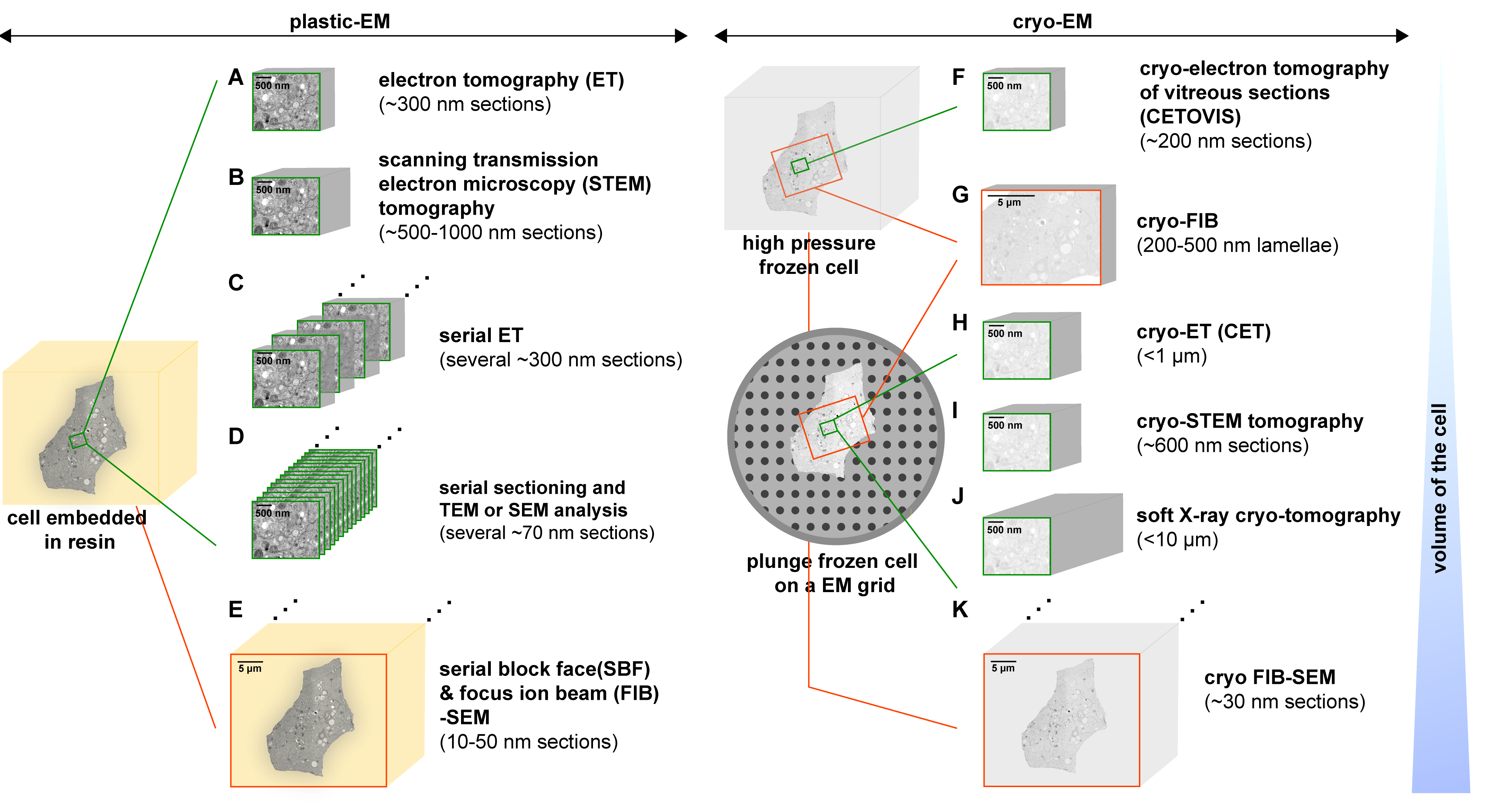
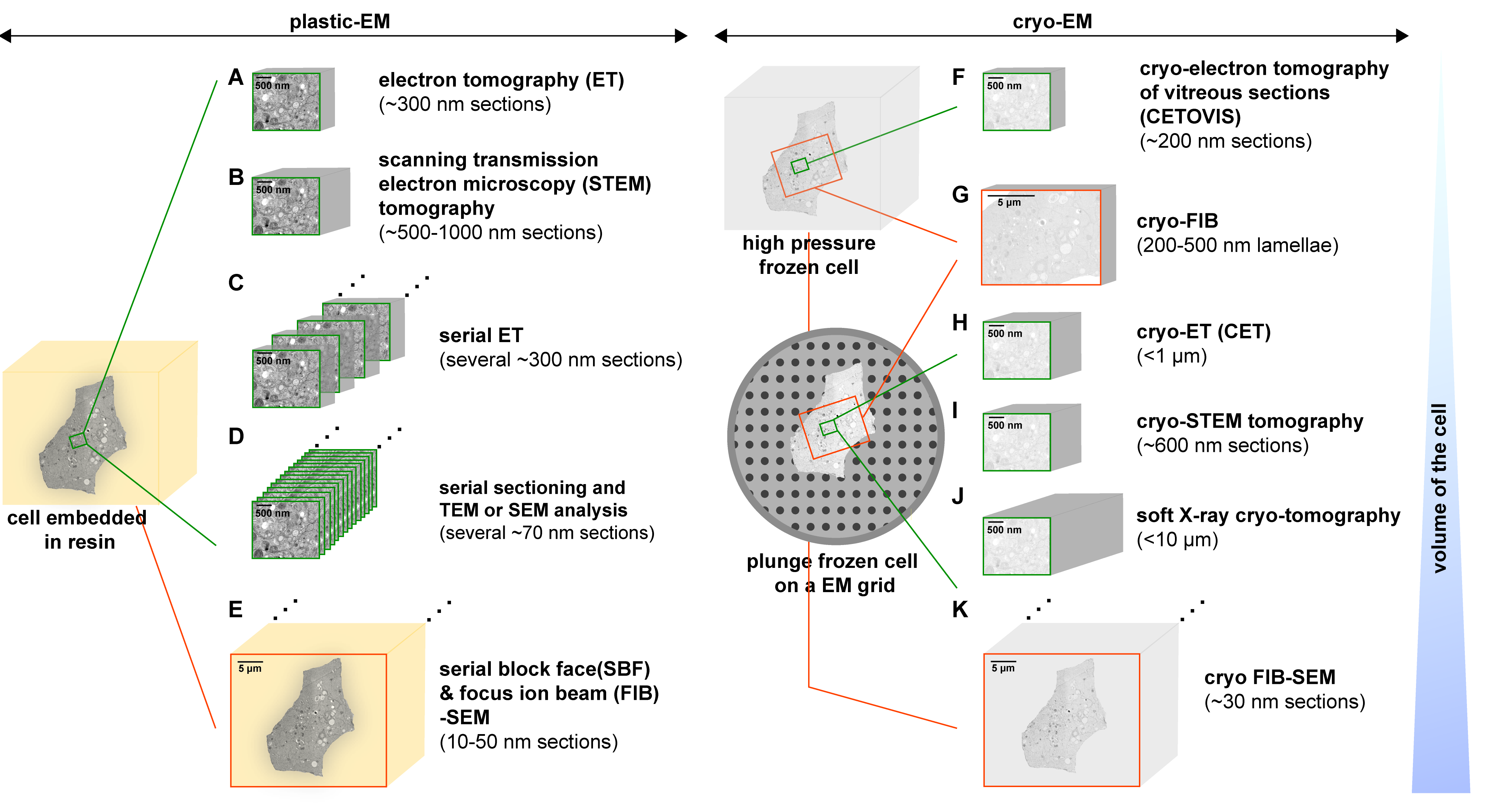
3. Methods to Study the 3D Architecture of Virus-Infected Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3D-EM Method | Virus | Cells * | Step of the Virus Life Cycle Analyzed | Reference |
|---|---|---|---|---|
| Electron Tomography (ET) | Flock House Virus (FHV) | Drosophila S2 | Replication | [91] |
| Herpes Simplex Virus type 1 (HSV-1) | HEp-2 | Assembly (Envelopment) | [92] | |
| Rice Dwarf Virus (RDV) | Leafhopper vector | Release | [93] | |
| Human Immunodeficiency Virus-1 (HIV)-1 & Simian Immunodeficiency Virus (SIV) | T cells | Entry | [94] | |
| SARS-Coronavirus | Vero E6 | Replication | [95] | |
| HIV-1 | MT-4 & HeLa | Assembly & Budding | [96] | |
| Human T-Lymphotropic Virus 1 (HTLV-1) | MS9 & Jurkat | Spread | [97] | |
| Vaccinia Virus (VV) | HeLa | Assembly | [98] | |
| Dengue Virus (DENV) | Huh-7 | Replication & Assembly | [99] | |
| VV | HeLa spinner | Assembly | [100] | |
| Rice Gall Dwarf Virus (RGDV) | VCM | Release | [101] | |
| Murine Gammaherpesvirus | NIH 3T3 | Attachment, Entry, Assembly & Egress | [102] | |
| West Nile Virus (WNV) | Vero | Replication | [103] | |
| DENV | C6/36, Vero & SK Hep1 | Replication | [104] | |
| SARS-Coronavirus | Vero E6 | Replication | [105] | |
| Rubella Virus | BHK-21 | Replication | [54] | |
| Marburg Virus (MARV) | Huh-7 | Budding & Release | [106] | |
| Semliki Forest Virus (SFV) | BHK-21 | Budding | [107] | |
| Coxsackievirus B3 (CVB3) | Vero E6 | Replication | [108] | |
| RGDV | NC-24 | Replication | [109] | |
| Equine Arterivirus (EAV) | Vero E6 | Replication | [110] | |
| Poliovirus type I | HeLa | Replication | [111] | |
| Hepatitis C Virus (HCV) | Huh 7.5 | Replication | [20] | |
| Langat Virus | Vero & ISE6 | Replication & Assembly | [112] | |
| Tick Borne Encephalitis Virus (TBEV) | BHK-21 | Replication & Assembly | [113] | |
| Infectious Bronchitis Virus (IBV) | CK | Replication | [114] | |
| VV & Mimivirus | HeLa & A. polyphaga | Assembly | [115] | |
| WNV | BHK | Assembly | [116] | |
| DENV | C6/36 | Replication & Assembly | [117] | |
| Beet Black Scorch Virus (BBSV) | Nicotiana benthamiana | Replication | [118] | |
| HIV-1 | Gut-associated lymphoid tissue | Budding, Release & Cell-to-cell transmission | [119] | |
| WNV | Vero | Replication & Assembly | [120] | |
| Autographa californica Multiple Nucleopolyhedrovirus (AcMNPV) | Sf9 | Assembly (Envelopment) | [121] | |
| TBEV | HN | Replication & Assembly | [122] | |
| African Swine Fever Virus (ASFV) | COS | Replication & Assembly | [123] | |
| Scanning Transmission Electron Microscopy (STEM) tomography | Mimivirus | Acanthamoeba polyphaga | Replication & Assembly | [124] |
| ASFV | COS | Replication & Assembly | [123] | |
| Paramecium Bursaria Chlorella Virus 1 (PBCV-1) | Chlorella variabilis | Replication & Assembly | [125] | |
| Serial Sectioning | VV | PtK2 | Replication & Assembly | [90] |
| Bunyavirus | BHK-21 | Replication & Assembly | [126] | |
| Varicella-Zoster Virus (VZV) | MeWo | Assembly | [127] | |
| HCV | Huh 7.5 | Replication & Assembly | [128] | |
| Human Cytomegalovirus –(HCMV) | Fibroblasts, human endothelial cells & macrophages | Assembly (Envelopment) | [50] | |
| Reovirus | HeLa | Assembly | [129] | |
| Zucchini Yellow Mosaic Virus (ZYMV) | Cucurbita pepo L. plant cells | Spread | [130] | |
| Focus Ion Beam-Scanning Electron Microscopy (FIB-SEM) | HIV-1 | Macrophages | Release | [131] |
| HIV-1 | T & dendritic cells | Cell-to-cell transmission | [132] | |
| HIV-1 | T cells & astrocytes | Cell-to-cell transmission | [133] | |
| PBCV-1 | Chlorella variabilis | Replication & Assembly | [125] | |
| Cryo-ET (CET) | VV | Ptk2 | Disassembly | [134] |
| HSV-1 | Vero, PtK2, and HFF | Entry | [135] | |
| HIV-1 | MDM | Release | [131] | |
| HIV-1 | U-87 MG and U-373 MG | Assembly & Budding | [136] | |
| MARV | Huh-7 | Assembly & Budding | [137] | |
| HSV-1 | Hippocampal neurons | Assembly (Envelopment) | [138] | |
| Influenza A Virus | MDCK | Budding | [139] | |
| Bacteriophage T7 | Escherichia coli | Entry | [140] | |
| RDV | NC24 | Egress | [141] | |
| Syn5 Cyanophage | WH8109 | Assembly | [142] | |
| Baculovirus | B16 | Spread | [143] | |
| HIV-1 | HUVEC | Release | [144] | |
| Soft X-ray Cryo Tomography | VV | PtK2 | Replication & Assembly | [90] |
| Pseudorabies Virus (PrV) | EFN-R | Assembly (Envelopment) | [145] |
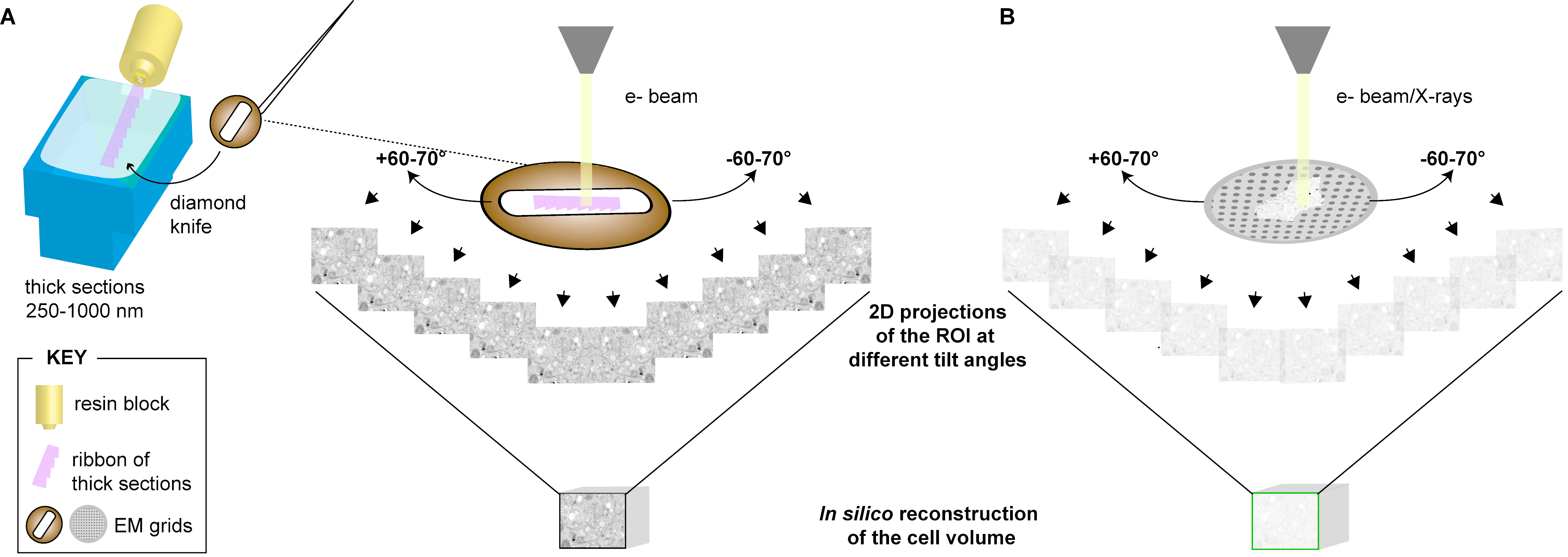
3.1. Electron Tomography (ET)
3.1.1. Description
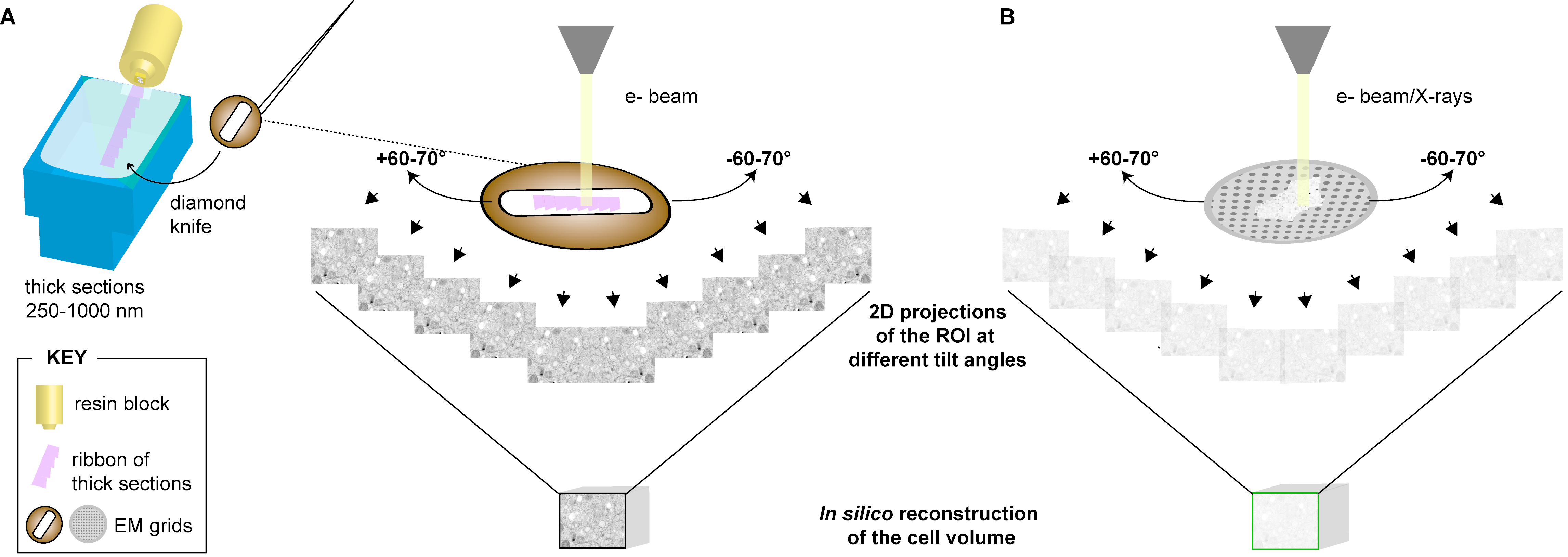
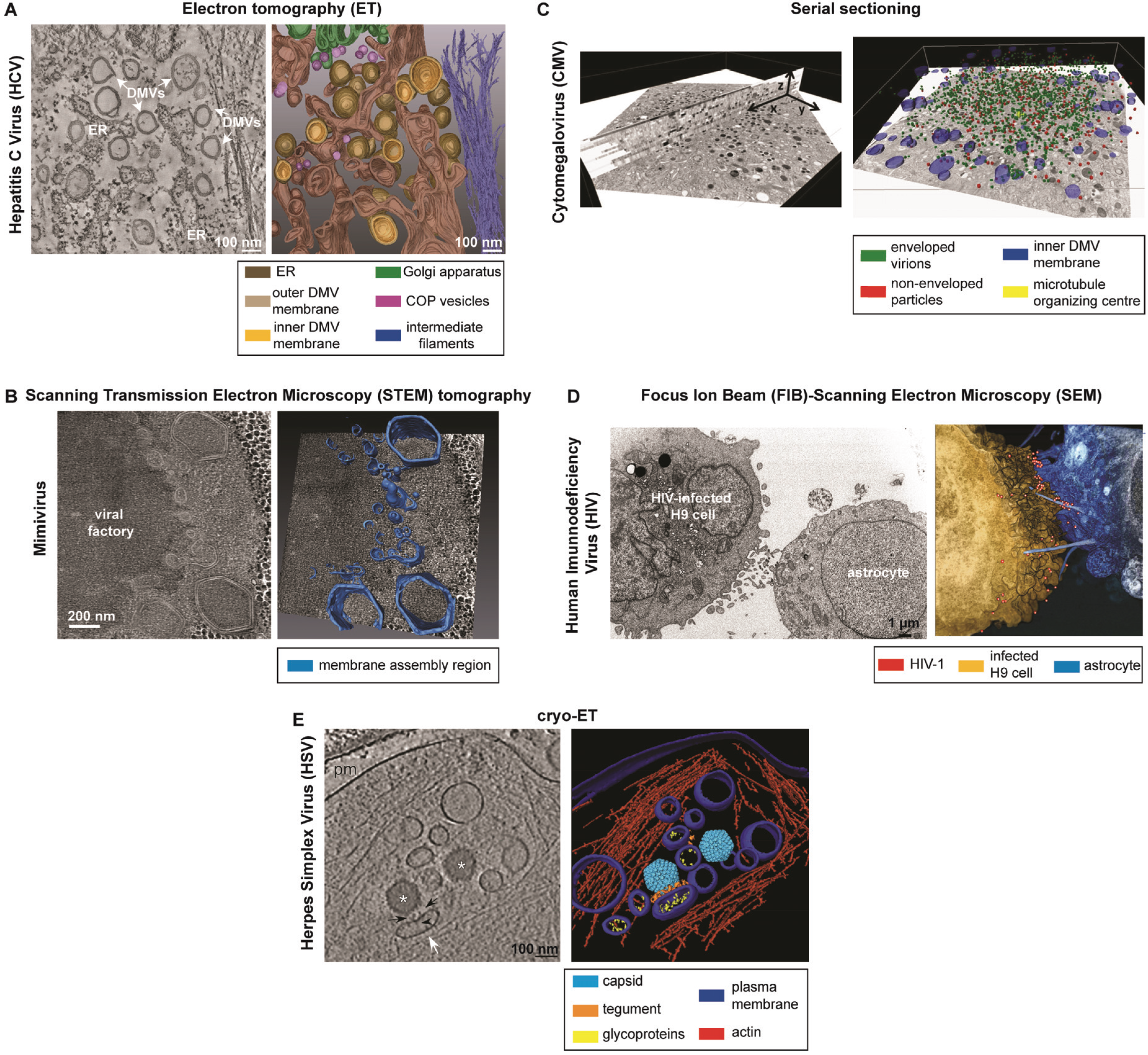
3.1.2. Applications to the Study of Virus-Infected Cells
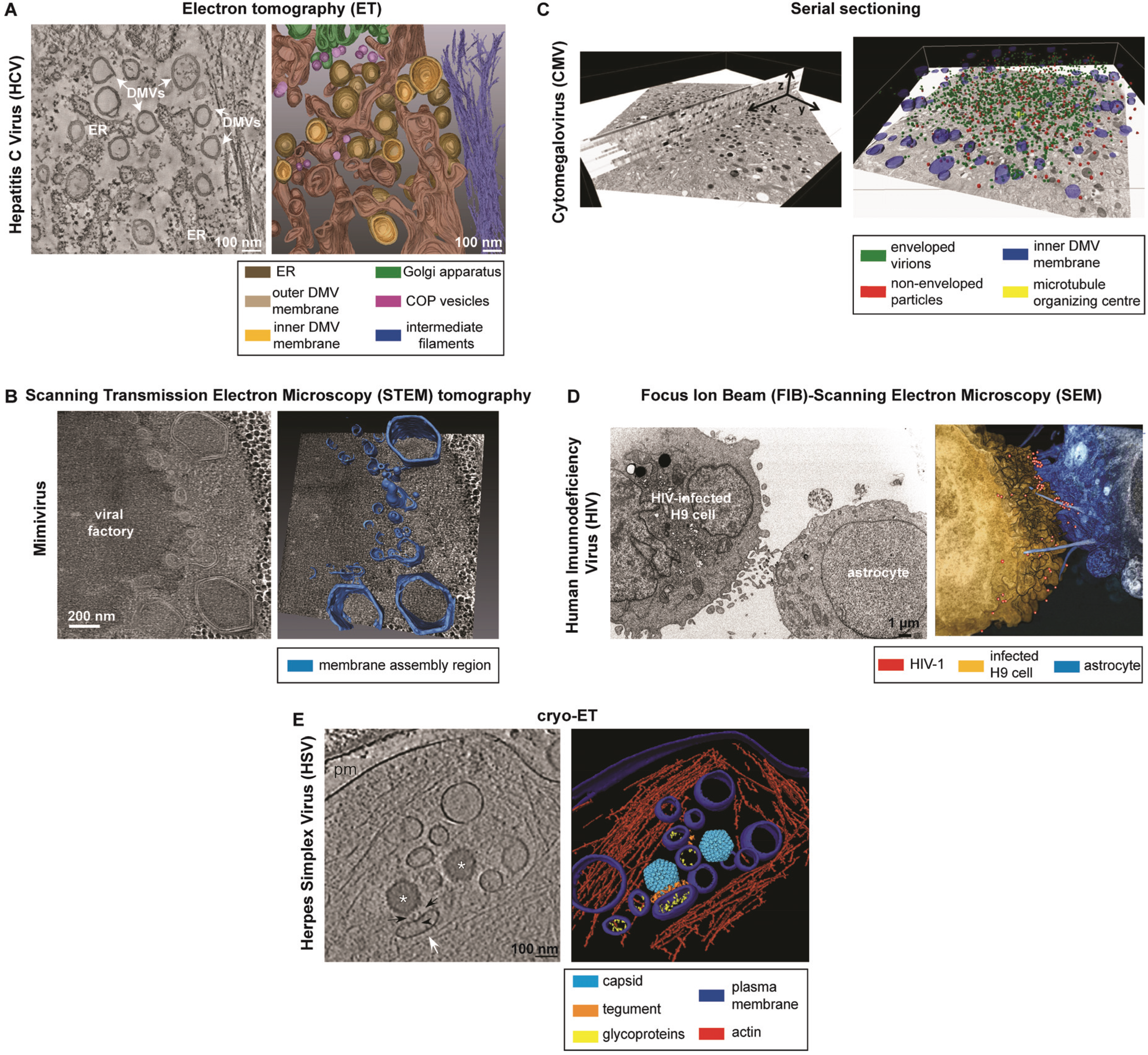
3.2. Scanning Transmission Electron Microscopy (STEM) Tomography
3.2.1. Description
3.2.2. Applications to the Study of Virus-Infected Cells
3.3. Serial Sectioning
3.3.1. Description
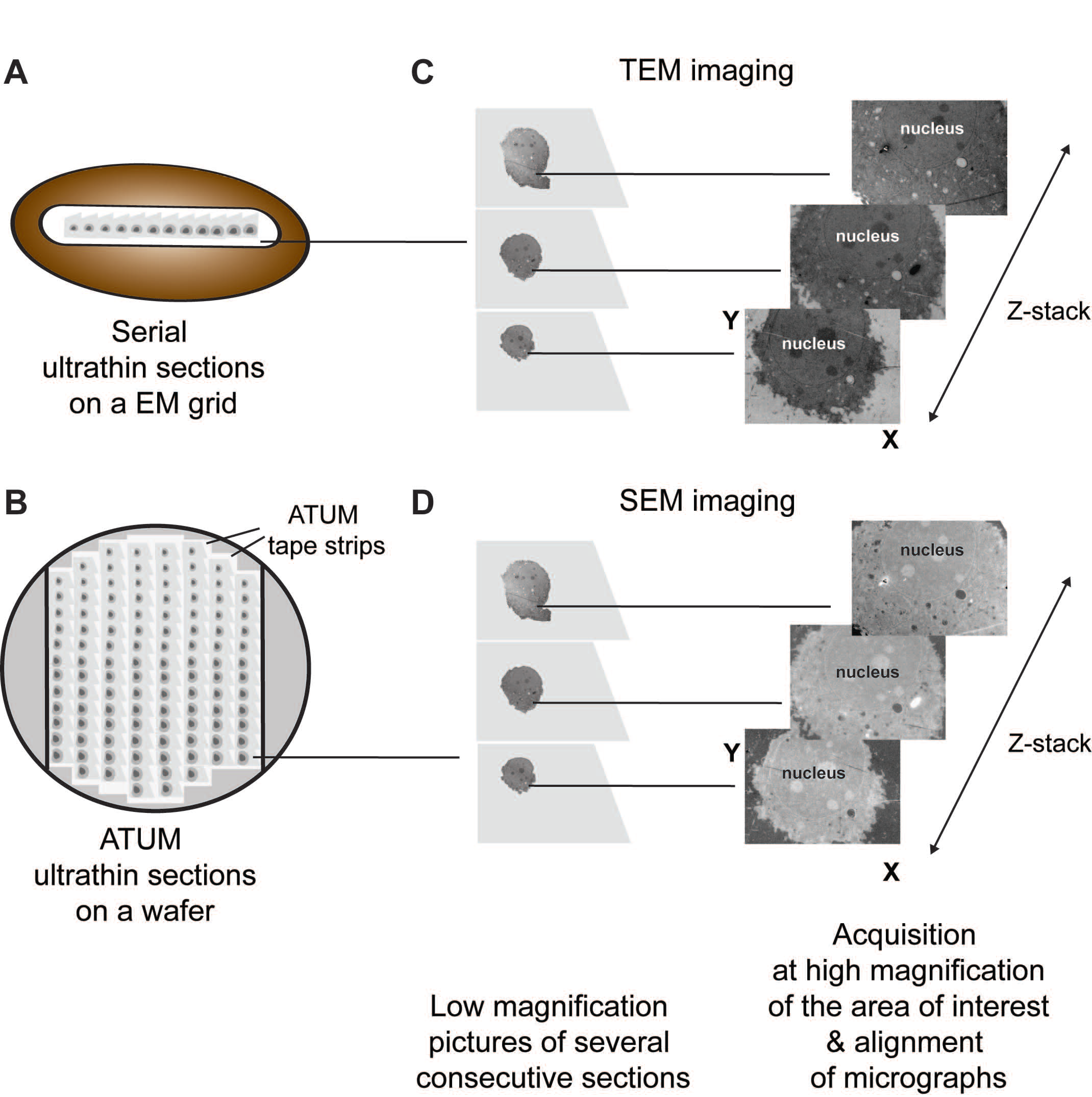
3.3.2. Applications to the Study of Virus-Infected Cells
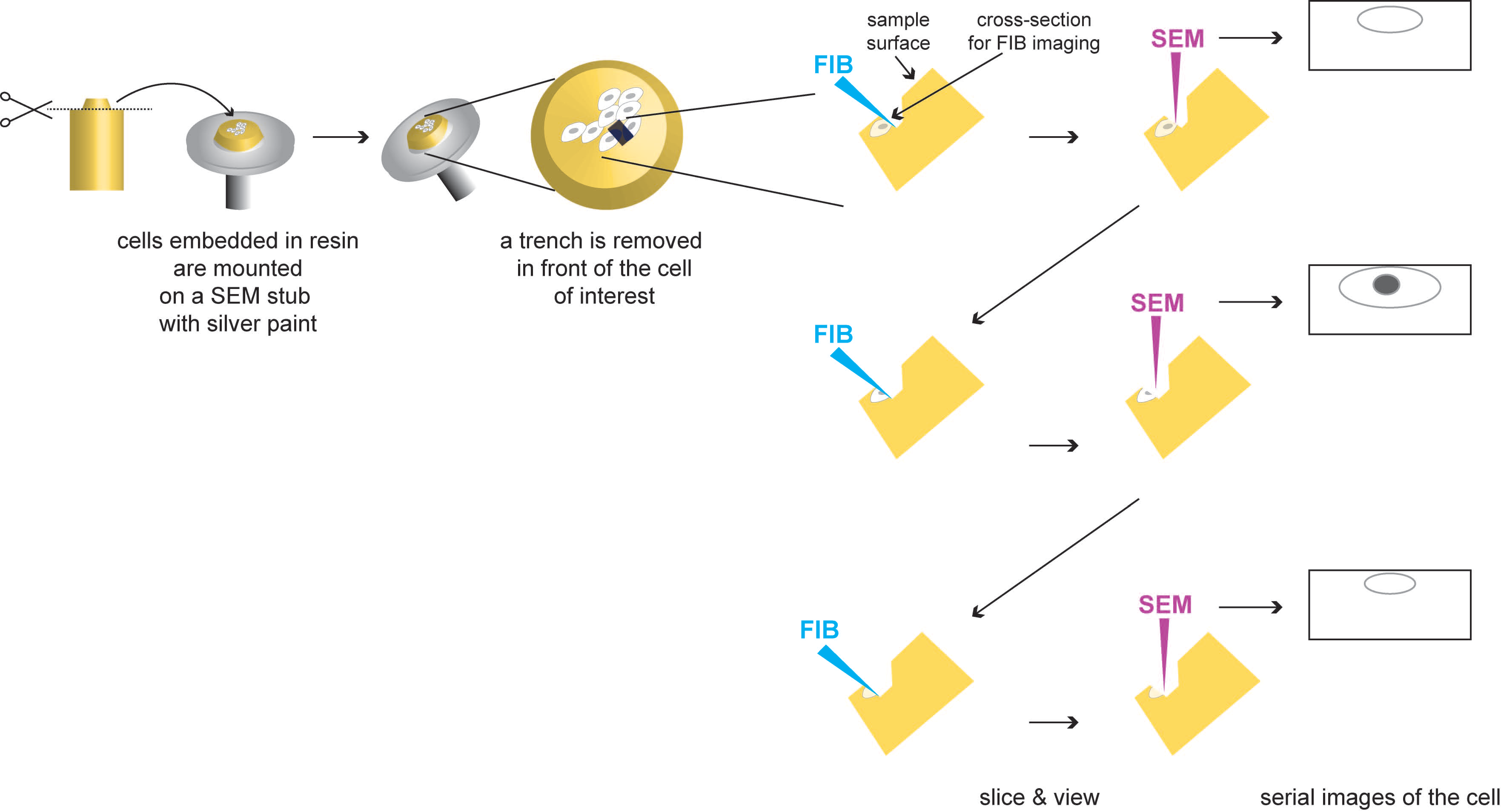
3.4. Serial Block Face (SBF) and Focus Ion Beam (FIB)-Scanning Electron Microscopy (SEM)
3.4.1. Description
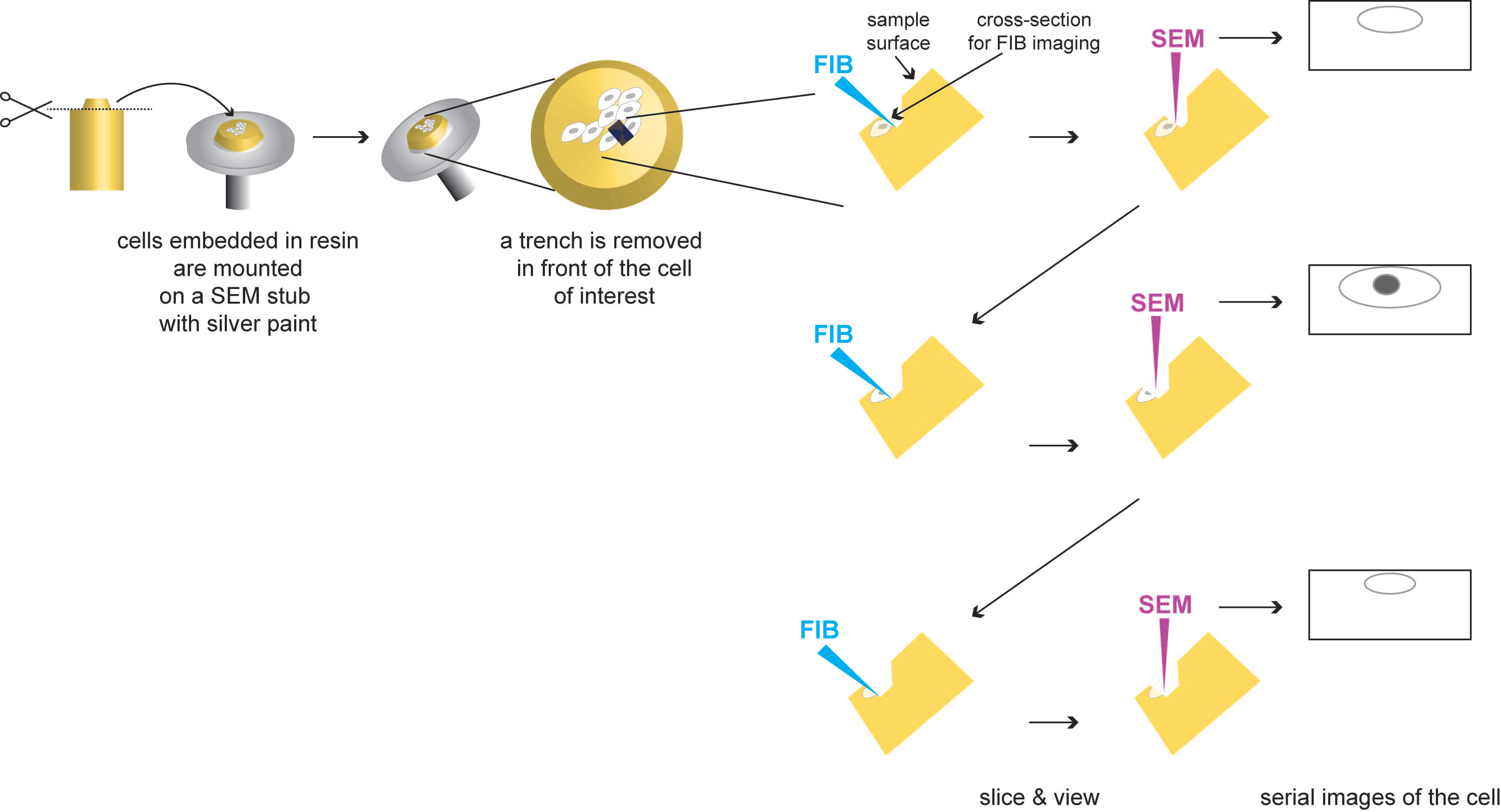
3.4.2. Applications to the Study of Virus-Infected Cells
3.5. Cryo-Methods
3.5.1. Cryo-Electron Tomography of Vitreous Sections (CETOVIS)
3.5.2. Cryo-FIB
3.5.3. Cryo-ET (CET)
3.5.4. Cryo-STEM Tomography
3.5.5. Soft X-ray Cryo-Tomography
3.5.6. Cryo FIB-SEM
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, Z.H. Towards atomic resolution structural determination by single-particle cryo-electron microscopy. Curr. Opin. Struct. Biol. 2008, 18, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Walz, T. The advent of near-atomic resolution in single-particle electron microscopy. Annu. Rev. Biochem. 2009, 78, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Garcea, R.L.; Grigorieff, N.; Harrison, S.C. Subunit interactions in bovine papillomavirus. Proc. Natl. Acad. Sci. USA 2010, 107, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Davey, N.E.; Ulbrich, P.; Riches, J.D.; de Marco, A.; Rumlova, M.; Sachse, C.; Ruml, T.; Briggs, J.A. Structure of the immature retroviral capsid at 8 A resolution by cryo-electron microscopy. Nature 2012, 487, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Ruska, E. Nobel lecture. The development of the electron microscope and of electron microscopy. Biosci. Rep. 1987, 7, 607–629. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.S.; Gelderblom, H.R. Diagnostic electron microscopy is still a timely and rewarding method. J. Clin. Virol. 1999, 13, 105–119. [Google Scholar] [CrossRef]
- Curry, A.; Appleton, H.; Dowsett, B. Application of transmission electron microscopy to the clinical study of viral and bacterial infections: Present and future. Micron 2006, 37, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.S.; Miller, S.E. Modern uses of electron microscopy for detection of viruses. Clin. Microbiol. Rev. 2009, 22, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Gelderblom, H.R. Electron microscopy in rapid viral diagnosis: An update. New Microbiol. 2014, 37, 403–422. [Google Scholar] [PubMed]
- Hazelton, P.R.; Gelderblom, H.R. Electron microscopy for rapid diagnosis of infectious agents in emergent situations. Emerg. Infect. Dis. 2003, 9, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Roingeard, P. Viral detection by electron microscopy: Past, present and future. Biol. Cell 2008, 100, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Biel, S.S.; Madeley, D. Diagnostic virology—The need for electron microscopy: A discussion paper. J. Clin. Virol. 2001, 22, 1–9. [Google Scholar] [CrossRef]
- Risco, C.; de Castro, I.F.; Sanz-Sanchez, L.; Narayan, K.; Grandinetti, G.; Subramaniam, S. Three-dimensional imaging of viral infections. Annu. Rev. Virol. 2014, 1, 453–473. [Google Scholar] [CrossRef]
- Griffiths, G. Fine Structure Immunocytochemistry; Springer: Berlin, Germany; Heidelberg, Germany, 1993. [Google Scholar]
- Hayat, M.A. Fixation for Electron Microscopy; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Steinbrecht, R.A.; Zierold, K. Cryotechniques in Biological Electron Microscopy; Springer-Verlag: Berlin, Germany; Heidelberg, Germany, 1987. [Google Scholar]
- Verkleij, A.J.; Leunissen, J.L.M. Immuno-Gold Labeling in Cell Biology; CRC Press, Inc.: Boca Raton, FL, USA, 1989. [Google Scholar]
- Laue, M. Electron microscopy of viruses. Methods Cell Biol. 2010, 96, 1–20. [Google Scholar] [PubMed]
- Flitney, F.W. The time course of the fixation of albumin by formaldehyde, glutaraldehyde, acrolein and other higher aldehydes. J. R. Micros. Soc. 1966, 85, 353–364. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Hayat, M.A. The production of artifacts. Ultrastruct. Pathol. 1981, 2, 93. [Google Scholar] [PubMed]
- McDowall, A.W.; Chang, J.J.; Freeman, R.; Lepault, J.; Walter, C.A.; Dubochet, J. Electron microscopy of frozen hydrated sections of vitreous ice and vitrified biological samples. J. Microsc. 1983, 131 Pt 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McDowall, A.; Gruenberg, J.; Romisch, K.; Griffiths, G. The structure of organelles of the endocytic pathway in hydrated cryosections of cultured cells. Eur. J. Cell Biol. 1989, 49, 281–294. [Google Scholar] [PubMed]
- Small, J.V. Organization of actin in the leading edge of cultured cells: Influence of osmium tetroxide and dehydration on the ultrastructure of actin meshworks. J. Cell Biol. 1981, 91 3 Pt 1, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Studer, D.; Humbel, B.M.; Chiquet, M. Electron microscopy of high pressure frozen samples: Bridging the gap between cellular ultrastructure and atomic resolution. Histochem. Cell Biol. 2008, 130, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Adrian, M.; Dubochet, J.; Lepault, J.; McDowall, A.W. Cryo-electron microscopy of viruses. Nature 1984, 308, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Dubochet, J.; Adrian, M.; Chang, J.J.; Homo, J.C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Meister, N.; Moor, H. Freezing in a propane jet and its application in freeze-fracturing. Mikroskopie 1980, 36, 129–140. [Google Scholar] [PubMed]
- Giddings, T.H., Jr.; Staehelin, L.A. Ribosome binding sites visualized on freeze-fractured membranes of the rough endoplasmic reticulum. J. Cell Biol. 1980, 85, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Espevik, T.; Elgsaeter, A. In situ liquid propane jet-freezing and freeze-etching of monolayer cell cultures. J. Microsc. 1981, 123 Pt 1, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Swales, L.S.; Lane, N.J. Insect intercellular junctions: Rapid freezing by jet propane. J. Cell Sci. 1983, 62, 223–236. [Google Scholar] [PubMed]
- Haggis, G.H. Study of the conditions necessary for propane-jet freezing of fresh biological tissues without detectable ice formation. J. Microsc. 1986, 143 Pt 3, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Galway, M.E.; Heckman, J.W., Jr.; Hyde, G.J.; Fowke, L.C. Advances in high-pressure and plunge-freeze fixation. Methods Cell Biol. 1995, 49, 3–19. [Google Scholar] [PubMed]
- Nitta, K.; Kaneko, Y. Simple plunge freezing applied to plant tissues for capturing the ultrastructure close to the living state. J. Electron Microsc. (Tokyo) 2004, 53, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Richter, T.; Biel, S.S.; Sattler, M.; Wenck, H.; Wittern, K.P.; Wiesendanger, R.; Wepf, R. Pros and cons: Cryo-electron microscopic evaluation of block faces versus cryo-sections from frozen-hydrated skin specimens prepared by different techniques. J. Microsc. 2007, 225 Pt 2, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Shimoni, E.; Muller, M. On optimizing high-pressure freezing: From heat transfer theory to a new microbiopsy device. J. Microsc. 1998, 192 Pt 3, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Dubochet, J. The physics of rapid cooling and its implications for cryoimmobilization of cells. Methods Cell Biol. 2007, 79, 7–21. [Google Scholar] [PubMed]
- Studer, D.; Michel, M.; Wohlwend, M.; Hunziker, E.B.; Buschmann, M.D. Vitrification of articular cartilage by high-pressure freezing. J. Microsc. 1995, 179 Pt 3, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.P.; Daw, C.S.; Rorvik, M.C. The construction and operation of a simple inexpensive slam freezing device for electron microscopy. J. Microsc. 1987, 147 Pt 1, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Meryman, H.T. Cryopreservation of living cells: Principles and practice. Transfusion 2007, 47, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Moor, H. Theory and practice of high pressure freezing. In Cryotechniques in Biological Electron Microscopy; Steinbrecht, R.A., Zierold, K., Eds.; Sringer: Berlin, Germany; Heidelberg, Germany, 1987; pp. 175–191. [Google Scholar]
- Studer, D.; Michel, M.; Muller, M. High pressure freezing comes of age. Scanning Microsc. Suppl. 1989, 3, 253–268. [Google Scholar] [PubMed]
- Vanhecke, D.; Graber, W.; Studer, D. Close-to-native ultrastructural preservation by high pressure freezing. Methods Cell Biol. 2008, 88, 151–164. [Google Scholar] [PubMed]
- Al-Amoudi, A.; Dubochet, J.; Studer, D. Amorphous solid water produced by cryosectioning of crystalline ice at 113 K. J. Microsc. 2002, 207 Pt 2, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Eppenberger-Eberhardt, M.; Riesinger, I.; Messerli, M.; Schwarb, P.; Muller, M.; Eppenberger, H.M.; Wallimann, T. Adult rat cardiomyocytes cultured in creatine-deficient medium display large mitochondria with paracrystalline inclusions, enriched for creatine kinase. J. Cell Biol. 1991, 113, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Verkade, P. Moving EM: The rapid transfer system as a new tool for correlative light and electron microscopy and high throughput for high-pressure freezing. J. Microsc. 2008, 230 Pt 2, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, R.E.; Cole, N.L. Preparation of cultured mammalian cells for transmission and scanning electron microscopy using Aclar film. J. Electron Microsc. Tech. 1988, 10, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Masurovsky, E.B.; Bunge, R.P. Aclar film in biological electron mircoscopy. J. Electron Microsc. Tech. 1989, 12, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, N.; Humbel, B.M.; van Donselaar, E.; Verkleij, A.J.; Burger, K.N. Aclar discs: A versatile substrate for routine high-pressure freezing of mammalian cell monolayers. J. Microsc. 2006, 221 Pt 3, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Schauflinger, M.; Villinger, C.; Mertens, T.; Walther, P.; von Einem, J. Analysis of human cytomegalovirus secondary envelopment by advanced electron microscopy. Cell Microbiol. 2013, 15, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, H.; Mannweiler, K.; Muller, M. High-pressure freezing of cell suspensions in cellulose capillary tubes. J. Microsc. 1994, 175 Pt 1, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.B.; Trujillo, J.; Leahy, I.; Razmus, D.; Dehate, R.; Lorcheim, P.; Czarneski, M.A.; Zimmerman, D.; Newton, J.T.; Haddow, A.D.; et al. Construction and organization of a BSL-3 cryo-electron microscopy laboratory at UTMB. J. Struct. Biol. 2013, 181, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Sosinsky, G.E.; Crum, J.; Jones, Y.Z.; Lanman, J.; Smarr, B.; Terada, M.; Martone, M.E.; Deerinck, T.J.; Johnson, J.E.; Ellisman, M.H. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. J. Struct. Biol. 2008, 161, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Lopez-Iglesias, C.; Tzeng, W.P.; Frey, T.K.; Fernandez, J.J.; Risco, C. Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Murk, J.L.; Posthuma, G.; Koster, A.J.; Geuze, H.J.; Verkleij, A.J.; Kleijmeer, M.J.; Humbel, B.M. Influence of aldehyde fixation on the morphology of endosomes and lysosomes: Quantitative analysis and electron tomography. J. Microsc. 2003, 212 Pt 1, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Vale, F.F.; Correia, A.C.; Matos, B.; Moura Nunes, J.F.; Alves de Matos, A.P. Applications of transmission electron microscopy to virus detection and identification. In Microscopy: Science. Technology, Applications and Education; Mendez-Vilas, A., Diaz, J., Eds.; FORMATEX: Badajoz, Spain, 2010; pp. 128–136. [Google Scholar]
- Stoeckenius, W.; Mahr, S.C. Studies on the reaction of osmium tetroxide with lipids and related compounds. Lab. Invest. 1965, 14, 1196–1207. [Google Scholar] [PubMed]
- Korn, E.D. Structure of biological membranes. Science 1966, 153, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T.; Guerra, F.C.; Magalhaes, M.M. The fixative action of uranyl acetate in electron microscopy. Experientia 1968, 24, 1074. [Google Scholar] [CrossRef] [PubMed]
- Nielson, A.J.; Griffith, W.P. Tissue fixation by osmium tetroxide. A possible role for proteins. J. Histochem. Cytochem. 1979, 27, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Maupin-Szamier, P.; Pollard, T.D. Actin filament destruction by osmium tetroxide. J. Cell Biol. 1978, 77, 837–852. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, M.; Zollinger, M. Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J. Histochem. Cytochem. 1983, 31, 101–109. [Google Scholar] [CrossRef] [PubMed]
- FINCK, H. Epoxy resins in electron microscopy. J. Biophys. Biochem. Cytol. 1960, 7, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Hawes, P.C. Preparation of cultured cells using high-pressure freezing and freeze substitution for subsequent 2D or 3D visualization in the transmission electron microscope. Methods Mol. Biol. 2015, 1282, 271–282. [Google Scholar] [PubMed]
- Hunziker, E.B.; Herrmann, W.; Schenk, R.K.; Mueller, M.; Moor, H. Cartilage ultrastructure after high pressure freezing, freeze substitution, and low temperature embedding. I. Chondrocyte ultrastructure—Implications for the theories of mineralization and vascular invasion. J. Cell Biol. 1984, 98, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Humbel, B.; Müller, M. Freeze-substitution and low temperature embeding. In The Science of Biological Specimen Preparation; O’Hare, A.M.F., Müller, M., Becker, R.P., Boyde, A., Wolosewick, J.J., Eds.; SEM Inc. AMF O’Hare: Chicago, IL, USA, 1986; pp. 175–183. [Google Scholar]
- Walther, P.; Ziegler, A. Freeze substitution of high-pressure frozen samples: The visibility of biological membranes is improved when the substitution medium contains water. J. Microsc. 2002, 208 Pt 1, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Petsko, G.A. Protein crystallography at sub-zero temperatures: Cryo-protective mother liquors for protein crystals. J. Mol. Biol. 1975, 96, 381–392. [Google Scholar] [CrossRef]
- Carlemalm, E.; Armbruster, B.L.; Chiovetti, R.; Garavito, R.M.; Hobot, J.A.; Villiger, W.; Kellenberger, E. Perspectives for achieving improved information by the observation of thin sections in the electron microscope. Tokai J. Exp. Clin. Med. 1982, 7, 33–42. [Google Scholar] [PubMed]
- Newman, G.R.; Jasani, B.; Williams, E.D. A simple post-embedding system for the rapid demonstration of tissue antigens under the electron microscope. Histochem. J. 1983, 15, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H.; Humbel, B.M. Influence of fixatives and embedding media on immunolabelling of freeze-substituted cells. Scanning Microsc. Suppl. 1989, 3, 57–63. [Google Scholar] [PubMed]
- Luby-Phelps, K.; Ning, G.; Fogerty, J.; Besharse, J.C. Visualization of identified GFP-expressing cells by light and electron microscopy. J. Histochem. Cytochem. 2003, 51, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Nixon, S.J.; Webb, R.I.; Floetenmeyer, M.; Schieber, N.; Lo, H.P.; Parton, R.G. A single method for cryofixation and correlative light, electron microscopy and tomography of zebrafish embryos. Traffic 2009, 10, 131–136. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.L.; Webb, R.I. Freeze substitution in 3 hours or less. J. Microsc. 2011, 243, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Peddie, C.J.; Blight, K.; Wilson, E.; Melia, C.; Marrison, J.; Carzaniga, R.; Domart, M.C.; O’Toole, P.; Larijani, B.; Collinson, L.M. Correlative and integrated light and electron microscopy of in-resin GFP fluorescence, used to localise diacylglycerol in mammalian cells. Ultramicroscopy 2014, 143, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kizilyaprak, C.; Longo, G.; Daraspe, J.; Humbel, B.M. Investigation of resins suitable for the preparation of biological sample for 3-D electron microscopy. J. Struct. Biol. 2015, 189, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Briggman, K.L.; Bock, D.D. Volume electron microscopy for neuronal circuit reconstruction. Curr. Opin. Neurobiol. 2012, 22, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.L.; Sarbash, V.I.; Ataullakhanov, F.I.; McIntosh, J.R.; Nicastro, D. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. J. Microsc. 2007, 227 Pt 2, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moran, H.; Dahl, A.O. Electron microscopy of ultrathin frozen sections of pollen grains. Science 1952, 116, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Richter, K. Aspects of cryofixation and cryosectioning for the observation of bulk biological samples in the hydrated state by cryoelectron microscopy. Scanning Microsc. Suppl. 1996, 10, 375–385. [Google Scholar] [PubMed]
- Frederik, P.M.; Busing, W.M.; Persson, A. Concerning the nature of the cryosectioning process. J. Microsc. 1982, 125 Pt 2, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Frederik, P.M.; Busing, W.M.; Persson, A. Surface defects on thin cryosections. Scan Electron Microsc. 1984, Volume Pt 1, 433–443. [Google Scholar]
- Frederik, P.M.; Bomans, P.H.; Stuart, M.C. The ultrastructure of cryo-sections and intact vitrified cells—The effects of cryoprotectants and acceleration voltage on beam induced bubbling. Scanning Microsc. Suppl. 1991, 5, S43–S51. [Google Scholar] [PubMed]
- Sitte, H. Advanced instrumentation and methodology related to cryoultramicrotomy: A review. Scanning Microsc. Suppl. 1996, 10, 387–463. [Google Scholar] [PubMed]
- Al-Amoudi, A.; Chang, J.J.; Leforestier, A.; McDowall, A.; Salamin, L.M.; Norlen, L.P.; Richter, K.; Blanc, N.S.; Studer, D.; Dubochet, J. Cryo-electron microscopy of vitreous sections. EMBO J. 2004, 23, 3583–3588. [Google Scholar] [CrossRef] [PubMed]
- Chlanda, P.; Sachse, M. Cryo-electron microscopy of vitreous sections. Methods Mol. Biol. 2014, 1117, 193–214. [Google Scholar] [PubMed]
- Pierson, J.; Fernandez, J.J.; Bos, E.; Amini, S.; Gnaegi, H.; Vos, M.; Bel, B.; Adolfsen, F.; Carrascosa, J.L.; Peters, P.J. Improving the technique of vitreous cryo-sectioning for cryo-electron tomography: Electrostatic charging for section attachment and implementation of an anti-contamination glove box. J. Struct. Biol. 2010, 169, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Elias, H. Identification of structure by the common-sense approach. J. Microsc. 1972, 95, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Chichon, F.J.; Rodriguez, M.J.; Pereiro, E.; Chiappi, M.; Perdiguero, B.; Guttmann, P.; Werner, S.; Rehbein, S.; Schneider, G.; Esteban, M.; et al. Cryo X-ray nano-tomography of vaccinia virus infected cells. J. Struct. Biol. 2012, 177, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Kopek, B.G.; Perkins, G.; Miller, D.J.; Ellisman, M.H.; Ahlquist, P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 2007, 5, e220. [Google Scholar] [CrossRef] [PubMed]
- Baines, J.D.; Hsieh, C.E.; Wills, E.; Mannella, C.; Marko, M. Electron tomography of nascent herpes simplex virus virions. J. Virol. 2007, 81, 2726–2735. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Wei, T.; Omura, T.; Takagi, J.; Iwasaki, K. Three-dimensional architecture of virus-packed tubule. J. Electron Microsc. (Tokyo) 2007, 56, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Sougrat, R.; Bartesaghi, A.; Lifson, J.D.; Bennett, A.E.; Bess, J.W.; Zabransky, D.J.; Subramaniam, S. Electron tomography of the contact between T cells and SIV/HIV-1: Implications for viral entry. PLoS Pathog. 2007, 3, e63. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; Briggs, J.A.; Glass, B.; Riches, J.D.; Simon, M.N.; Johnson, M.C.; Muller, B.; Grunewald, K.; Krausslich, H.G. Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host. Microb. 2008, 4, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Majorovits, E.; Nejmeddine, M.; Tanaka, Y.; Taylor, G.P.; Fuller, S.D.; Bangham, C.R. Human T-lymphotropic virus-1 visualized at the virological synapse by electron tomography. PLoS ONE 2008, 3, e2251. [Google Scholar] [CrossRef] [PubMed]
- Chichon, F.J.; Rodriguez, M.J.; Risco, C.; Fraile-Ramos, A.; Fernandez, J.J.; Esteban, M.; Carrascosa, J.L. Membrane remodelling during vaccinia virus morphogenesis. Biol. Cell 2009, 101, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host. Microb. 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Chlanda, P.; Carbajal, M.A.; Cyrklaff, M.; Griffiths, G.; Krijnse-Locker, J. Membrane rupture generates single open membrane sheets during vaccinia virus assembly. Cell Host. Microb. 2009, 6, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Uehara-Ichiki, T.; Miyazaki, N.; Hibino, H.; Iwasaki, K.; Omura, T. Association of Rice gall dwarf virus with microtubules is necessary for viral release from cultured insect vector cells. J. Virol. 2009, 83, 10830–10835. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Ryazantsev, S.; Sun, R.; Zhou, Z.H. Three-dimensional visualization of gammaherpesvirus life cycle in host cells by electron tomography. Structure 2010, 18, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Gangodkar, S.; Jain, P.; Dixit, N.; Ghosh, K.; Basu, A. Dengue virus-induced autophagosomes and changes in endomembrane ultrastructure imaged by electron tomography and whole-mount grid-cell culture techniques. J. Electron Microsc. (Tokyo) 2010, 59, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Swett-Tapia, C.; van den Worm, S.H.; Te Velthuis, A.J.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J.; Kikkert, M. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 2010, 84, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Kolesnikova, L.; Krahling, V.; Riches, J.D.; Becker, S.; Briggs, J.A. Electron tomography reveals the steps in filovirus budding. PLoS Pathog. 2010, 6, e1000875. [Google Scholar] [CrossRef] [PubMed]
- Soonsawad, P.; Xing, L.; Milla, E.; Espinoza, J.M.; Kawano, M.; Marko, M.; Hsieh, C.; Furukawa, H.; Kawasaki, M.; Weerachatyanukul, W.; et al. Structural evidence of glycoprotein assembly in cellular membrane compartments prior to Alphavirus budding. J. Virol. 2010, 84, 11145–11151. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Barcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Miyazaki, N.; Uehara-Ichiki, T.; Hibino, H.; Shimizu, T.; Netsu, O.; Kikuchi, A.; Sasaya, T.; Iwasaki, K.; Omura, T. Three-dimensional analysis of the association of viral particles with mitochondria during the replication of Rice gall dwarf virus. J. Mol. Biol. 2011, 410, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Barcena, M.; Limpens, R.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural characterization of arterivirus replication structures: Reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS ONE 2012, 7, e47912. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. MBio 2013, 4, e00801–e00813. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.; Welsch, S.; Chlanda, P.; Hagen, W.; Hoppe, S.; Kolovou, A.; Pagnier, I.; Raoult, D.; Krijnse, L.J. Open membranes are the precursors for assembly of large DNA viruses. Cell Microbiol. 2013, 15, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, A.; Gillespie, L.; Morgan, G.; van der Heide, P.; Khromykh, A.; Mackenzie, J. The West Nile virus assembly process evades the conserved antiviral mechanism of the interferon-induced MXA protein. Virology 2014, 448, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Jin, X.; Zhang, X.; Li, Y.; Wang, C.; Wang, X.; Hong, J.; Wang, X.; Li, D.; Zhang, Y. Morphogenesis of Endoplasmic Reticulum Membrane-Invaginated Vesicles during Beet Black Scorch Virus Infection: Role of Auxiliary Replication Protein and New Implications of Three-Dimensional Architecture. J. Virol. 2015, 89, 6184–6195. [Google Scholar] [CrossRef] [PubMed]
- Ladinsky, M.S.; Kieffer, C.; Olson, G.; Deruaz, M.; Vrbanac, V.; Tager, A.M.; Kwon, D.S.; Bjorkman, P.J. Electron tomography of HIV-1 infection in gut-associated lymphoid tissue. PLoS Pathog. 2014, 10, e1003899. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.C.; Popov, V.; Sherman, M.B.; Wen, J.; Barrett, A.D. Attenuated West Nile virus mutant NS1130-132QQA/175A/207A exhibits virus-induced ultrastructural changes and accumulation of protein in the endoplasmic reticulum. J. Virol. 2015, 89, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Li, K.; Tang, P.; Li, Y.; Zhou, Q.; Yang, K.; Zhang, Q. Three-dimensional visualization of the Autographa californica multiple nucleopolyhedrovirus occlusion-derived virion envelopment process gives new clues as to its mechanism. Virology 2015, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Bily, T.; Palus, M.; Eyer, L.; Elsterova, J.; Vancova, M.; Ruzek, D. Electron Tomography Analysis of Tick-Borne Encephalitis Virus Infection in Human Neurons. Sci. Rep. 2015, 5, 10745. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.; Andres, G.; Kolovou, A.; Hoppe, S.; Salas, M.L.; Walther, P.; Krijnse, L.J. African swine fever virus assembles a single membrane derived from rupture of the endoplasmic reticulum. Cell Microbiol. 2015, 17, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Mutsafi, Y.; Shimoni, E.; Shimon, A.; Minsky, A. Membrane assembly during the infection cycle of the giant Mimivirus. PLoS Pathog. 2013, 9, e1003367. [Google Scholar] [CrossRef] [PubMed]
- Milrot, E.; Mutsafi, Y.; Fridmann-Sirkis, Y.; Shimoni, E.; Rechav, K.; Gurnon, J.R.; van Etten, J.L.; Minsky, A. Virus-host interactions: Insights from the replication cycle of the large Paramecium bursaria chlorella virus. Cell Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Lopez-Montero, N.; Elliott, R.M.; Fernandez, J.J.; Risco, C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cell Microbiol. 2008, 10, 2012–2028. [Google Scholar] [CrossRef] [PubMed]
- Reichelt, M.; Joubert, L.; Perrino, J.; Koh, A.L.; Phanwar, I.; Arvin, A.M. 3D reconstruction of VZV infected cell nuclei and PML nuclear cages by serial section array scanning electron microscopy and electron tomography. PLoS Pathog. 2012, 8, e1002740. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell Mol. Life Sci. 2013, 70, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, d.C.I.; Zamora, P.F.; Ooms, L.; Fernandez, J.J.; Lai, C.M.; Mainou, B.A.; Dermody, T.S.; Risco, C. Reovirus forms neo-organelles for progeny particle assembly within reorganized cell membranes. MBio 2014, 5. [Google Scholar]
- Zellnig, G.; Pockl, M.H.; Mostl, S.; Zechmann, B. Two and three dimensional characterization of Zucchini Yellow Mosaic Virus induced structural alterations in Cucurbita pepo L. plants. J. Struct. Biol. 2014, 186, 245–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bennett, A.E.; Narayan, K.; Shi, D.; Hartnell, L.M.; Gousset, K.; He, H.; Lowekamp, B.C.; Yoo, T.S.; Bliss, D.; Freed, E.O.; Subramaniam, S. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog. 2009, 5, e1000591. [Google Scholar] [CrossRef] [PubMed]
- Felts, R.L.; Narayan, K.; Estes, J.D.; Shi, D.; Trubey, C.M.; Fu, J.; Hartnell, L.M.; Ruthel, G.T.; Schneider, D.K.; Nagashima, K.; et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13336–13341. [Google Scholar] [CrossRef] [PubMed]
- Do, T.; Murphy, G.; Earl, L.A.; del Prete, G.Q.; Grandinetti, G.; Li, G.H.; Estes, J.D.; Rao, P.; Trubey, C.M.; Thomas, J.; et al. Three-dimensional imaging of HIV-1 virological synapses reveals membrane architectures involved in virus transmission. J. Virol. 2014, 88, 10327–10339. [Google Scholar] [CrossRef] [PubMed]
- Cyrklaff, M.; Linaroudis, A.; Boicu, M.; Chlanda, P.; Baumeister, W.; Griffiths, G.; Krijnse-Locker, J. Whole cell cryo-electron tomography reveals distinct disassembly intermediates of vaccinia virus. PLoS ONE 2007, 2, e420. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.E.; Sodeik, B.; Grunewald, K. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. USA 2008, 105, 10559–10564. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; de Marco, A.; Oberwinkler, H.; Habermann, A.; Briggs, J.A.; Krausslich, H.G.; Grunewald, K. Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog. 2010, 6, e1001173. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Riches, J.D.; Kolesnikova, L.; Welsch, S.; Krahling, V.; Davey, N.; Parsy, M.L.; Becker, S.; Briggs, J.A. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol. 2011, 9, e1001196. [Google Scholar] [CrossRef] [PubMed]
- Ibiricu, I.; Huiskonen, J.T.; Dohner, K.; Bradke, F.; Sodeik, B.; Grunewald, K. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 2011, 7, e1002406. [Google Scholar] [CrossRef] [PubMed]
- Vijayakrishnan, S.; Loney, C.; Jackson, D.; Suphamungmee, W.; Rixon, F.J.; Bhella, D. Cryotomography of budding influenza A virus reveals filaments with diverse morphologies that mostly do not bear a genome at their distal end. PLoS Pathog. 2013, 9, e1003413. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. The bacteriophage t7 virion undergoes extensive structural remodeling during infection. Science 2013, 339, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, N.; Akita, F.; Nakagawa, A.; Murata, K.; Omura, T.; Iwasaki, K. Cryo-electron tomography: Moving towards revealing the viral life cycle of Rice dwarf virus. J. Synchrotron. Radiat. 2013, 20 Pt 6, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Fu, C.; Raytcheva, D.; Flanagan, J.; Khant, H.A.; Liu, X.; Rochat, R.H.; Haase-Pettingell, C.; Piret, J.; Ludtke, S.J.; et al. Visualizing virus assembly intermediates inside marine cyanobacteria. Nature 2013, 502, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.; Pfanzelter, J.; Winkler, C.; Narita, A.; Le, C.C.; Nemethova, M.; Carlier, M.F.; Maeda, Y.; Welch, M.D.; Ohkawa, T.; et al. Electron tomography and simulation of baculovirus actin comet tails support a tethered filament model of pathogen propulsion. PLoS Biol. 2014, 12, e1001765. [Google Scholar] [CrossRef] [PubMed]
- Woodward, C.L.; Cheng, S.N.; Jensen, G.J. Electron cryotomography studies of maturing HIV-1 particles reveal the assembly pathway of the viral core. J. Virol. 2015, 89, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Guttmann, P.; Klupp, B.; Werner, S.; Rehbein, S.; Mettenleiter, T.C.; Schneider, G.; Grunewald, K. Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. J. Struct. Biol. 2012, 177, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.J.; Grimm, R.; Typke, D.; Hegerl, R.; Stoschek, A.; Walz, J.; Baumeister, W. Perspectives of molecular and cellular electron tomography. J. Struct. Biol. 1997, 120, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W.; Grimm, R.; Walz, J. Electron tomography of molecules and cells. Trends Cell Biol. 1999, 9, 81–85. [Google Scholar] [CrossRef]
- Lucic, V.; Forster, F.; Baumeister, W. Structural studies by electron tomography: From cells to molecules. Annu. Rev. Biochem. 2005, 74, 833–865. [Google Scholar] [CrossRef] [PubMed]
- De Rosier, D.J.; Klug, A. Reconstruction of three dimensional structures from electron micrographs. Nature 1968, 217, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Hart, R.G. Electron microscopy of unstained biological material: The polytropic montage. Science 1968, 159, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Radon, J. Über die Bestimmung von Funktionen durch ihre Integralwerte längs gewisser Mannigfaltigkeiten. Sächs. Akad. Wiss. Leipzig Math. Phys. Kl. 1917, 69, 262–277. [Google Scholar]
- McDonald, K.L.; Auer, M. High-pressure freezing, cellular tomography, and structural cell biology. Biotechniques 2006, 41, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Penczek, P.; Marko, M.; Buttle, K.; Frank, J. Double-tilt electron tomography. Ultramicroscopy 1995, 60, 393–410. [Google Scholar] [CrossRef]
- Mastronarde, D.N. Dual-axis tomography: An approach with alignment methods that preserve resolution. J. Struct. Biol. 1997, 120, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Hoog, J.L.; Schwartz, C.; Noon, A.T.; O'Toole, E.T.; Mastronarde, D.N.; McIntosh, J.R.; Antony, C. Organization of interphase microtubules in fission yeast analyzed by electron tomography. Dev. Cell 2007, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Noske, A.B.; Costin, A.J.; Morgan, G.P.; Marsh, B.J. Expedited approaches to whole cell electron tomography and organelle mark-up in situ in high-pressure frozen pancreatic islets. J. Struct. Biol. 2008, 161, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Luther, P.K.; Lawrence, M.C.; Crowther, R.A. A method for monitoring the collapse of plastic sections as a function of electron dose. Ultramicroscopy 1988, 24, 7–18. [Google Scholar] [CrossRef]
- Iwasaki, K.; Omura, T. Electron tomography of the supramolecular structure of virus-infected cells. Curr. Opin. Struct. Biol. 2010, 20, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Membranous replication factories induced by plus-strand RNA viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef] [PubMed]
- Biskupek, J.; Leschner, J.; Walther, P.; Kaiser, U. Optimization of STEM tomography acquisition—A comparison of convergent beam and parallel beam STEM tomography. Ultramicroscopy 2010, 110, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Carlemalm, E.; Villiger, W.; Wurtz, M.; Mory, C.; Colliex, C. Z-contrast in biology. A comparison with other imaging modes. Ann. N. Y. Acad. Sci. 1986, 483, 202–228. [Google Scholar] [CrossRef] [PubMed]
- Yakushevska, A.E.; Lebbink, M.N.; Geerts, W.J.; Spek, L.; van Donselaar, E.G.; Jansen, K.A.; Humbel, B.M.; Post, J.A.; Verkleij, A.J.; Koster, A.J. STEM tomography in cell biology. J. Struct. Biol. 2007, 159, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Takagi, T.; Hirase, A.; Miyazawa, A. STEM tomography for thick biological specimens. Ultramicroscopy 2008, 109, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Hohmann-Marriott, M.F.; Sousa, A.A.; Azari, A.A.; Glushakova, S.; Zhang, G.; Zimmerberg, J.; Leapman, R.D. Nanoscale 3D cellular imaging by axial scanning transmission electron tomography. Nat. Methods 2009, 6, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Hohn, K.; Sailer, M.; Wang, L.; Lorenz, M.; Schneider, M.E.; Walther, P. Preparation of cryofixed cells for improved 3D ultrastructure with scanning transmission electron tomography. Histochem. Cell Biol. 2011, 135, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.A.; Leapman, R.D. Development and application of STEM for the biological sciences. Ultramicroscopy 2012, 123, 38–49. [Google Scholar] [CrossRef] [PubMed]
- SJOSTRAND, F.S. Ultrastructure of retinal rod synapses of the guinea pig eye as revealed by three-dimensional reconstructions from serial sections. J. Ultrastruct. Res. 1958, 2, 122–170. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef] [PubMed]
- Bumbarger, D.J.; Riebesell, M.; Rodelsperger, C.; Sommer, R.J. System-wide rewiring underlies behavioral differences in predatory and bacterial-feeding nematodes. Cell 2013, 152, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Micheva, K.D.; Smith, S.J. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron 2007, 55, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Saalfeld, S.; Cardona, A.; Hartenstein, V.; Tomancak, P. As-rigid-as-possible mosaicking and serial section registration of large ssTEM datasets. Bioinformatics. 2010, 26, i57–i63. [Google Scholar] [CrossRef] [PubMed]
- Kaynig, V.; Fischer, B.; Muller, E.; Buhmann, J.M. Fully automatic stitching and distortion correction of transmission electron microscope images. J. Struct. Biol. 2010, 171, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Hayworth, K.J.; Kasthuri, N.; Schalek, R.; Lichtman, J.W. Automating the Collection of Ultrathin Serial Sections for Large Volume TEM Reconstructions. Microsc. Microanal. 2006, 12 (Suppl. S2), 86–87. [Google Scholar] [CrossRef]
- Hayworth, K.J.; Morgan, J.L.; Schalek, R.; Berger, D.R.; Hildebrand, D.G.; Lichtman, J.W. Imaging ATUM ultrathin section libraries with WaferMapper: A multi-scale approach to EM reconstruction of neural circuits. Front. Neural. Circuits 2014, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Schauflinger, M.; Villinger, C.; Walther, P. Three-dimensional visualization of virus-infected cells by serial sectioning: An electron microscopic study using resin embedded cells. Methods Mol. Biol. 2013, 1064, 227–237. [Google Scholar] [PubMed]
- Risco, C.; Fernandez, D.C.I. Virus morphogenesis in the cell: Methods and observations. Subcell. Biochem. 2013, 68, 417–440. [Google Scholar] [PubMed]
- Denk, W.; Horstmann, H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol. 2004, 2, e329. [Google Scholar] [CrossRef] [PubMed]
- Heymann, J.A.; Hayles, M.; Gestmann, I.; Giannuzzi, L.A.; Lich, B.; Subramaniam, S. Site-specific 3D imaging of cells and tissues with a dual beam microscope. J. Struct. Biol. 2006, 155, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Hekking, L.H.; Lebbink, M.N.; de Winter, D.A.; Schneijdenberg, C.T.; Brand, C.M.; Humbel, B.M.; Verkleij, A.J.; Post, J.A. Focused ion beam-scanning electron microscope: Exploring large volumes of atherosclerotic tissue. J. Microsc. 2009, 235, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Villinger, C.; Schauflinger, M.; Gregorius, H.; Kranz, C.; Hohn, K.; Nafeey, S.; Walther, P. Three-dimensional imaging of adherent cells using FIB/SEM and STEM. Methods Mol. Biol. 2014, 1117, 617–638. [Google Scholar] [PubMed]
- Narayan, K.; Danielson, C.M.; Lagarec, K.; Lowekamp, B.C.; Coffman, P.; Laquerre, A.; Phaneuf, M.W.; Hope, T.J.; Subramaniam, S. Multi-resolution correlative focused ion beam scanning electron microscopy: Applications to cell biology. J. Struct. Biol. 2014, 185, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.; Rosset, S.; Cantoni, M. Focussed ion beam milling and scanning electron microscopy of brain tissue. J. Vis. Exp. 2011, 53, e2588. [Google Scholar] [CrossRef] [PubMed]
- Villinger, C.; Gregorius, H.; Kranz, C.; Hohn, K.; Munzberg, C.; von, W.G.; Mizaikoff, B.; Wanner, G.; Walther, P. FIB/SEM tomography with TEM-like resolution for 3D imaging of high-pressure frozen cells. Histochem. Cell Biol. 2012, 138, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Medalia, O.; Weber, I.; Frangakis, A.S.; Nicastro, D.; Gerisch, G.; Baumeister, W. Macromolecular architecture in eukaryotic cells visualized by cryoelectron tomography. Science 2002, 298, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W. Mapping molecular landscapes inside cells. Biol. Chem. 2004, 385, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.E.; Marko, M.; Frank, J.; Mannella, C.A. Electron tomographic analysis of frozen-hydrated tissue sections. J. Struct. Biol. 2002, 138, 63–73. [Google Scholar] [CrossRef]
- Frank, J.; Wagenknecht, T.; McEwen, B.F.; Marko, M.; Hsieh, C.E.; Mannella, C.A. Three-dimensional imaging of biological complexity. J. Struct. Biol. 2002, 138, 85–91. [Google Scholar] [CrossRef]
- Zhang, P.; Weis, R.M.; Peters, P.J.; Subramaniam, S. Electron tomography of bacterial chemotaxis receptor assemblies. Methods Cell Biol. 2007, 79, 373–384. [Google Scholar] [PubMed]
- Al-Amoudi, A.; Diez, D.C.; Betts, M.J.; Frangakis, A.S. The molecular architecture of cadherins in native epidermal desmosomes. Nature 2007, 450, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Norlen, L.; Oktem, O.; Skoglund, U. Molecular cryo-electron tomography of vitreous tissue sections: Current challenges. J. Microsc. 2009, 235, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Zuber, B.; Nikonenko, I.; Klauser, P.; Muller, D.; Dubochet, J. The mammalian central nervous synaptic cleft contains a high density of periodically organized complexes. Proc. Natl. Acad. Sci. USA 2005, 102, 19192–19197. [Google Scholar] [CrossRef] [PubMed]
- Gruska, M.; Medalia, O.; Baumeister, W.; Leis, A. Electron tomography of vitreous sections from cultured mammalian cells. J. Struct. Biol. 2008, 161, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Al-Amoudi, A.; Castano-Diez, D.; Devos, D.P.; Russell, R.B.; Johnson, G.T.; Frangakis, A.S. The three-dimensional molecular structure of the desmosomal plaque. Proc. Natl. Acad. Sci. USA 2011, 108, 6480–6485. [Google Scholar] [CrossRef] [PubMed]
- Al-Amoudi, A.; Studer, D.; Dubochet, J. Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. J. Struct. Biol. 2005, 150, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Dubochet, J. Cryo-EM—The first thirty years. J. Microsc. 2012, 245, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.; Hsieh, C.; Schalek, R.; Frank, J.; Mannella, C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat. Methods 2007, 4, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Rigort, A.; Bauerlein, F.J.; Villa, E.; Eibauer, M.; Laugks, T.; Baumeister, W.; Plitzko, J.M. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4449–4454. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Schmelzer, T.; Kishchenko, G.; Wagenknecht, T.; Marko, M. Practical workflow for cryo focused-ion-beam milling of tissues and cells for cryo-TEM tomography. J. Struct. Biol. 2014, 185, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, J.; Schampers, R.; Persoon, H.; Hyman, A.A.; Baumeister, W.; Plitzko, J.M. A focused ion beam milling and lift-out approach for site-specific preparation of frozen-hydrated lamellas from multicellular organisms. J. Struct. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Koning, R.I.; Koster, A.J. Cellular nanoimaging by cryo electron tomography. Methods Mol. Biol. 2013, 950, 227–251. [Google Scholar] [PubMed]
- Grunewald, K.; Cyrklaff, M. Structure of complex viruses and virus-infected cells by electron cryo tomography. Curr. Opin. Microbiol. 2006, 9, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Zeev-Ben-Mordehai, T.; Hagen, C.; Grunewald, K. A cool hybrid approach to the herpesvirus “life” cycle. Curr. Opin. Virol. 2014, 5, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.V.; Leonard, K.R. Scanning transmission electron microscopy of unstained biological sections. Nature 1978, 271, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hasegawa, T. Observations of unstained biological specimens using a low-energy, high-resolution STEM. J. Electron Microsc. (Tokyo) 2006, 55, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Buban, J.P.; Ramasse, Q.; Gipson, B.; Browning, N.D.; Stahlberg, H. High-resolution low-dose scanning transmission electron microscopy. J. Electron Microsc. (Tokyo) 2010, 59, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.G.; Houben, L.; Elbaum, M. Cryo-scanning transmission electron tomography of vitrified cells. Nat. Methods 2014, 11, 423–428. [Google Scholar] [CrossRef] [PubMed]
- McDermott, G.; Fox, D.M.; Epperly, L.; Wetzler, M.; Barron, A.E.; Le Gros, M.A.; Larabell, C.A. Visualizing and quantifying cell phenotype using soft X-ray tomography. Bioessays 2012, 34, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Guttmann, P.; Rehbein, S.; Werner, S.; Follath, R. Cryo X-ray microscope with flat sample geometry for correlative fluorescence and nanoscale tomographic imaging. J. Struct. Biol. 2012, 177, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Kirz, J.; Jacobsen, C.; Howells, M. Soft X-ray microscopes and their biological applications. Q. Rev. Biophys. 1995, 28, 33–130. [Google Scholar] [CrossRef] [PubMed]
- Schmahl, G.; Rudolph, D.; Niemann, B.; Guttmann, P.; Thieme, J.; Schneider, G. X-ray microscopy. Naturwissenschaften 1996, 83, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Ilse, W.; Hamamoto, D.; Nair, A.; Lelievre, S.A.; Denbeaux, G.; Johnson, L.; Pearson, A.L.; Yager, D.; Legros, M.A.; Larabell, C.A. High resolution protein localization using soft X-ray microscopy. J. Microsc. 2001, 201 Pt 3, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.N.; Jacobsen, C.; Williams, S. A characterisation of dark-field imaging of colloidal gold labels in a scanning transmission X-ray microscope. Ultramicroscopy 1996, 62, 191–213. [Google Scholar] [CrossRef]
- Weiss, D.; Schneider, G.; Niemann, B.; Guttmann, P.; Rudolph, D.; Schmahl, G. Computed tomography of cryogenic biological specimens based on X-ray microscopic images. Ultramicroscopy 2000, 84, 185–197. [Google Scholar] [CrossRef]
- Gu, W.; Etkin, L.D.; Le Gros, M.A.; Larabell, C.A. X-ray tomography of Schizosaccharomyces pombe. Differentiation 2007, 75, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.; Harteneck, B.D.; Liddle, J.A.; Anderson, E.H.; Attwood, D.T. Soft X-ray microscopy at a spatial resolution better than 15 nm. Nature 2005, 435, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Schertel, A.; Snaidero, N.; Han, H.M.; Ruhwedel, T.; Laue, M.; Grabenbauer, M.; Mobius, W. Cryo FIB-SEM: Volume imaging of cellular ultrastructure in native frozen specimens. J. Struct. Biol. 2013, 184, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Ke, D.; Debiec, K.; Zhao, G.; Meng, X.; Ambrose, Z.; Gibson, G.A.; Watkins, S.C.; Zhang, P. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure. 2011, 19, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.; Gatz, R.; Beck, F.; Rigort, A.; Baumeister, W.; Plitzko, J.M. Correlative microscopy: Bridging the gap between fluorescence light microscopy and cryo-electron tomography. J. Struct. Biol. 2007, 160, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.S.; Gunthert, M.; Gasser, P.; Lucas, F.; Wepf, R. Bridging microscopes: 3D correlative light and scanning electron microscopy of complex biological structures. Methods Cell Biol. 2012, 111, 325–356. [Google Scholar] [PubMed]
- De Boer, P.; Hoogenboom, J.P.; Giepmans, B.N. Correlated light and electron microscopy: Ultrastructure lights up! Nat. Methods 2015, 12, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Loussert, F.C.; Humbel, B.M. Correlative microscopy. Arch. Biochem. Biophys. 2015, 581, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, M.L.; Bailey, A.P.; Senyo, S.E.; Guillermier, C.; Perlstein, T.S.; Gould, A.P.; Lee, R.T.; Lechene, C.P. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature 2012, 481, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Saka, S.K.; Vogts, A.; Krohnert, K.; Hillion, F.; Rizzoli, S.O.; Wessels, J.T. Correlated optical and isotopic nanoscopy. Nat. Commun. 2014, 5, 3664. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A. Structural biology in situ--the potential of subtomogram averaging. Curr. Opin. Struct. Biol. 2013, 23, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Hoenger, A. High-resolution cryo-electron microscopy on macromolecular complexes and cell organelles. Protoplasma 2014, 251, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Bohm, J.; Frangakis, A.S.; Hegerl, R.; Nickell, S.; Typke, D.; Baumeister, W. Toward detecting and identifying macromolecules in a cellular context: Template matching applied to electron tomograms. Proc. Natl. Acad. Sci. USA 2000, 97, 14245–14250. [Google Scholar] [CrossRef] [PubMed]
- Frangakis, A.S.; Bohm, J.; Forster, F.; Nickell, S.; Nicastro, D.; Typke, D.; Hegerl, R.; Baumeister, W. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14153–14158. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.L.; Borgnia, M.J.; Bartesaghi, A.; Tran, E.E.; Earl, L.A.; Schauder, D.M.; Lengyel, J.; Pierson, J.; Patwardhan, A.; Subramaniam, S. Cryo-electron microscopy--a primer for the non-microscopist. FEBS J. 2013, 280, 28–45. [Google Scholar] [CrossRef] [PubMed]
- De Jonqe, N.; Ross, F.M. Electron microscopy of specimens in liquid. Nat. Nanotechnol. 2011, 6, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Dukes, M.J.; Gilmore, B.L.; Tanner, J.R.; McDonald, S.M.; Kelly, D.F. In situ TEM of biological assemblies in liquid. J. Vis. Exp. 2013, 82, 50936. [Google Scholar] [PubMed]
- Gilmore, B.L.; Showalter, S.P.; Dukes, M.J.; Tanner, J.R.; Demmert, A.C.; McDonald, S.M.; Kelly, D.F. Visualizing viral assemblies in a nanoscale biosphere. Lab Chip. 2013, 13, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Peckys, D.B.; de Jonge, N. Visualizing gold nanoparticle uptake in live cells with liquid scanning transmission electron microscopy. Nano. Lett. 2011, 11, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Brey, I.; Bartenschlager, R. Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses 2015, 7, 6316-6345. https://doi.org/10.3390/v7122940
Romero-Brey I, Bartenschlager R. Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses. 2015; 7(12):6316-6345. https://doi.org/10.3390/v7122940
Chicago/Turabian StyleRomero-Brey, Inés, and Ralf Bartenschlager. 2015. "Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells" Viruses 7, no. 12: 6316-6345. https://doi.org/10.3390/v7122940
APA StyleRomero-Brey, I., & Bartenschlager, R. (2015). Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses, 7(12), 6316-6345. https://doi.org/10.3390/v7122940