
Alphacoronaviruses Detected in French Bats Are Phylogeographically Linked to Coronaviruses of European Bats

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
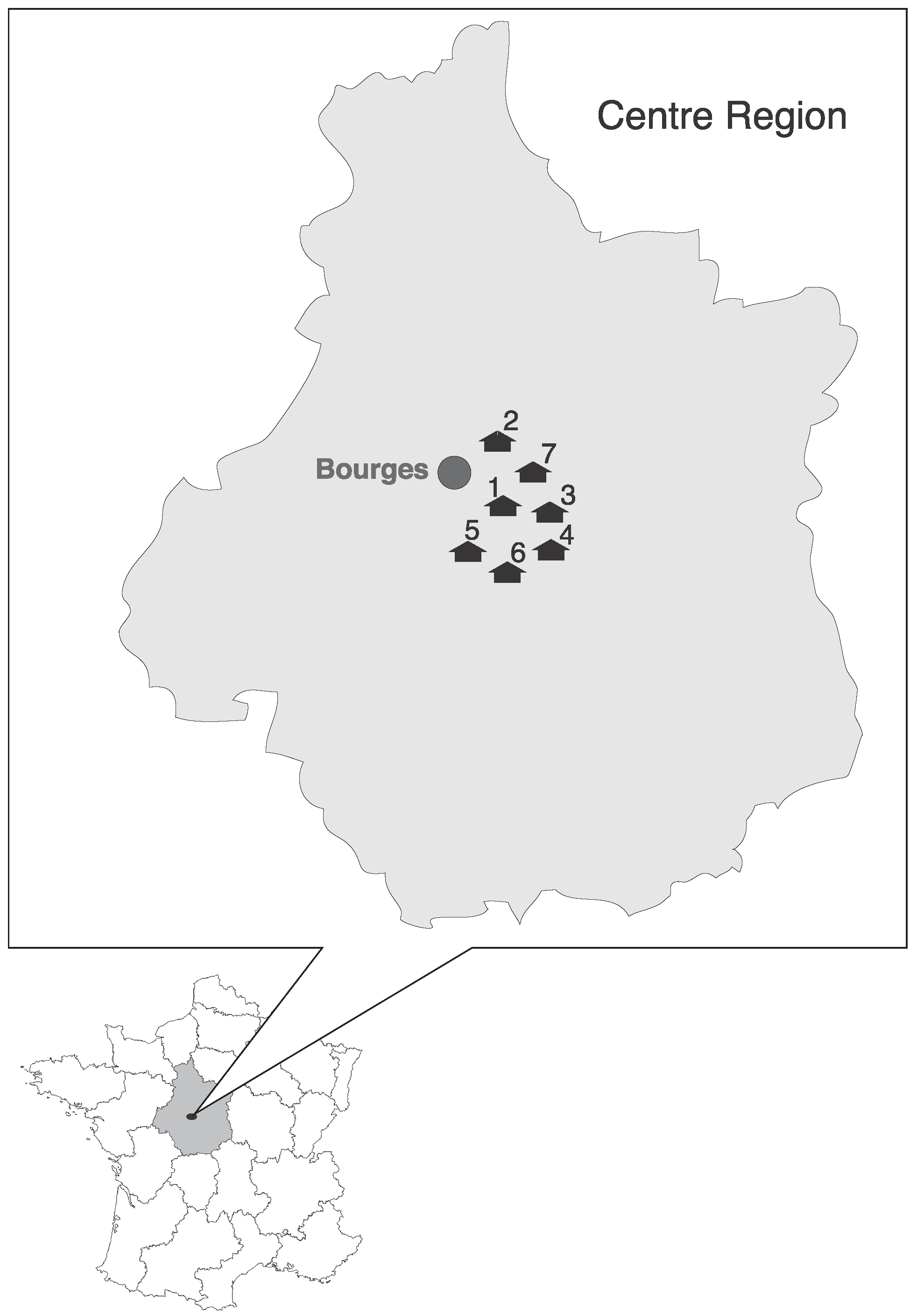
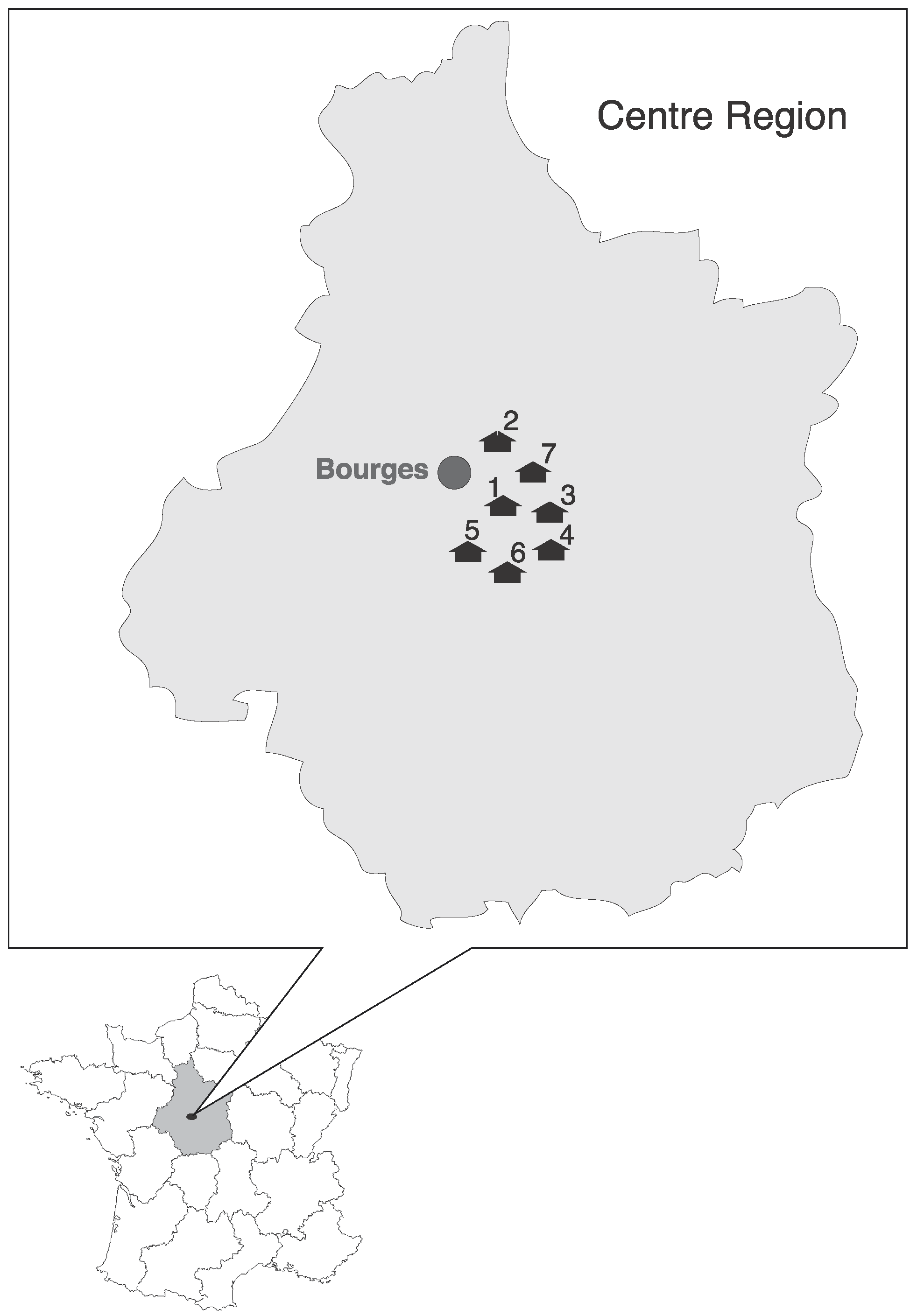
2.1. Study Area and Sampling

{kind=link}
{kind=link}
{kind=link}
| Positive Samples/Total of Tested Samples by Location | Total of Positive Samples/Total of Tested Samples | % of Positive | Sequences | Coronavirus Group | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bat Species | Size of Bat Colonies * | Day Roost | 1 | 2 | 3 | 4 | 5 | 6 | 7 | All Areas | |||
| Pipistrellus pipistrellus | Small and Medium | Attics, Barns | 3/53 | 2/20 | 0/10 | 0/35 | 5/118 | 4.2 | Ppip1_FR_2014, Ppip2_FR_2014, Ppip3_FR_2014 | α | |||
| Barbastella barbastellus | Small | Attic | 0/24 | 0 | |||||||||
| Myotis myotis | Small | Barn | 0/10 | 0 | |||||||||
| Eptesicus serotinus | Small | Attic | 0/10 | 0 | |||||||||
| Total | 5/162 | 3.1 | |||||||||||
2.2. Genome Detection and Sequencing
2.3. Sequence Analysis
2.4. Phylogeographic Analysis
| GenBank Accession Number | Group | Host Species | Geographic Origin | Name | Reference |
|---|---|---|---|---|---|
| AY903459 | β | Human | Belgium | OC43_BEL_2003 | [30] |
| KC243392 | β | Pipistrellus nathusii | Ukraine | Pnat_UKR_2011 | [31] |
| KC243391 | β | Pipistrellus nathusii | Romania | Pnat_ROM_2009 | [31] |
| KF906251 | β | Dromedary | United Arab Emirates | Dro_UAE_2013 | [14] |
| JX869059 | β | Human | Saudi Arabia | MERS_SAU_2012 | [32] |
| EF507780 | β | Human | France | HKU1_FR_2005 | [33] |
| GU190221 | β | Rhinolophus euryale | Bulgaria | Reur2_BLG_2008 | [17] |
| FJ588686 | β | Rhinolophus sinicus | China | Rsin_CHI_2006 | [34] |
| AY304488 | β | Civet | China | Civ_CHI_2003 | [35] |
| KJ652334 | α | Myotis daubentonii | Hungary | Mdau_HUN_2013 | [19] |
| KJ652333 | α | Myotis nattereri | Hungary | Mnat_HUN_2013 | [19] |
| KJ652332 | α | Pipistrellus pigmae | Hungary | Ppig_HUN_2013 | [3] |
| KJ652331 | α | Myotis myotis | Hungary | Mmyo_HUN_2013 | [3] |
| KJ652330 | α | Rhinolophus ferrumequinum | Hungary | Rfer_HUN_2013 | [3] |
| KJ652329 | α | Rhinolophus ferrumequinum | Hungary | Rfer_HUN_2013 | [3] |
| KF500949 | α | Pipistrellus kuhlii | Italy | Pkuh1_ITA_2010 | [18] |
| KF500945 | α | Pipistrellus kuhlii | Italy | Pkuh2_ITA_2011 | [18] |
| JF440366 | α | Myotis nattereri | United Kingdom | Mnat1_UK_2009 | [17] |
| JF440365 | α | Myotis nattereri | United Kingdom | Mnat2_UK_2009 | [17] |
| JF440353 | α | Myotis daubentonii | United Kingdom | Mdau1_UK_2009 | [12] |
| JF440351 | α | Myotis daubentonii | United Kingdom | Mdau2_UK_2009 | [12] |
| JF440349 | α | Myotis daubentonii | United Kingdom | Mdau3_UK_2009 | [17] |
| HQ184061 | α | Hypsugo savii | Spain | Hsav_SP_2007 | [16] |
| HQ184060 | α | Pipistrellus sp. | Spain | Psp_SP_2007 | [16] |
| HQ184058 | α | Pipistrellus kuhlii | Spain | Pkuh_SP_2007 | [16] |
| HQ184057 | α | Myotis myotis | Spain | Mmyo_SP_2007 | [16] |
| HQ184056 | α | Myotis daubentonii | Spain | Mdau_SP_2007 | [16] |
| HQ184051 | α | Nyctalus lasiopterus | Spain | Nlas_SP_2007 | [16] |
| GU190239 | α | Nyctalus leisleri | Bulgaria | Nlei_BLG_2008 | [36] |
| GU190237 | α | Rhinolophus euryale | Bulgaria | Reur1_BLG_2008 | [36] |
| GQ259966 | α | Myotis dasycneme | The Netherlands | Mdas_NLD_2006 | [15] |
| GQ259964 | α | Pipistrellus pipistrellus | The Netherlands | Ppip_NLD_2008 | [15] |
| GQ259967 | α | Myotis dasycneme | The Netherlands | Mdas_NLD_2007 | [15] |
| EU375871 | α | Myotis daubentonii | Germany | Mdau_GER_2007 | [14] |
| EU375869 | α | Pipistrellus nathusius | Germany | Pnat1_GER_2007 | [14] |
| EU375868 | α | Pipistrellus pigmae | Germany | Ppig1_GER-2007 | [14] |
| EU375867 | α | Pipistrellus pigmae | Germany | Ppig2_GER-2007 | [14] |
| EU375864 | α | Pipistrellus nathusius | Germany | Pnat2_GER_2007 | [14] |
| EU375863 | α | Myotis dasycneme | Germany | Mdas_GER_2007 | [14] |
| AY864196 | α | Miniopterus sp. | China | HKU8_CHI_2004 | [37] |
| DQ249228 | α | Miniopterus sp. | China | HKU8_CHI_2005 | [38] |
| NC009988 | α | Rhinolophus sp. | China | HKU2_CHI_2004 | [39] |
2.5. Statistical Analyses
2.6. Nucleotide Sequence Accession Numbers
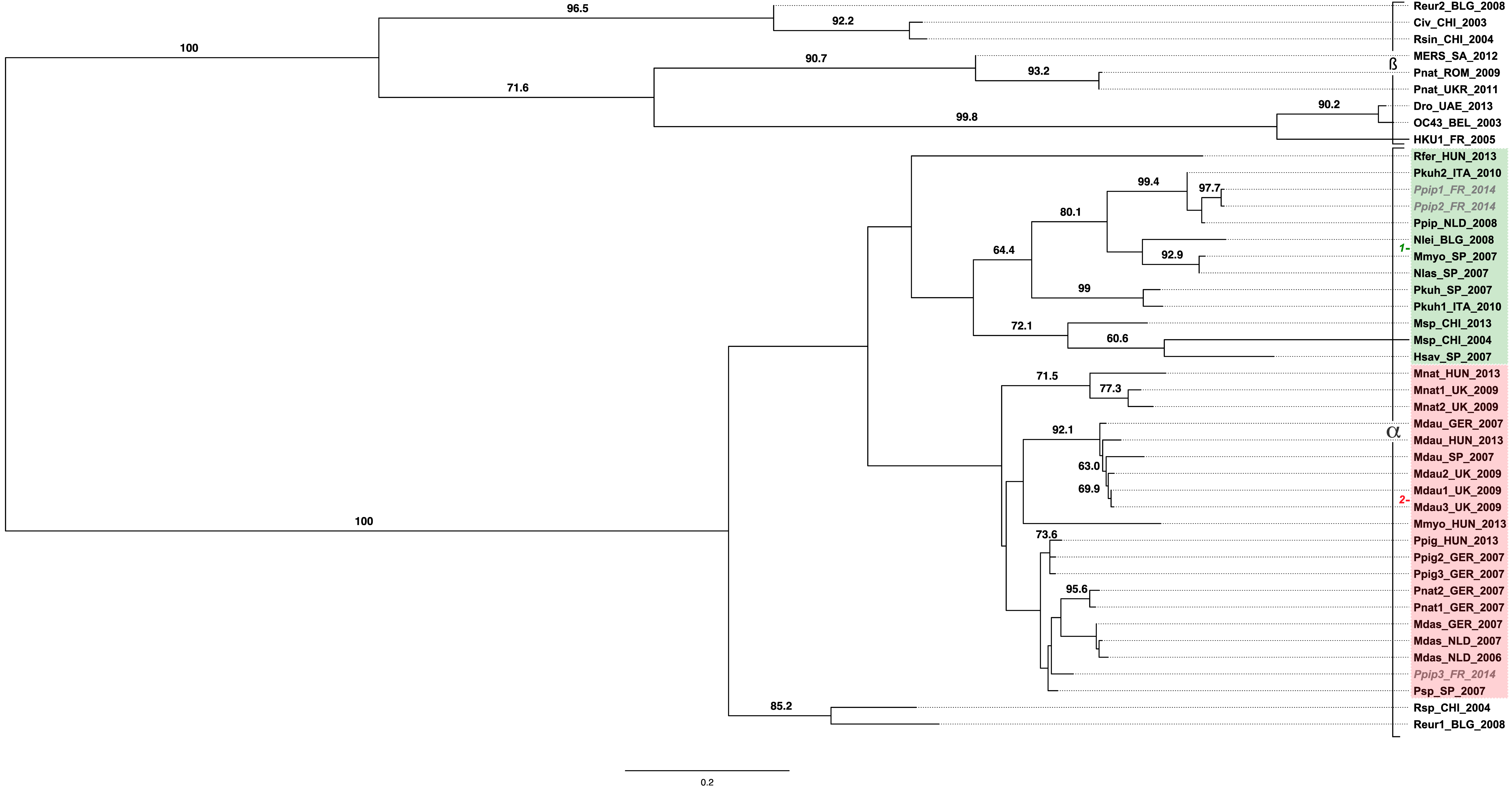
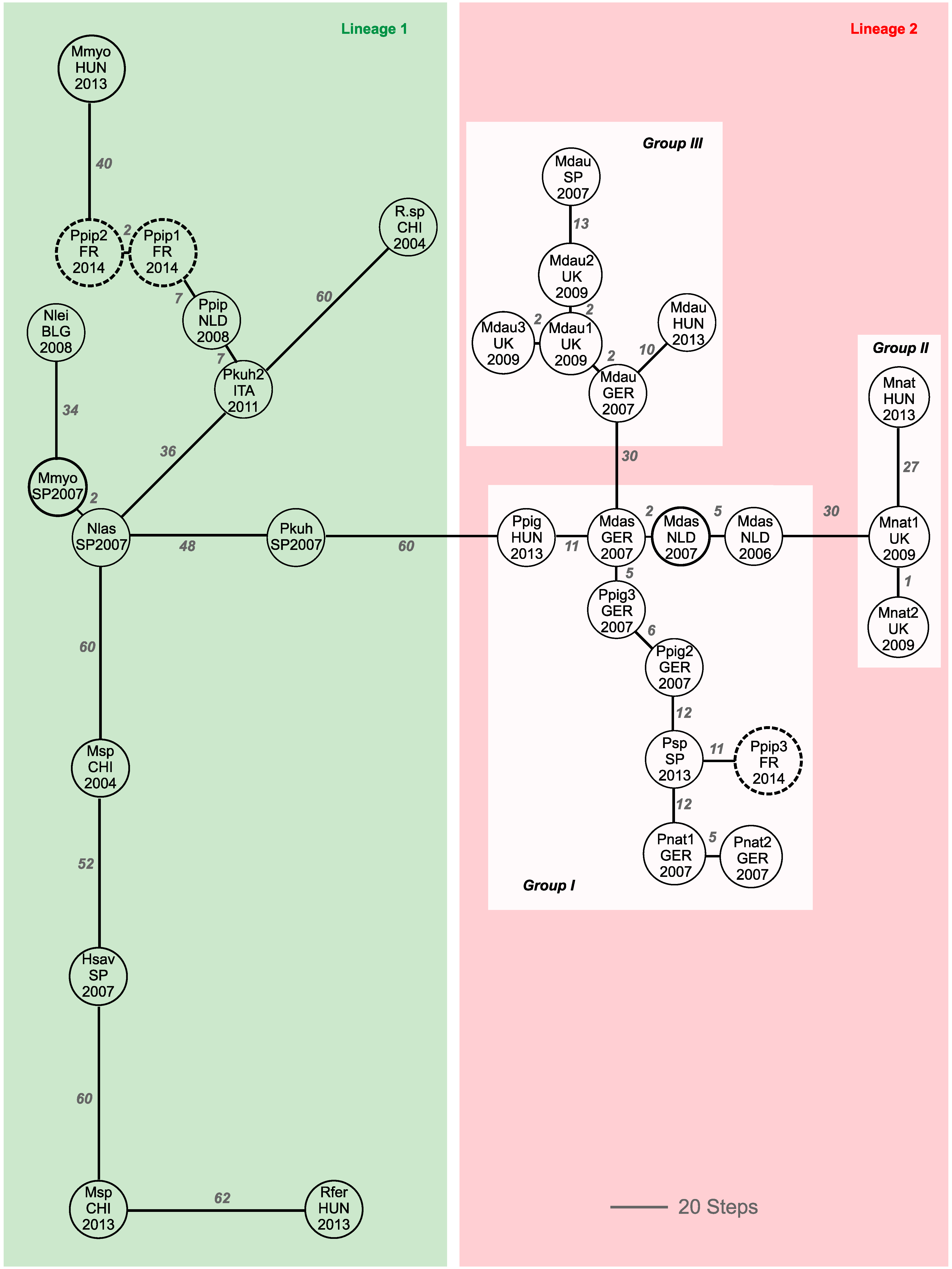
3. Results
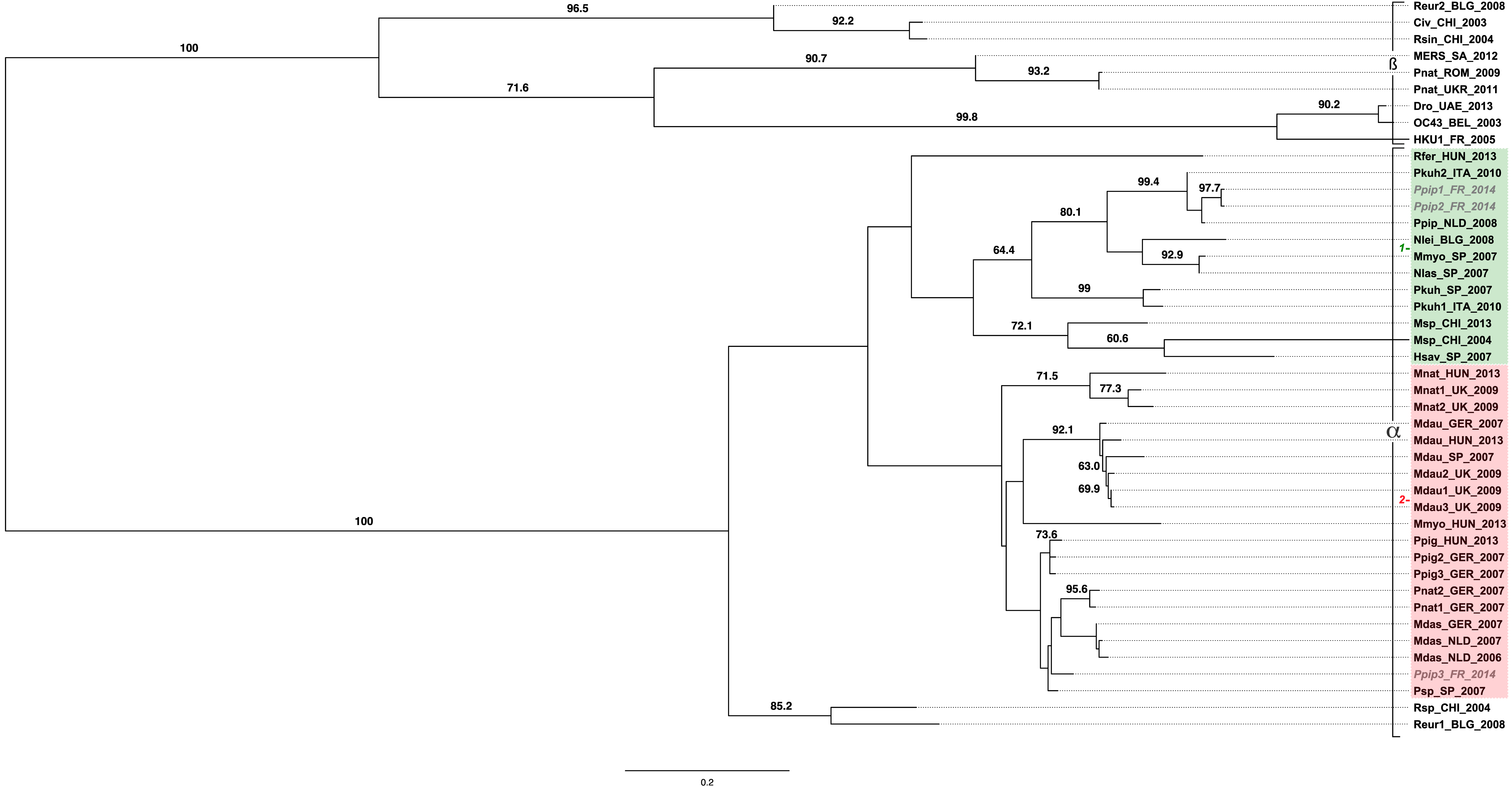
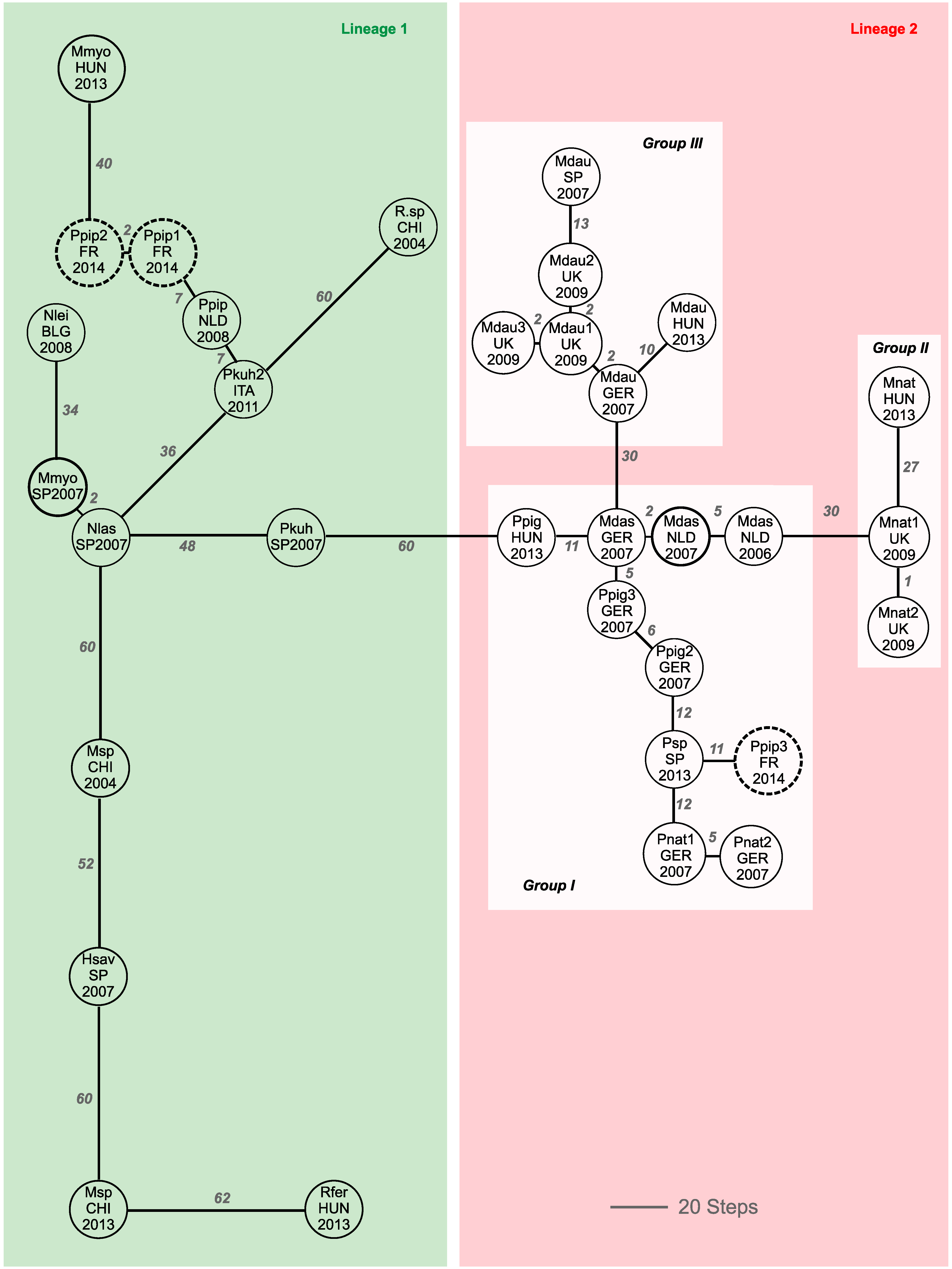
4. Discussion
4.1. Prevalence of Alpha-CoV in French Pipistrellus pipistrellus
4.2. Phylogeographic Relatedness among Alpha-CoVs Detected in European Bats
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [PubMed]
- Jones, K.E.; Purvis, A.; MacLarnon, A.; Bininda-Emonds, O.R.P.; Simmons, N.B. A phylogenetic supertree of the bats (Mammalia: Chiroptera). Biol. Rev. Camb. Philos. Soc. 2002, 77, 223–259. [Google Scholar] [CrossRef] [PubMed]
- Bisson, I.-A.; Safi, K.; Holland, R.A. Evidence for Repeated Independent Evolution of Migration in the Largest Family of Bats. PLoS ONE 2009, 4, e7504. [Google Scholar] [CrossRef] [PubMed]
- Protected Bat Species UNEP/EUROBATS. Available online: http://www.eurobats.org/about_eurobats/protected_bat_species (accessed on 15 July 2015).
- Brunet-Rossinni, A.K.; Austad, S.N. Ageing studies on bats: A review. Biogerontology 2004, 5, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-J.; Wen, H.-L.; Zhou, C.-M.; Chen, F.-F.; Luo, L.-M.; Liu, J.-W.; Yu, X.-J. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Bloomquist, E.W.; Lemey, P.; Suchard, M.A. Three roads diverged? Routes to phylogeographic inference. Trends Ecol. Evol. 2010, 25, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.H.; Shaw, G.M.; de Cock, K.M.; Sharp, P.M. AIDS as a zoonosis: Scientific and public health implications. Science 2000, 287, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Tan, Y.; Ghedin, E.; Wentworth, D.E.; st George, K.; Edelman, L.; Beck, E.T.; Fan, J.; Lam, T.T.-Y.; Kumar, S.; et al. Phylogeography of the spring and fall waves of the H1N1/09 pandemic influenza virus in the United States. J. Virol. 2011, 85, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.F.; de Carvalho-Dominguez-Souza, B.F.; Drexler, J.F.; Lima, R.S.; de Rosário, M.; de Oliveira-Hughes-Veiga-do-Rosário, M.; Netto, E.M. Theories about evolutionary origins of human hepatitis B virus in primates and humans. Braz. J. Infect. Dis. Off. Publ. Braz. Soc. Infect. Dis. 2014, 18, 535–543. [Google Scholar]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.E.M.; Lina, P.H.C.; Pielaat, A.; de Vries, A.; Dam-Deisz, C.; Adema, J.; Drexler, J.F.; Drosten, C.; Kooi, E.A. Circulation of group 2 coronaviruses in a bat species common to urban areas in Western Europe. Vector Borne Zoonotic Dis. 2010, 10, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Falcón, A.; Vázquez-Morón, S.; Casas, I.; Aznar, C.; Ruiz, G.; Pozo, F.; Perez-Breña, P.; Juste, J.; Ibáñez, C.; Garin, I.; et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol. 2011, 156, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- August, T.A.; Mathews, F.; Nunn, M.A. Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis. 2012, 12, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Lelli, D.; Papetti, A.; Sabelli, C.; Rosti, E.; Moreno, A.; Boniotti, M.B. Detection of coronaviruses in bats of various species in Italy. Viruses 2013, 5, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Dallos, B.; Görföl, T.; Boldogh, S.; Estók, P.; Kurucz, K.; Kutas, A.; Földes, F.; Oldal, M.; Németh, V.; et al. Molecular survey of RNA viruses in Hungarian bats: Discovering novel astroviruses, coronaviruses, and caliciviruses. Vector Borne Zoonotic Dis. 2014, 14, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Dietz, C.; von Helversen, O. Available online: http://scholar.google.fr/scholar_url?url=http://www.researchgate.net/profile/Christian_Dietz/publication/274838308_llustrated_Identification_key_to_the_Bats_of_Europe_-_complete_pdf/links/552b56a60cf2779ab7930be7.pdf&hl=fr&sa=X&scisig=AAGBfm1eq39Y0IQQR7Pv_3ffuC7mRbBiWw&nossl=1&oi=scholarr&ved=0CB4QgAMoADAAahUKEwj_84DXjPzIAhXH1hQKHWHaBkU (accessed on 6 November 2015).
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat Guano Virome: Predominance of Dietary Viruses from Insects and Plants plus Novel Mammalian Viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic Analysis of Viruses from Bat Fecal Samples Reveals Many Novel Viruses in Insectivorous Bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- De Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods) 4.0 Beta. Available online: http://www.sinauer.com/paup-phylogenetic-analysis-using-parsimony-and-other-methods-4-0-beta.html (accessed on 15 July 2015).
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- ATGC: SMS. Available online: http://www.atgc-montpellier.fr/sms/ (accessed on 15 July 2015).
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Schneider, S.; Roessli, D.; Excoffier, L. Arlequin: A Software for Population Genetics Data Analysis, 2000th ed.; User Manual Ver 2.000; University of Geneva: Geneva, Switzerland, 2000. [Google Scholar]
- Vijgen, L.; Keyaerts, E.; Lemey, P.; Moës, E.; Li, S.; Vandamme, A.-M.; van Ranst, M. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology 2005, 337, 85–92. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human Betacoronavirus 2c EMC/2012-related Viruses in Bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.M.E.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic Characterization of a Newly Discovered Coronavirus Associated with Acute Respiratory Distress Syndrome in Humans. mBio 2012, 3, e00473-12. [Google Scholar] [CrossRef] [PubMed]
- Vabret, A.; Dina, J.; Gouarin, S.; Petitjean, J.; Tripey, V.; Brouard, J.; Freymuth, F. Human (non-severe acute respiratory syndrome) coronavirus infections in hospitalised children in France. J. Paediatr. Child Health 2008, 44, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Hon, C.-C.; Li, Y.; Wang, D.; Xu, G.; Zhang, H.; Zhou, P.; Poon, L.L.M.; Lam, T.T.-Y.; Leung, F.C.-C.; et al. Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans. J. Gen. Virol. 2010, 91, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic Characterization of Severe Acute Respiratory Syndrome-Related Coronavirus in European Bats and Classification of Coronaviruses Based on Partial RNA-Dependent RNA Polymerase Gene Sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.M.; Chu, D.K.W.; Chan, K.H.; Wong, O.K.; Ellis, T.M.; Leung, Y.H.C.; Lau, S.K.P.; Woo, P.C.Y.; Suen, K.Y.; Yuen, K.Y.; et al. Identification of a novel coronavirus in bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Li, K.S.M.; Poon, R.W.S.; Wong, B.H.L.; Tsoi, H.; Yip, B.C.K.; Huang, Y.; Chan, K.; Yuen, K. Molecular diversity of coronaviruses in bats. Virology 2006, 351, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Wang, M.; Lam, C.S.F.; Xu, H.; Guo, R.; Chan, K.-H.; Zheng, B.-J.; et al. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 2007, 367, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.C.; Zhang, J.X.; Zhang, S.Y.; Wang, P.; Fan, X.H.; Li, L.F.; Li, G.; Dong, B.Q.; Liu, W.; Cheung, C.L.; et al. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 2006, 80, 7481–7490. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, P.; Marciano, S.; Scaravelli, D.; Priori, P.; Zecchin, B.; Capua, I.; Monne, I.; Cattoli, G. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes 2013, 48, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Baldwin, H.J.; Fumie Tateno, A.; Melim Zerbinati, R.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [PubMed]
- Demanche, C.; Deville, M.; Michaux, J.; Barriel, V.; Pinçon, C.; Aliouat-Denis, C.M.; Pottier, M.; Noël, C.; Viscogliosi, E.; Aliouat, E.M.; et al. What do Pneumocystis organisms tell us about the phylogeography of their hosts? The case of the woodmouse Apodemus sylvaticus in continental Europe and western Mediterranean islands. PLoS ONE 2015, 10, e0120839. [Google Scholar] [CrossRef] [PubMed]
- Realpe, T.; Correa, N.; Rozo, J.C.; Ferro, B.E.; Gomez, V.; Zapata, E.; Ribon, W.; Puerto, G.; Castro, C.; Nieto, L.M.; et al. Population structure among mycobacterium tuberculosis isolates from pulmonary tuberculosis patients in Colombia. PLoS ONE 2014, 9, e93848. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.B. Plate tectonics, seaways and climate in the historical biogeography of mammals. Mem. Inst. Oswaldo Cruz 2000, 95, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Carrington, C.V.F.; Foster, J.E.; Zhu, H.C.; Zhang, J.X.; Smith, G.J.D.; Thompson, N.; Auguste, A.J.; Ramkissoon, V.; Adesiyun, A.A.; Guan, Y. Detection and phylogenetic analysis of group 1 coronaviruses in South American bats. Emerg. Infect. Dis. 2008, 14, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Wacharapluesadee, S.; Duengkae, P.; Rodpan, A.; Kaewpom, T.; Maneeorn, P.; Kanchanasaka, B.; Yingsakmongkon, S.; Sittidetboripat, N.; Chareesaen, C.; Khlangsap, N.; et al. Diversity of coronavirus in bats from Eastern Thailand. Virol. J. 2015, 12, 57. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goffard, A.; Demanche, C.; Arthur, L.; Pinçon, C.; Michaux, J.; Dubuisson, J. Alphacoronaviruses Detected in French Bats Are Phylogeographically Linked to Coronaviruses of European Bats. Viruses 2015, 7, 6279-6290. https://doi.org/10.3390/v7122937
Goffard A, Demanche C, Arthur L, Pinçon C, Michaux J, Dubuisson J. Alphacoronaviruses Detected in French Bats Are Phylogeographically Linked to Coronaviruses of European Bats. Viruses. 2015; 7(12):6279-6290. https://doi.org/10.3390/v7122937
Chicago/Turabian StyleGoffard, Anne, Christine Demanche, Laurent Arthur, Claire Pinçon, Johan Michaux, and Jean Dubuisson. 2015. "Alphacoronaviruses Detected in French Bats Are Phylogeographically Linked to Coronaviruses of European Bats" Viruses 7, no. 12: 6279-6290. https://doi.org/10.3390/v7122937
APA StyleGoffard, A., Demanche, C., Arthur, L., Pinçon, C., Michaux, J., & Dubuisson, J. (2015). Alphacoronaviruses Detected in French Bats Are Phylogeographically Linked to Coronaviruses of European Bats. Viruses, 7(12), 6279-6290. https://doi.org/10.3390/v7122937