
An Evolutionarily Young Polar Bear (Ursus maritimus) Endogenous Retrovirus Identified from Next Generation Sequence Data


Abstract
:1. Introduction
2. Results
2.1. Identification of an Evolutionarily Young Gammaretroviral ERV in Polar Bears



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | Start | End | Savings | Seq pv |
|---|---|---|---|---|
| scaffold143_3,240,578-3,261,249_LTR-A_solitary | 311 | 311 | 8 | 0,000999 |
| scaffold162_1-18,384_LTR-A/B | 186 | 216 | 30 | 0,000999 |
| Polar Bear Scaffolds | TSD Sequence |
|---|---|
| Scaffold 1 | CATC/CATC |
| Scaffold 7 | ACTT/ACTT |
| Scaffold 162 | AGGC/AGGC |
| Scaffold 200 | ATGG/ATGG |
| Scaffold 182 | CTTC/CTTC |
| Scaffold 30 | CTCT/CTCT |
| Scaffold 80 | GTGT/GTGT |
| Scaffold 42 | ATCT/ATCT |
| Scaffold 52 | TATC/TATC |
| Scaffold 217 | GTAG/GTAG |
| Scaffold 143 | TGAA/TGAA |
| Scaffold 72 | GCAC/GCAC |
| Scaffold 155 | AGAC/AGAC |
| Scaffold 5 | GTAT/GTAT |
| Scaffold 18,513 | N/A |
| Scaffold 18,762 | N/A |
| Scaffold 23,372 | N/A |
| Scaffold 35,249 | N/A |
| Scaffold 62,681 | N/A |
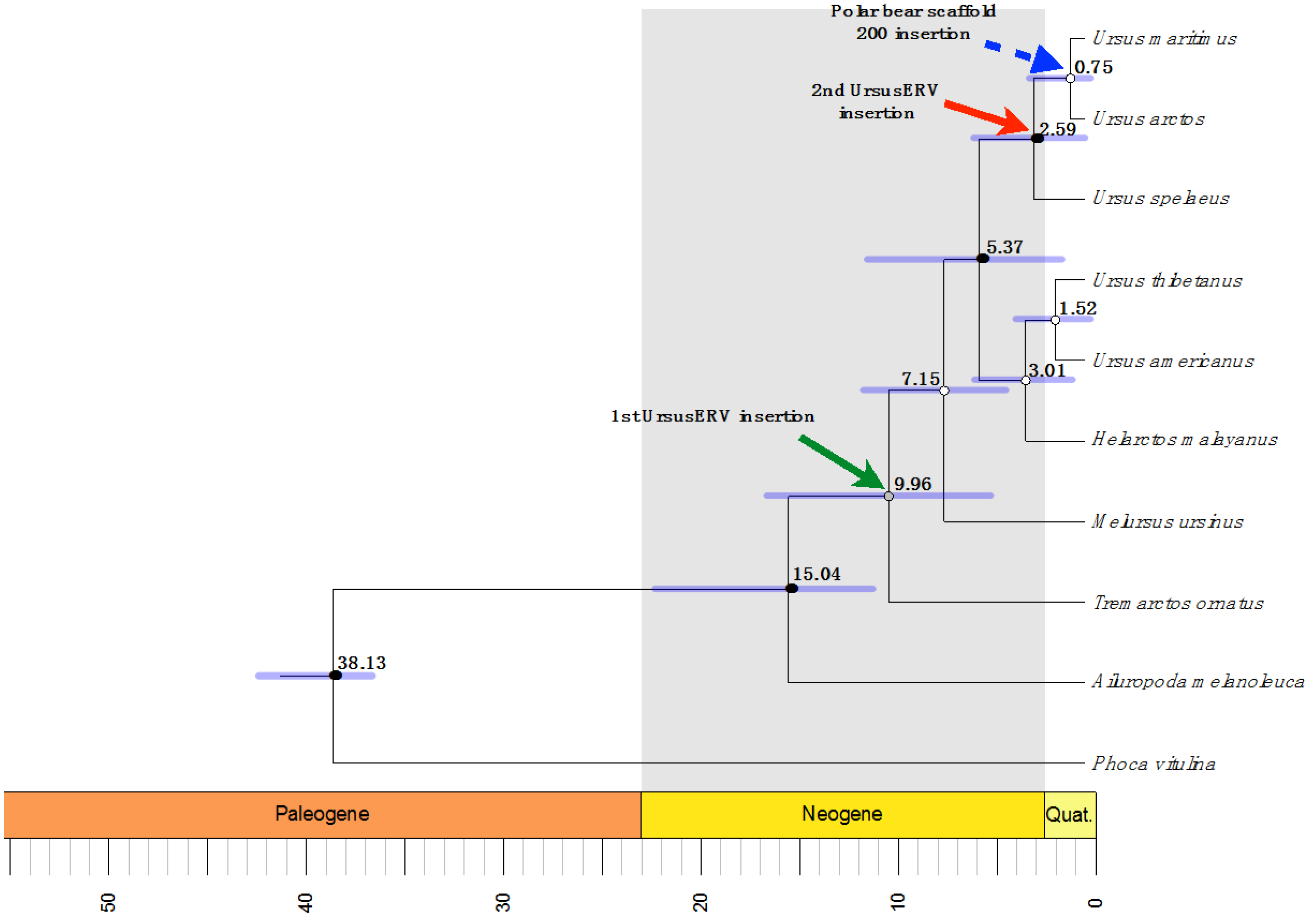
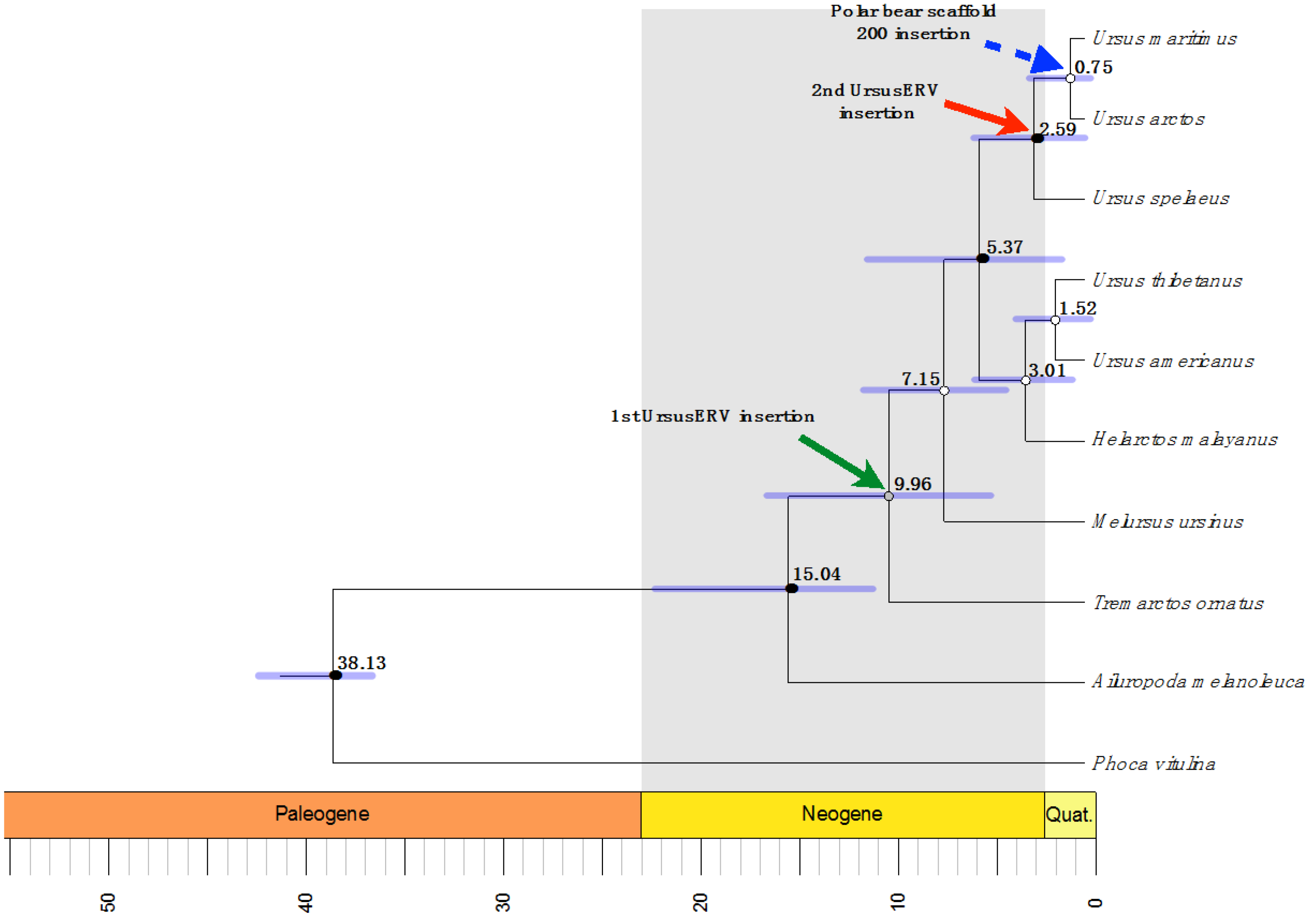
2.2. UrsusERV Age Estimation
| Polar Bear Scaffolds | 5′ LTR Location | 3′ LTR Location | Overall Mean K2P Distance | Age Estimation Mya |
|---|---|---|---|---|
| Scaffold 1 | 66,674,296–66,674,797 | 66,681,308–66,681,790 | 0.002 | 0.625 |
| Scaffold 7 | 30,626,500–30,627,187 | 30,633,692–30,634,278 | 0.007 | 2.1875 |
| Scaffold 162 | 7152–7643 | 13,824–14,441 | ND | ND |
| Scaffold 200 | 145,219–145,715 | 152,850–153,350 | 0 | 0 |
2.3. UrsusERV Molecular and Genome Screening of Bear Species
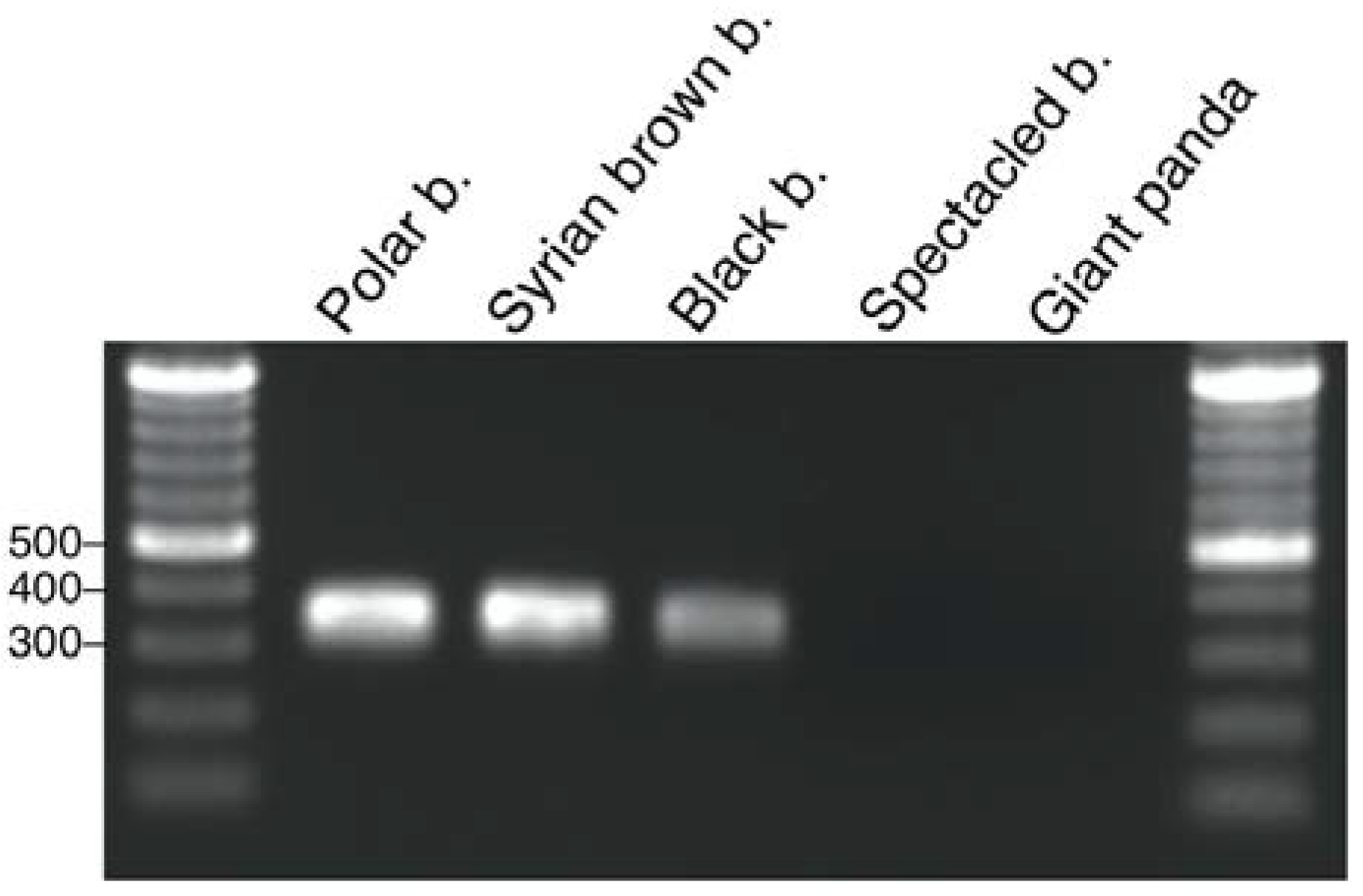
2.4. UrsusERV Phylogenetic Analysis
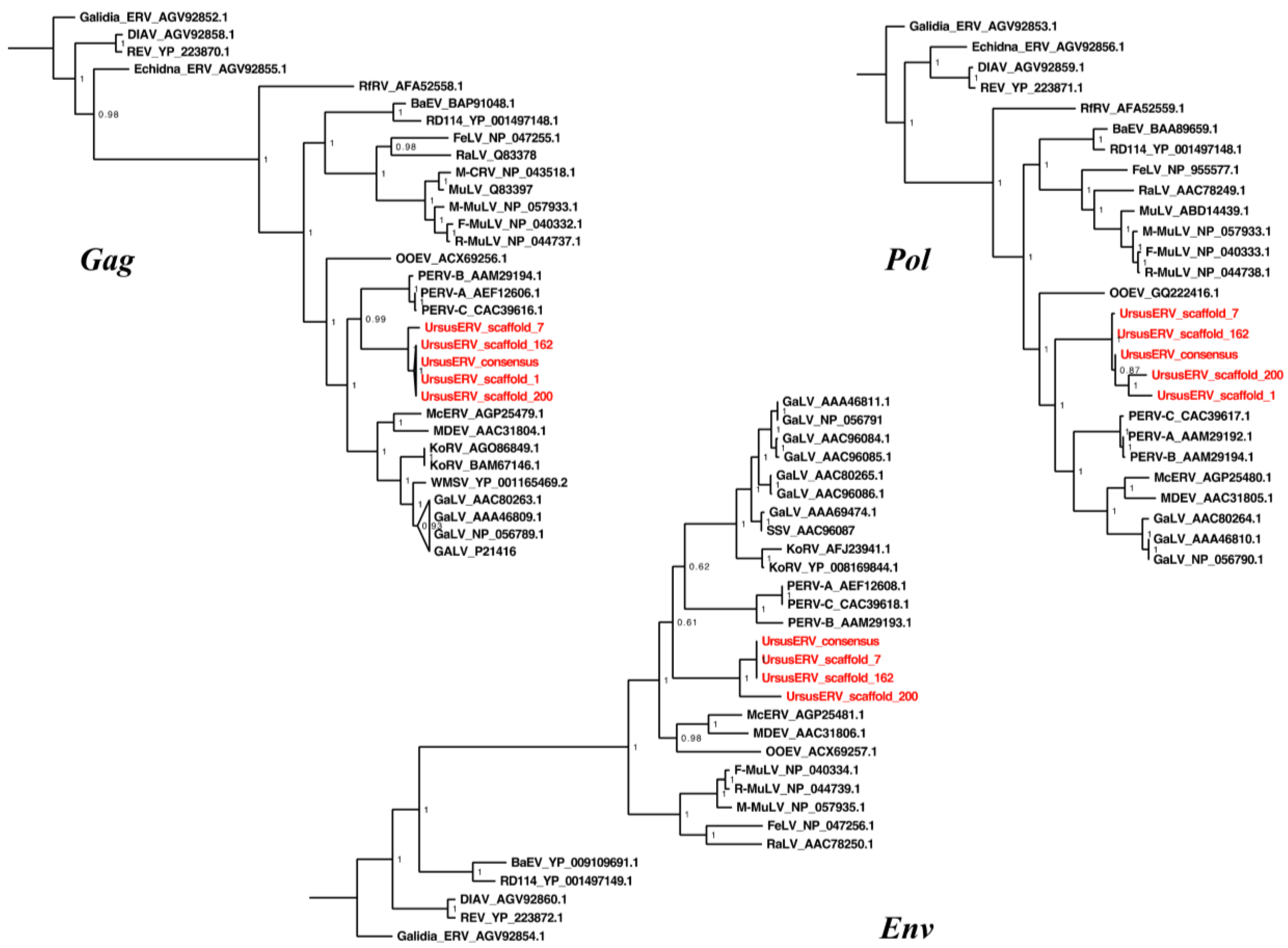

3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Nucleic Acid Preparation and Next Generation Sequencing
4.3. UrsusERV Polymerase Chain Reaction in Multiple Bear Species
4.4. Mining of UrsusERV Sequences in the Polar Bear Genome
4.5. Generation of UrsusERV Provirus and LTR Consensus Sequences
4.6. Recombination Inference Analysis
4.7. Sequence Read Archive and Bioinformatic Analysis
4.8. Phylogenetic Analysis
4.9. Age Estimation of UrsusERV Proviral Sequences
4.10. Estimation of Divergence Times
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lamere, S.A.; st Leger, J.A.; Schrenzel, M.D.; Anthony, S.J.; Rideout, B.A.; Salomon, D.R. Molecular characterization of a novel gammaretrovirus in killer whales (Orcinus orca). J. Virol. 2009, 83, 12956–12967. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Q.; He, G.; Rossiter, S.J.; Holmes, E.C.; Cui, J. Ancient invasion of an extinct γ retrovirus in cetaceans. Virology 2013, 441, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.J. Evolution at the host-retrovirus interface. BioEssays 2006, 28, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Rambaut, A.; Pybus, O.G. The evolutionary dynamics of endogenous retroviruses. TIM 2005, 13, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Tsangaras, K.; Heeger, F.; Avila-Arcos, M.; Stenglein, M.D.; Chen, W.; Sun, W.; Mazzoni, C.J.; Osterrieder, N.; Greenwood, A.D. A novel endogenous betaretrovirus group characterized from polar bears (Ursus maritimus) and giant pandas (Ailuropoda melanoleuca). Virology 2013, 443, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, A.M.; Gifford, R.J. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 2013, 11, e1001642. [Google Scholar] [CrossRef] [PubMed]
- Elleder, D.; Kim, O.; Padhi, A.; Bankert, J.G.; Simeonov, I.; Schuster, S.C.; Wittekindt, N.E.; Motameny, S.; Poss, M. Polymorphic integrations of an endogenous γ retrovirus in the mule deer genome. J. Virol. 2012, 86, 2787–2796. [Google Scholar] [CrossRef] [PubMed]
- Heidmann, O.; Vernochet, C.; Dupressoir, A.; Heidmann, T. Identification of an endogenous retroviral envelope gene with fusogenic activity and placenta-specific expression in the rabbit: A new “syncytin” in a third order of mammals. Retrovirology 2009, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Meese, E. Human endogenous retroviruses in the primate lineage and their influence on host genomes. Cytogenet. Genome Res. 2005, 110, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Tarlinton, R.E.; Meers, J.; Young, P.R. Retroviral invasion of the koala genome. Nature 2006, 442, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, F.; Caporale, M.; Varela, M.; Biek, R.; Chessa, B.; Alberti, A.; Golder, M.; Mura, M.; Zhang, Y.P.; Yu, L.; et al. A paradigm for virus-host coevolution: Sequential counter-adaptations between endogenous and exogenous retroviruses. PLoS Pathog. 2007, 3, e170. [Google Scholar] [CrossRef] [PubMed]
- Avila-Arcos, M.C.; Ho, S.Y.; Ishida, Y.; Nikolaidis, N.; Tsangaras, K.; Honig, K.; Medina, R.; Rasmussen, M.; Fordyce, S.L.; Calvignac-Spencer, S.; et al. One hundred twenty years of Koala retrovirus evolution determined from museum skins. Mol. Biol. Evol. 2013, 30, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, C.; Simmonds, P. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J. Mol. Evol. 2004, 59, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Sistiaga-Poveda, M.; Jugo, B.M. Evolutionary dynamics of endogenous jaagsiekte sheep retroviruses proliferation in the domestic sheep, mouflon and Pyrenean chamois. Heredity 2014, 112, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Stoye, J.P. Koala retrovirus: A genome invasion in real time. Genome Biol. 2006, 7, 241. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsangaras, K.; Siracusa, M.C.; Nikolaidis, N.; Ishida, Y.; Cui, P.; Vielgrader, H.; Helgen, K.M.; Roca, A.L.; Greenwood, A.D. Hybridization capture reveals evolution and conservation across the entire Koala retrovirus genome. PLoS ONE 2014, 9, e95633. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Zhao, K.; Greenwood, A.D.; Roca, A.L. Proliferation of endogenous retroviruses in the early stages of a host germ line invasion. Mol. Biol. Evol. 2015, 32, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, N.M.; Farrell, K.B.; Eiden, M.V. In vitro characterization of a Koala retrovirus. J. Virol. 2006, 80, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Escalera-Zamudio, M.; Mendoza, M.L.; Heeger, F.; Loza-Rubio, E.; Rojas-Anaya, E.; Mendez-Ojeda, M.L.; Taboada, B.; Mazzoni, C.J.; Arias, C.F.; Greenwood, A.D. A novel endogenous betaretrovirus in the common vampire bat (Desmodus rotundus) suggests multiple independent infection and cross-species transmission events. J. Virol. 2015, 89, 5180–5184. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, J.; Tachedjian, G.; Tachedjian, M.; Holmes, E.C.; Zhang, S.; Wang, L.F. Identification of diverse groups of endogenous γ retroviruses in mega- and microbats. J. Gen. Virol. 2012, 93, 2037–2045. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, G.; Willerslev, E.; Wang, J.; Wang, J. Genomic data from the polar bear (Ursus maritimus). GigaScience 2011, 10, 100008. [Google Scholar]
- Li, R.; Fan, W.; Tian, G.; Zhu, H.; He, L.; Cai, J.; Huang, Q.; Cai, Q.; Li, B.; Bai, Y.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Szentiks, C.A.; Tsangaras, K.; Abendroth, B.; Scheuch, M.; Stenglein, M.D.; Wohlsein, P.; Heeger, F.; Hoveler, R.; Chen, W.; Sun, W.; et al. Polar bear encephalitis: Establishment of a comprehensive next-generation pathogen analysis pipeline for captive and free-living wildlife. J. Comp. Pathol. 2014, 150, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Pages, M.; Calvignac, S.; Klein, C.; Paris, M.; Hughes, S.; Hanni, C. Combined analysis of fourteen nuclear genes refines the Ursidae phylogeny. Mol. Phylogenet. Evol. 2008, 47, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Hailer, F.; Kutschera, V.E.; Hallstrom, B.M.; Klassert, D.; Fain, S.R.; Leonard, J.A.; Arnason, U.; Janke, A. Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science 2012, 336, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, Q.W.; Ryder, O.A.; Zhang, Y.P. Phylogeny of the bears (Ursidae) based on nuclear and mitochondrial genes. Mol. Phylogenet. Evol. 2004, 32, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, 2010. pdb prot5448. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Schuster, S.C.; Welch, A.J.; Ratan, A.; Bedoya-Reina, O.C.; Zhao, F.; Kim, H.L.; Burhans, R.C.; Drautz, D.I.; Wittekindt, N.E.; et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. USA 2012, 109, E2382–E2390. [Google Scholar] [CrossRef] [PubMed]
- Scobie, L.; Takeuchi, Y. Porcine endogenous retrovirus and other viruses in xenotransplantation. Curr. Opin. Organ Transpl. 2009, 14, 175–179. [Google Scholar] [CrossRef]
- Lindqvist, C.; Schuster, S.C.; Sun, Y.; Talbot, S.L.; Qi, J.; Ratan, A.; Tomsho, L.P.; Kasson, L.; Zeyl, E.; Aars, J.; et al. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proc. Natl. Acad. Sci. USA 2010, 107, 5053–5057. [Google Scholar] [CrossRef] [PubMed]
- Repeatmasker Open-3.0. Available online: http://www.repeatmasker.org (accessed on 20 December 2014).
- Sperber, G.; Lovgren, A.; Eriksson, N.E.; Benachenhou, F.; Blomberg, J. Retrotector online, a rational tool for analysis of retroviral elements in small and medium size vertebrate genomic sequences. BMC Bioinform. 2009, 10, S4. [Google Scholar] [CrossRef] [PubMed]
- Tempel, S. Using and understanding repeatmasker. Methods Mol. Biol. 2012, 859, 29–51. [Google Scholar] [PubMed]
- Hanger, J.J.; Bromham, L.D.; McKee, J.J.; O’Brien, T.M.; Robinson, W.F. The nucleotide sequence of Koala (Phascolarctos cinereus) retrovirus: A novel type C endogenous virus related to gibbon ape leukemia virus. J. Virol. 2000, 74, 4264–4272. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Sperber, G.O.; Airola, T.; Jern, P.; Blomberg, J. Automated recognition of retroviral sequences in genomic data–retrotector. Nucleic Acids Res. 2007, 35, 4964–4976. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Etherington, G.J.; Dicks, J.; Roberts, I.N. Recombination analysis tool (RAT): A program for the high-throughput detection of recombination. Bioinformatics 2005, 21, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Gard: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Maydt, J.; Lengauer, T. Recco: Recombination analysis using cost optimization. Bioinformatics 2006, 22, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Dorman, K.S.; Fang, F.; Suchard, M.A. Dual multiple change-point model leads to more accurate recombination detection. Bioinformatics 2005, 21, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Serrao, E.; Ballandras-Colas, A.; Cherepanov, P.; Maertens, G.N.; Engelman, A.N. Key determinants of target DNA recognition by retroviral intasomes. Retrovirology 2015, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Belshaw, R.; Watson, J.; Katzourakis, A.; Howe, A.; Woolven-Allen, J.; Burt, A.; Tristem, M. Rate of recombinational deletion among human endogenous retroviruses. J. Virol. 2007, 81, 9437–9442. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.F.; Coffin, J.M. Human endogenous retroviral elements as indicators of ectopic recombination events in the primate genome. Genetics 2005, 171, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Stoye, J.P. Endogenous retroviruses: Still active after all these years? Curr. Biol. 2001, 11, R914–R916. [Google Scholar] [CrossRef]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC table browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Krause, J.; Unger, T.; Nocon, A.; Malaspinas, A.S.; Kolokotronis, S.O.; Stiller, M.; Soibelzon, L.; Spriggs, H.; Dear, P.H.; Briggs, A.W.; et al. Mitochondrial genomes reveal an explosive radiation of extinct and extant bears near the miocene-pliocene boundary. BMC Evol. Biol. 2008, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.A.; Stirling, I.; Kistler, L.; Salamzade, R.; Ersmark, E.; Fulton, T.L.; Stiller, M.; Green, R.E.; Shapiro, B. Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Mol. Ecol. 2015, 24, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. Mafft: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Hayward, A.; Grabherr, M.; Jern, P. Broad-scale phylogenomics provides insights into retrovirus-host evolution. Proc. Natl. Acad. Sci. USA 2013, 110, 20146–20151. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Tonjes, R.R.; Niebert, M. Relative age of proviral porcine endogenous retrovirus sequences in Sus scrofa based on the molecular clock hypothesis. J. Virol. 2003, 77, 12363–12368. [Google Scholar] [CrossRef] [PubMed]
- Magiorkinis, G.; Gifford, R.J.; Katzourakis, A.; de Ranter, J.; Belshaw, R. Env-less endogenous retroviruses are genomic superspreaders. Proc. Natl. Acad. Sci. USA 2012, 109, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Flockerzi, A.; Burkhardt, S.; Schempp, W.; Meese, E.; Mayer, J. Human endogenous retrovirus HERV-K14 families: Status, variants, evolution, and mobilization of other cellular sequences. J. Virol. 2005, 79, 2941–2949. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.; Tristem, M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 2003, 26, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Schrago, C.G.; Russo, C.A. Timing the origin of new world monkeys. Mol. Biol. Evol. 2003, 20, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- Marcel, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Prottest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. Mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. Beast 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Heath, T.A.; Huelsenbeck, J.P.; Stadler, T. The fossilized birth-death process for coherent calibration of divergence-time estimates. Proc. Natl. Acad. Sci. USA 2014, 111, E2957–E2966. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ho, S.Y.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- R: A Language and Environment for Statistical Computing. R foundation for Statistical Computing: Vienna, Austria. Available online: http://www.R-project.org2013 (accessed on 1 September 2015).
- Revell, L.J. Phytools: An r package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Heibl, C. Phyloch: R language tree plotting tools and interfaces to diverse phylogenetic software packages. 2008. [Google Scholar]
- Bell, M.A.; Lloyd, G.T. Strap: An R package for plotting phylogenies against stratigraphy and assessing their stratigraphic congruence. Palaeo 2015, 58, 379–389. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsangaras, K.; Mayer, J.; Alquezar-Planas, D.E.; Greenwood, A.D. An Evolutionarily Young Polar Bear (Ursus maritimus) Endogenous Retrovirus Identified from Next Generation Sequence Data. Viruses 2015, 7, 6089-6107. https://doi.org/10.3390/v7112927
Tsangaras K, Mayer J, Alquezar-Planas DE, Greenwood AD. An Evolutionarily Young Polar Bear (Ursus maritimus) Endogenous Retrovirus Identified from Next Generation Sequence Data. Viruses. 2015; 7(11):6089-6107. https://doi.org/10.3390/v7112927
Chicago/Turabian StyleTsangaras, Kyriakos, Jens Mayer, David E. Alquezar-Planas, and Alex D. Greenwood. 2015. "An Evolutionarily Young Polar Bear (Ursus maritimus) Endogenous Retrovirus Identified from Next Generation Sequence Data" Viruses 7, no. 11: 6089-6107. https://doi.org/10.3390/v7112927
APA StyleTsangaras, K., Mayer, J., Alquezar-Planas, D. E., & Greenwood, A. D. (2015). An Evolutionarily Young Polar Bear (Ursus maritimus) Endogenous Retrovirus Identified from Next Generation Sequence Data. Viruses, 7(11), 6089-6107. https://doi.org/10.3390/v7112927