
Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
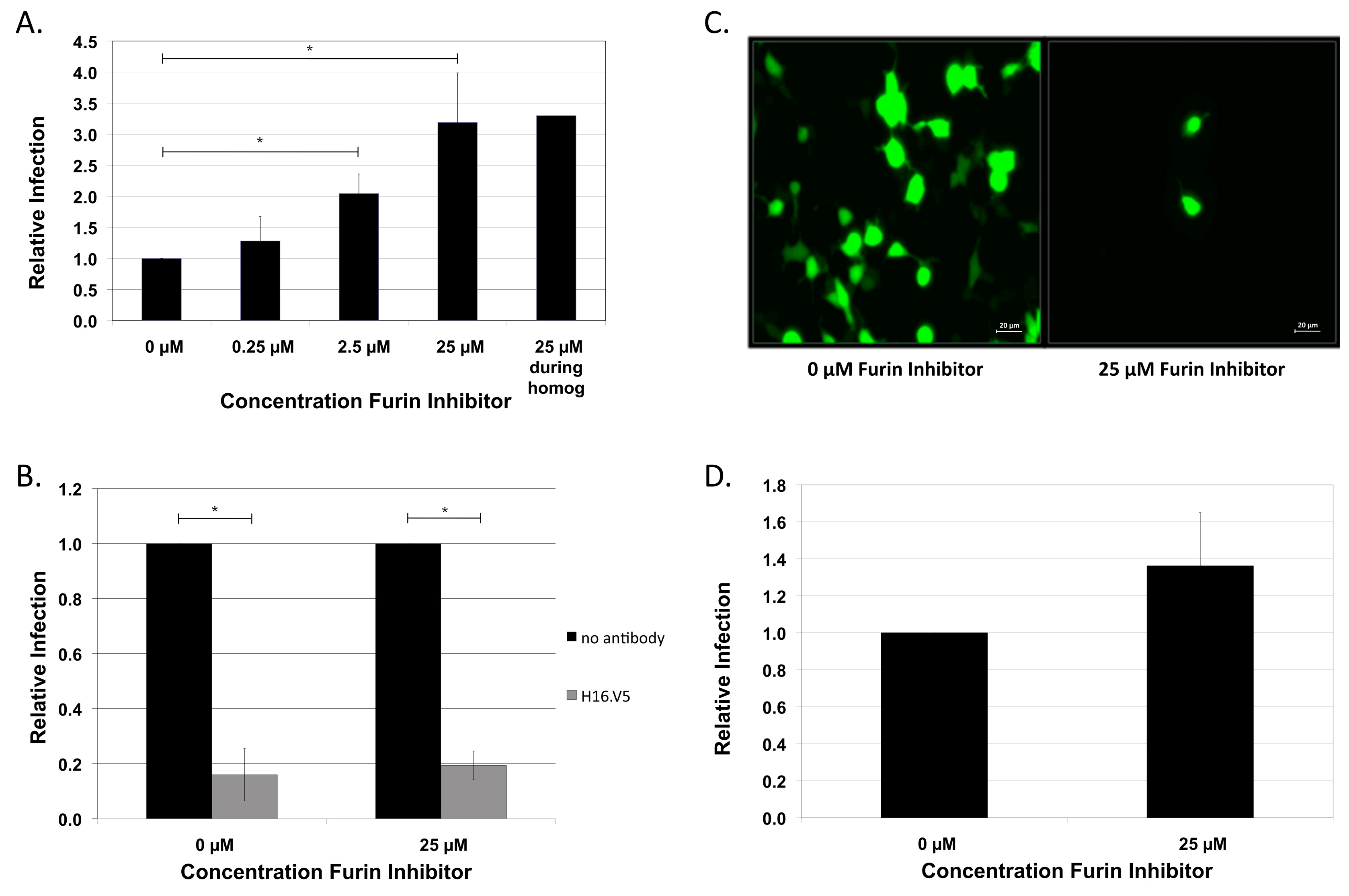
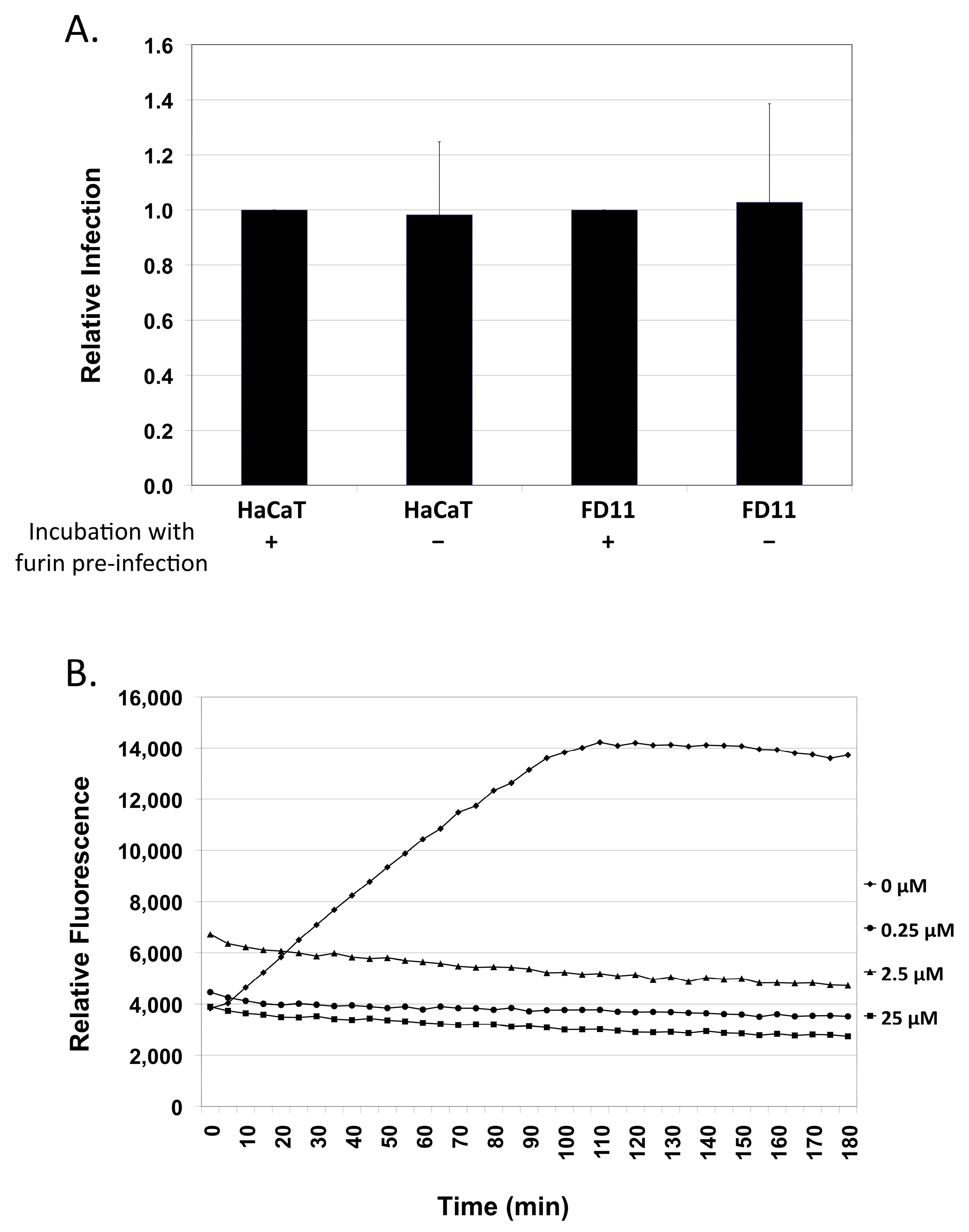
2.1. HPV16 NV Is Independent of Cellular Furin and Furin-Related Proprotein Convertases for Infection of HaCaT Keratinocytes
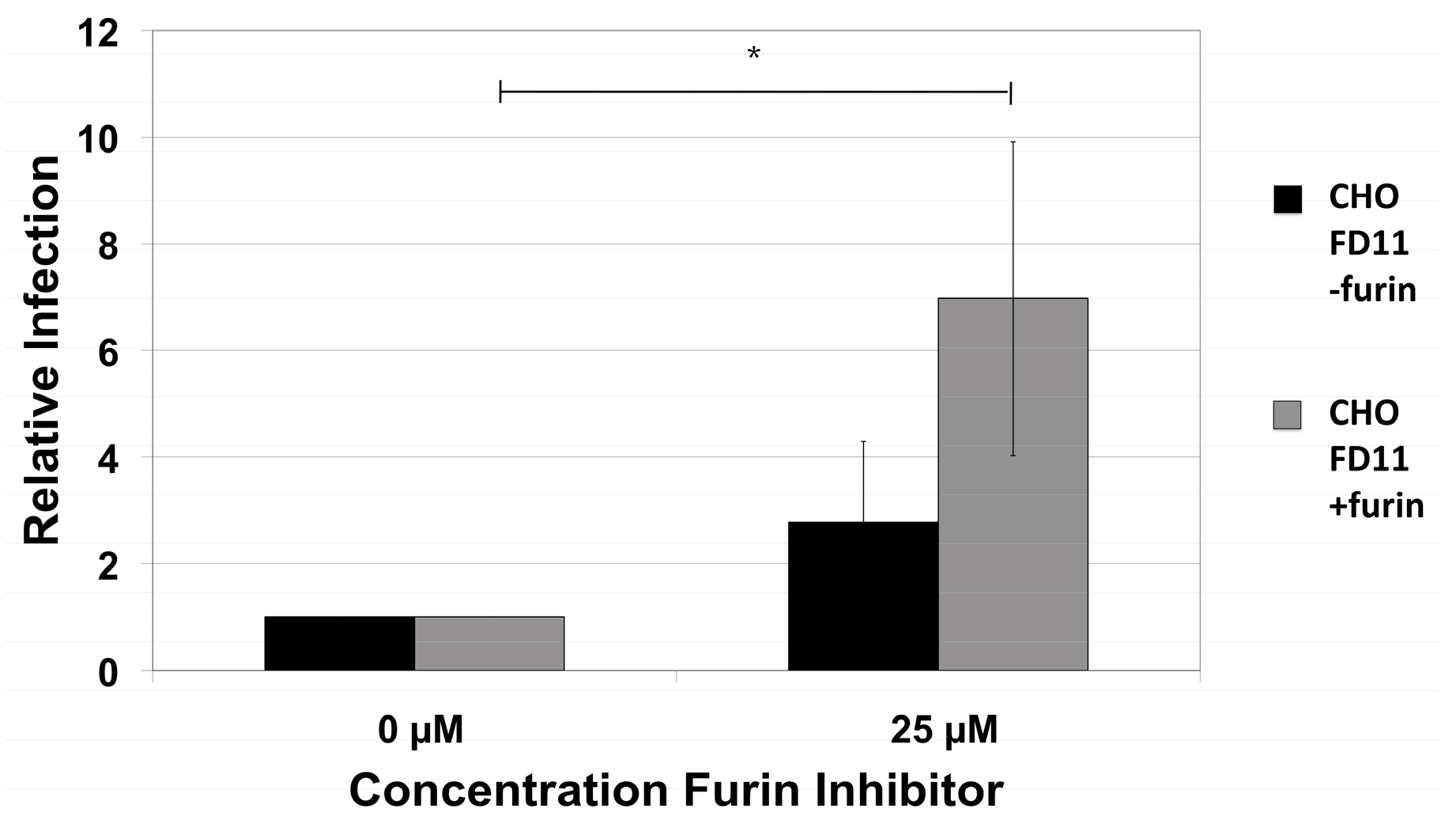
2.2. HPV16 NV Can Infect Furin-Deficient Cells
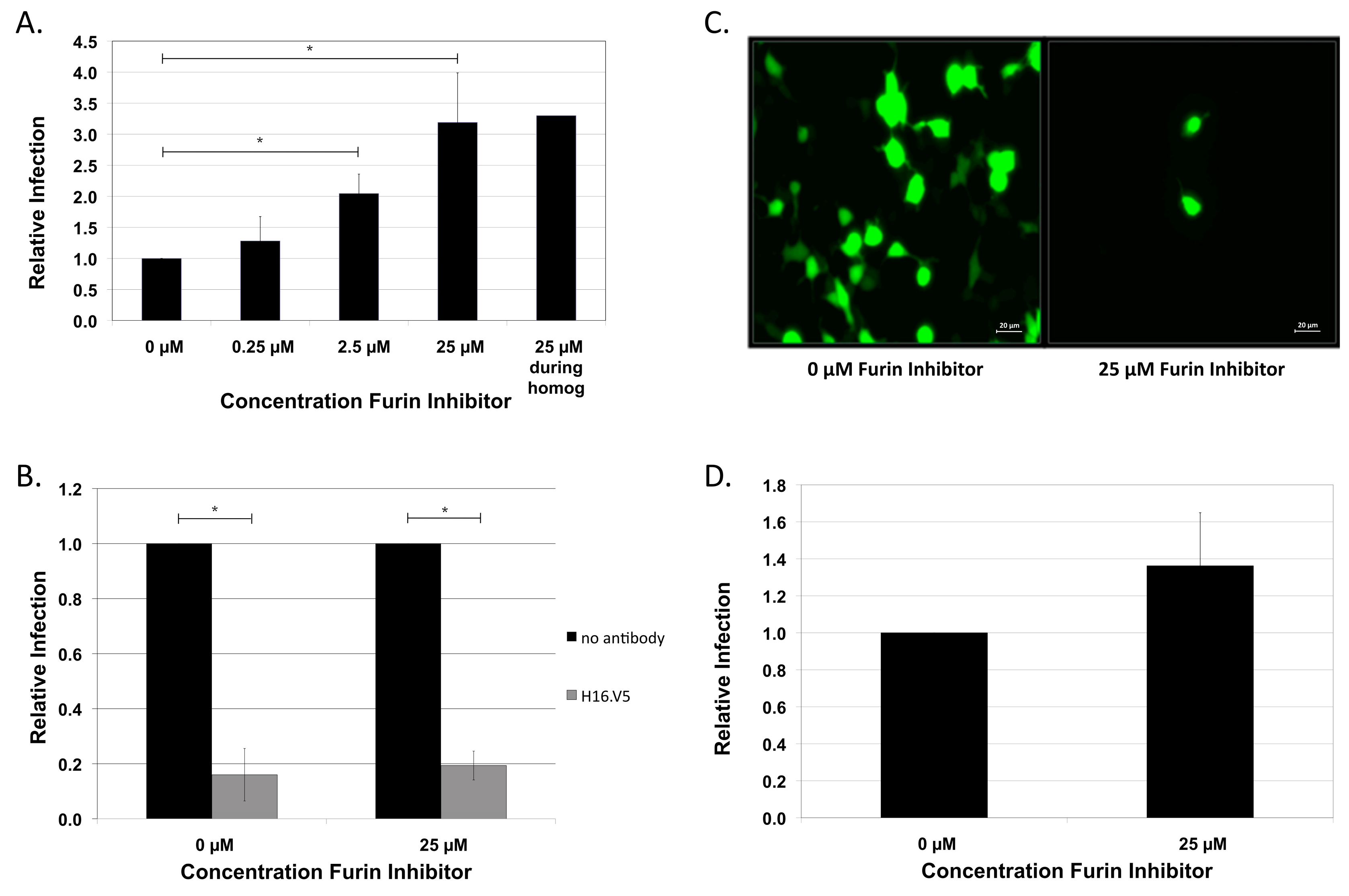
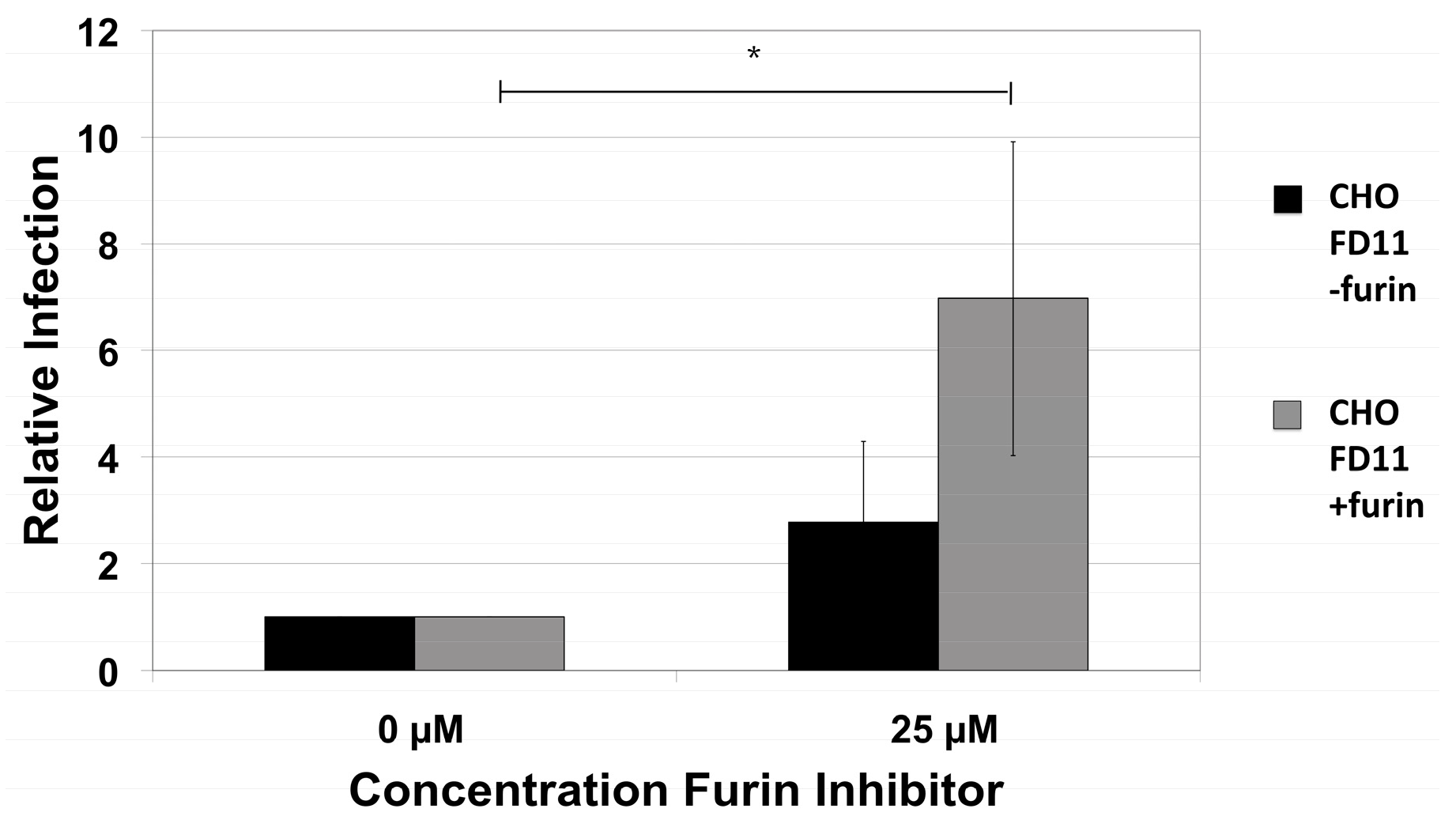
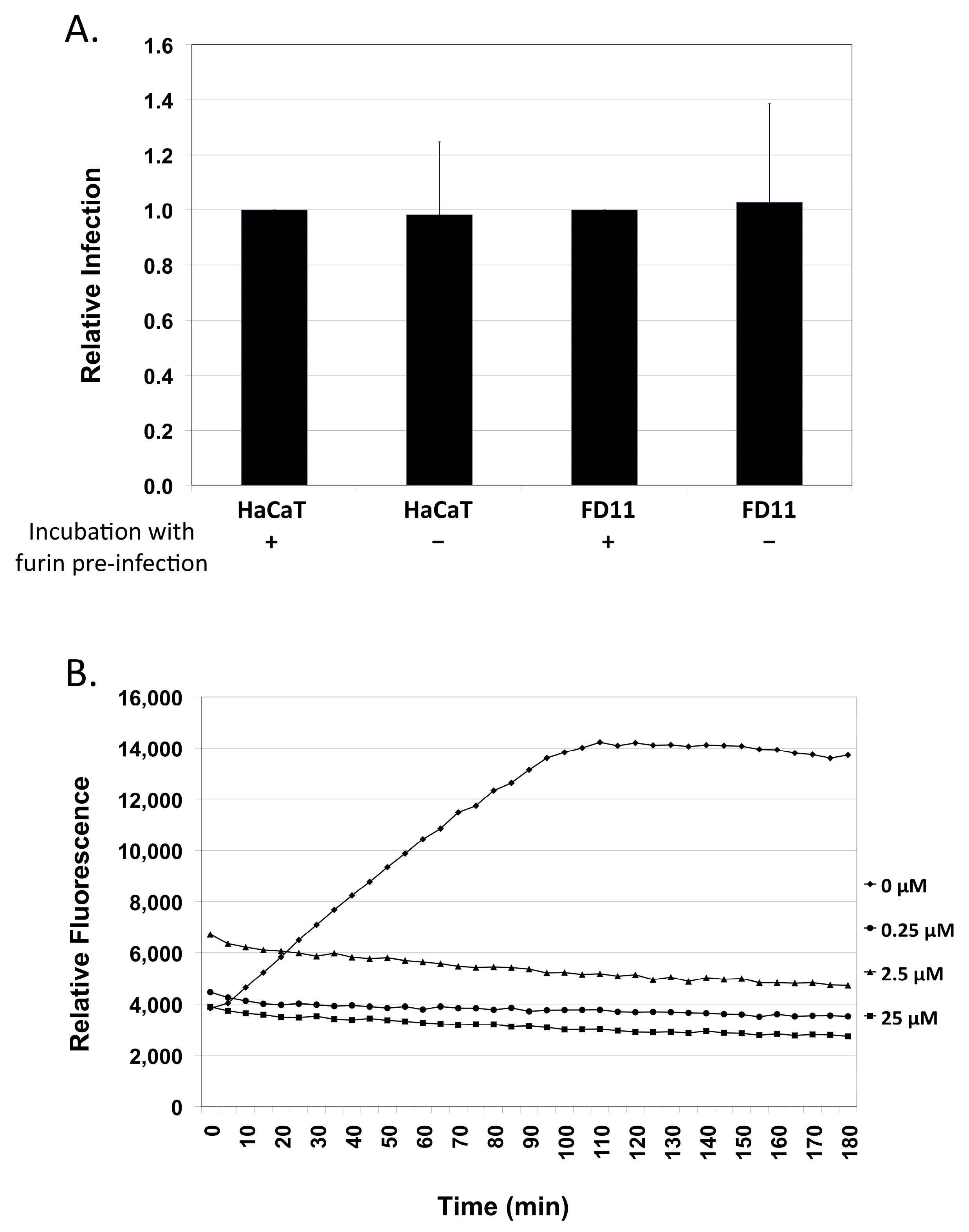
2.3. Exogenous Furin Has No Impact on Infection
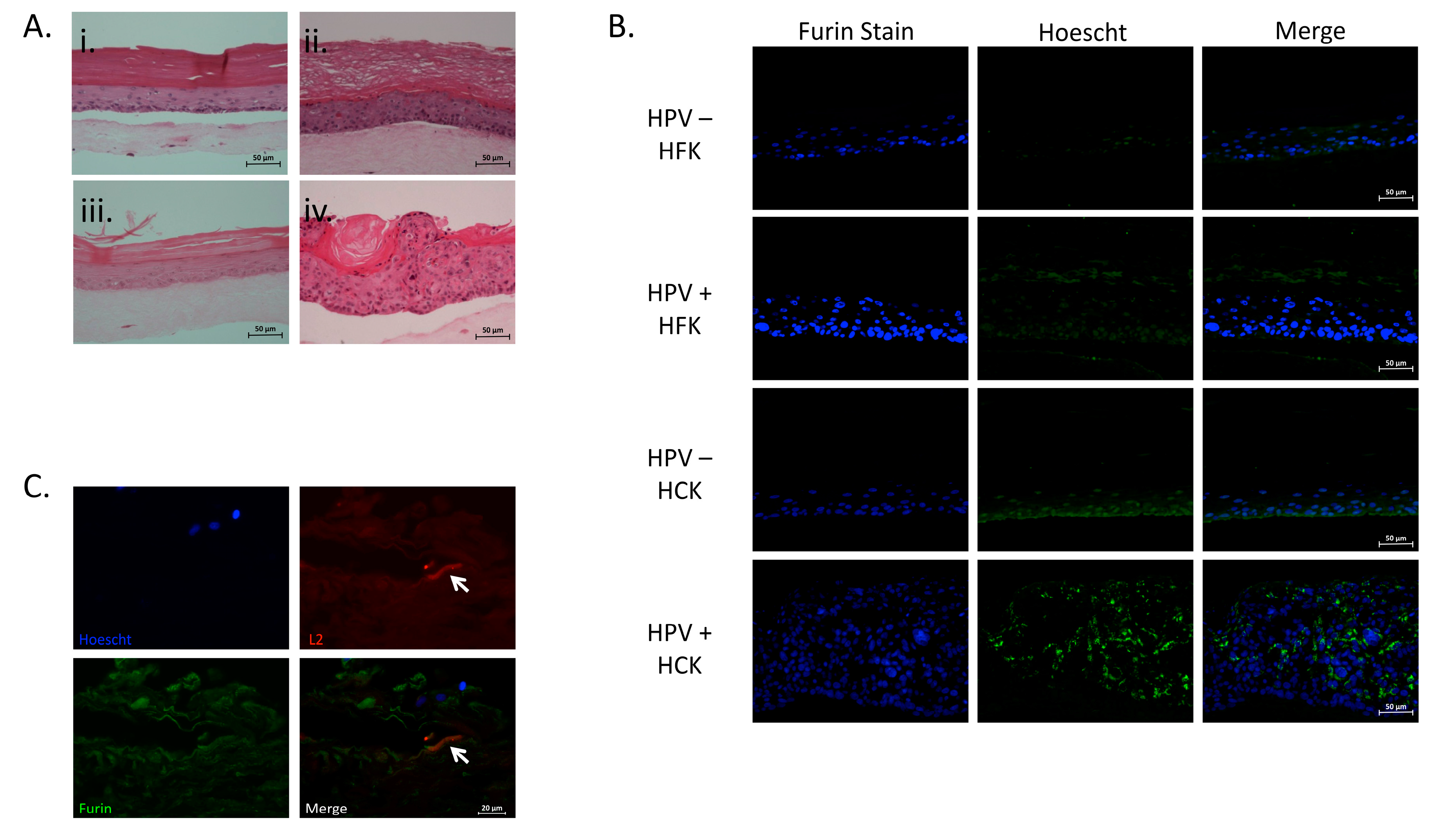
2.4. Expression of Furin in Organotypic Foreskin and Cervical Cultures

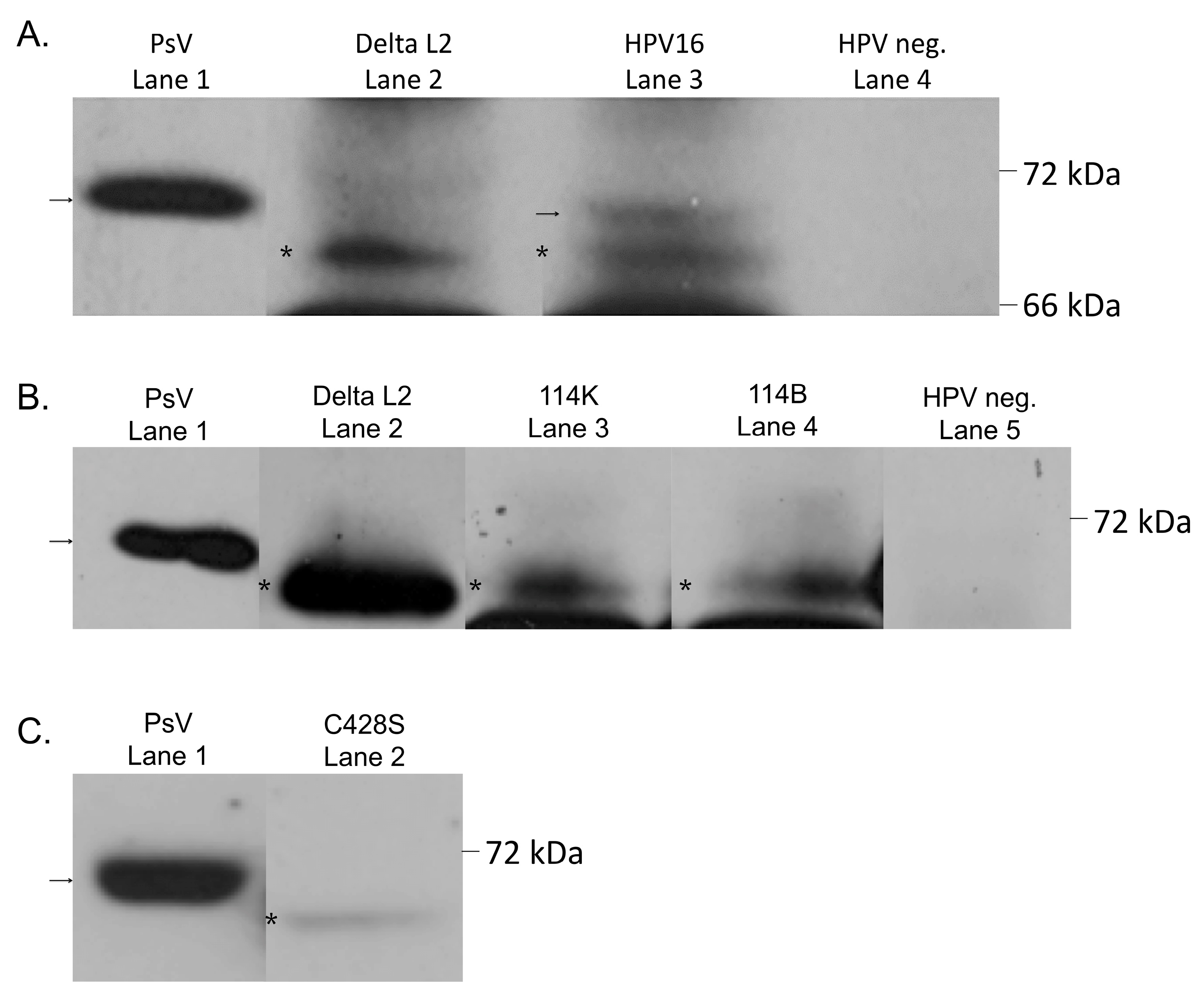
2.5. Cleavage of HPV16 during Virion Morphogenesis
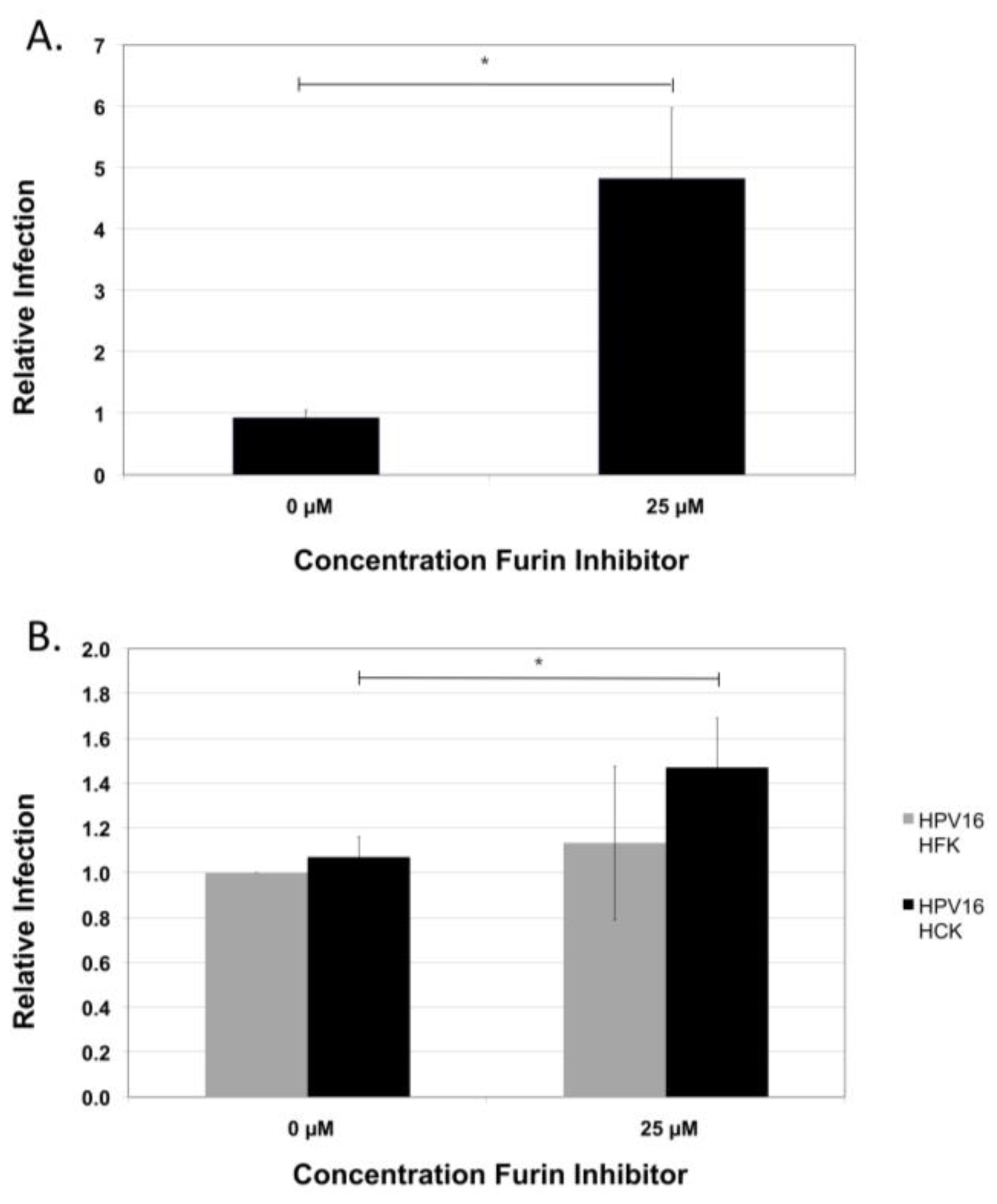
2.6. Infection of Primary Foreskin and Cervical Keratinocytes by HPV16 NV Derived from Either Tissue
2.7. Conservation of the Furin Cleavage Site in L2
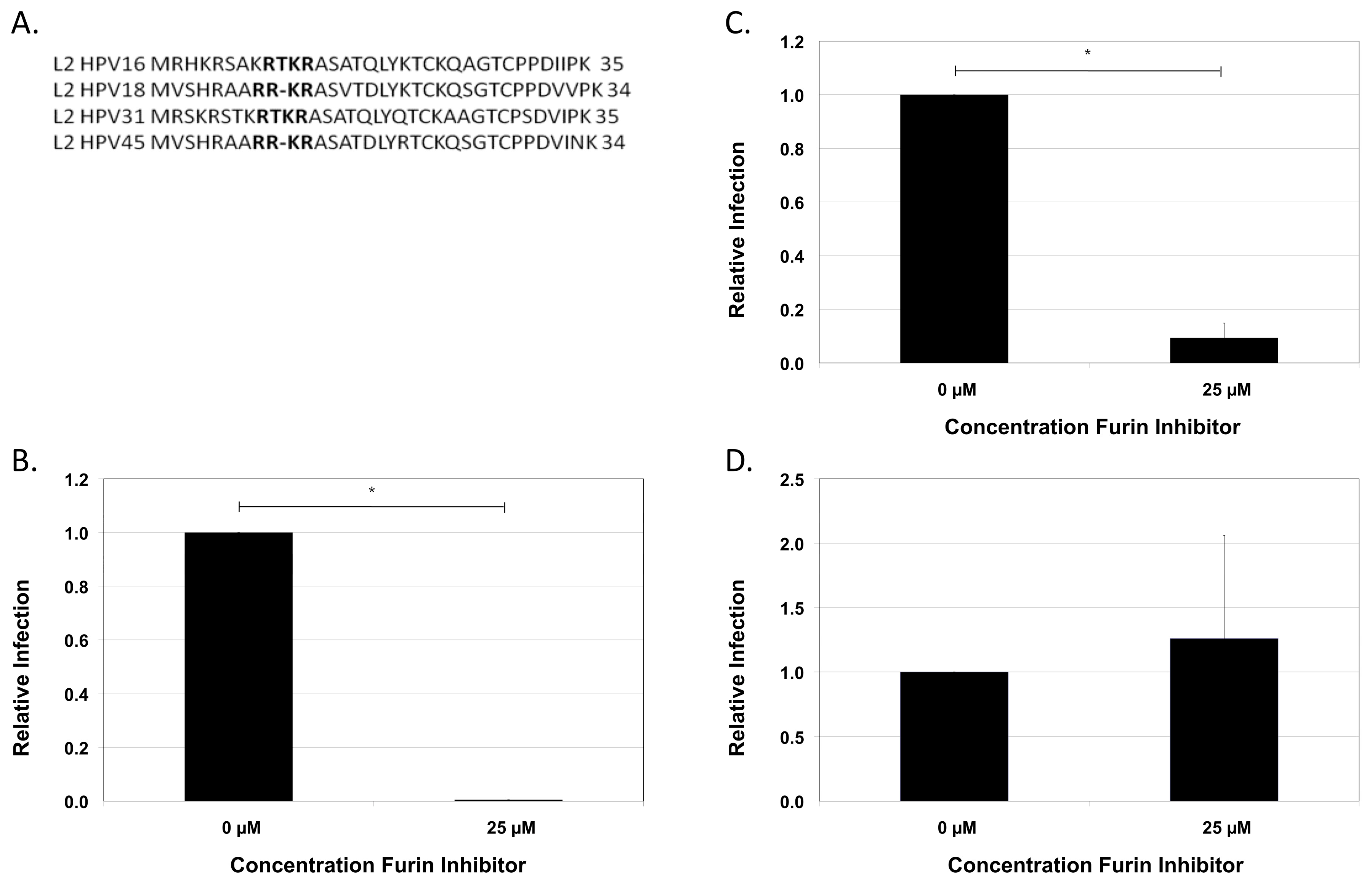
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Virus Production
4.2. Virus Titers
4.3. Infectivity Assays
4.4. In Vitro Furin Enzyme-Treatment of Virus Preps and Fluorogenic Furin Assay
4.5. Histology and Furin Immunofluorescence Staining
4.6. SDS-PAGE and L2 Western Blot
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schiffman, M.; Kjaer, S.K. Natural history of anogenital human papillomavirus infection and neoplasia. J. Natl. Cancer Inst. Monogr. 2003, 31, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.S.; Newcomb, W.W.; Olson, N.H.; Cowsert, L.M.; Olson, C.; Brown, J.C. Structures of bovine and human papillomaviruses. Analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys. J. 1991, 60, 1445–1456. [Google Scholar] [CrossRef]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Knappe, M.; Bodevin, S.; Selinka, H.C.; Spillmann, D.; Streeck, R.E.; Chen, X.S.; Lindahl, U.; Sapp, M. Surface-exposed amino acid residues of hpv16 L1 protein mediating interaction with cell surface heparan sulfate. J. Biol. Chem. 2007, 282, 27913–27922. [Google Scholar] [CrossRef] [PubMed]
- Volpers, C.; Unckell, F.; Schirmacher, P.; Streeck, R.E.; Sapp, M. Binding and internalization of human papillomavirus type 33 virus-like particles by eukaryotic cells. J. Virol. 1995, 69, 3258–3264. [Google Scholar] [PubMed]
- Trus, B.L.; Roden, R.B.; Greenstone, H.L.; Vrhel, M.; Schiller, J.T.; Booy, F.P. Novel structural features of bovine papillomavirus capsid revealed by a three-dimensional reconstruction to 9 Å resolution. Nat. Struct. Biol. 1997, 4, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the papillomavirus capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [PubMed]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Greenstone, H.L.; Kirnbauer, R.; Booy, F.P.; Jessie, J.; Lowy, D.R.; Schiller, J.T. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 1996, 70, 5875–5883. [Google Scholar] [PubMed]
- Zhao, K.N.; Sun, X.Y.; Frazer, I.H.; Zhou, J. Dna packaging by L1 and L2 capsid proteins of bovine papillomavirus type 1. Virology 1998, 243, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Ozaki, S.; Tanaka, K.; Kanda, T. Human papillomavirus 16 minor capsid protein L2 helps capsomeres assemble independently of intercapsomeric disulfide bonding. Virus Genes 2005, 31, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Sapp, M.; Spoden, G.A. Host-cell factors involved in papillomavirus entry. Med. Microbiol. Immunol. 2012, 201, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Kawana, Y.; Kawana, K.; Yoshikawa, H.; Taketani, Y.; Yoshiike, K.; Kanda, T. Human papillomavirus type 16 minor capsid protein L2 N-terminal region containing a common neutralization epitope binds to the cell surface and enters the cytoplasm. J. Virol. 2001, 75, 2331–2336. [Google Scholar] [PubMed]
- Yang, R.; Day, P.M.; Yutzy, W.H.T.; Lin, K.Y.; Hung, C.F.; Roden, R.B. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J. Virol. 2003, 77, 3531–3541. [Google Scholar] [CrossRef]
- Woodham, A.W.; da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE 2012, 7, e43519. [Google Scholar] [CrossRef] [PubMed]
- Kawana, K.; Matsumoto, K.; Yoshikawa, H.; Taketani, Y.; Kawana, T.; Yoshiike, K.; Kanda, T. A surface immunodeterminant of human papillomavirus type 16 minor capsid protein L2. Virology 1998, 245, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef] [PubMed]
- Bergant Marusic, M.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Bossis, I.; Roden, R.B.; Gambhira, R.; Yang, R.; Tagaya, M.; Howley, P.M.; Meneses, P.I. Interaction of tsnare syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J. Virol. 2005, 79, 6723–6731. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yutzy, W.H.T.; Viscidi, R.P.; Roden, R.B. Interaction of L2 with β-actin directs intracellular transport of papillomavirus and infection. J. Biol. Chem. 2003, 278, 12546–12553. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Becker, K.A.; Lambert, C.; Nowak, T.; Sapp, C.; Strand, D.; Streeck, R.E.; Sapp, M. Identification of a dynein interacting domain in the papillomavirus minor capsid protein L2. J. Virol. 2006, 80, 6691–6696. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J. Virol. 1998, 72, 142–150. [Google Scholar] [PubMed]
- Kondo, K.; Ishii, Y.; Mori, S.; Shimabukuro, S.; Yoshikawa, H.; Kanda, T. Nuclear location of minor capsid protein L2 is required for expression of a reporter plasmid packaged in HPV51 pseudovirions. Virology 2009, 394, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low ph primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar] [PubMed]
- Nakayama, K. Furin: A mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 1997, 327(Pt. 3), 625–635. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y. Protease-dependent virus tropism and pathogenicity. Trends Microbiol. 1993, 1, 81–87. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Raff, A.B.; Woodham, A.W.; Raff, L.M.; Skeate, J.G.; Yan, L.; da Silva, D.M.; Schelhaas, M.; Kast, W.M. The evolving field of human papillomavirus receptor research: A review of binding and entry. J. Virol. 2013, 87, 6062–6072. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 2003, 307, 1–11. [Google Scholar] [CrossRef]
- Embers, M.E.; Budgeon, L.R.; Culp, T.D.; Reed, C.A.; Pickel, M.D.; Christensen, N.D. Differential antibody responses to a distinct region of human papillomavirus minor capsid proteins. Vaccine 2004, 22, 670–680. [Google Scholar] [PubMed]
- Gambhira, R.; Karanam, B.; Jagu, S.; Roberts, J.N.; Buck, C.B.; Bossis, I.; Alphs, H.; Culp, T.; Christensen, N.D.; Roden, R.B. A protective and broadly cross-neutralizing epitope of human papillomavirus L2. J. Virol. 2007, 81, 13927–13931. [Google Scholar] [PubMed]
- White, W.I.; Wilson, S.D.; Palmer-Hill, F.J.; Woods, R.M.; Ghim, S.J.; Hewitt, L.A.; Goldman, D.M.; Burke, S.J.; Jenson, A.B.; Koenig, S.; et al. Characterization of a major neutralizing epitope on human papillomavirus type 16 L1. J. Virol. 1999, 73, 4882–4889. [Google Scholar] [PubMed]
- Kirnbauer, R.; Booy, F.; Cheng, N.; Lowy, D.R.; Schiller, J.T. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. USA 1992, 89, 12180–12184. [Google Scholar] [PubMed]
- Biryukov, J.; Meyers, C. Papillomavirus infectious pathways: A comparison of systems. Viruses 2015, 7, 4303–4325. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Alam, S.; Ryndock, E.J.; Cruz, L.; Christensen, N.D.; Roden, R.B.; Meyers, C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J. Virol. 2009, 83, 10515–10526. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Differentiation-dependent interpentameric disulfide bond stabilizes native human papillomavirus type 16. PLoS ONE 2011, 6, e22427. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Maturation of papillomavirus capsids. J. Virol. 2005, 79, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Moseley, P.L.; Lowe, S.L.; Ozbun, M.A. Inducible heat shock protein 70 enhances HPV31 viral genome replication and virion production during the differentiation-dependent life cycle in human keratinocytes. Virus Res. 2010, 147, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chromy, L.R.; Pipas, J.M.; Garcea, R.L. Chaperone-mediated in vitro assembly of polyomavirus capsids. Proc. Natl. Acad. Sci. USA 2003, 100, 10477–10482. [Google Scholar] [PubMed]
- Chromy, L.R.; Oltman, A.; Estes, P.A.; Garcea, R.L. Chaperone-mediated in vitro disassembly of polyoma- and papillomaviruses. J. Virol. 2006, 80, 5086–5091. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Cross-neutralization potential of native human papillomavirus N-terminal L2 epitopes. PLoS ONE 2011, 6, e16405. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Gambhira, R.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Mechanisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J. Virol. 2008, 82, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Meyers, C. Differential dependence on host cell glycosaminoglycans for infection of epithelial cells by high-risk HPV types. PLoS ONE 2013, 8, e68379. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.F.; Mukherjee, S.; Bienkowska-Haba, M.; Pang, J.; Sapp, M. Human papillomavirus species-specific interaction with the basement membrane-resident non-heparan sulfate receptor. Viruses 2014, 6, 4856–4879. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 2008, 82, 12565–12568. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Armstrong, A.; Haderer, P.; Christensen, N.D.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T.; Kirnbauer, R. Characterization of a human papillomavirus type 16 variant-dependent neutralizing epitope. J. Virol. 1997, 71, 6247–6252. [Google Scholar] [PubMed]
- Christensen, N.D.; Dillner, J.; Eklund, C.; Carter, J.J.; Wipf, G.C.; Reed, C.A.; Cladel, N.M.; Galloway, D.A. Surface conformational and linear epitopes on HPV-16 and HPV-18 L1 virus-like particles as defined by monoclonal antibodies. Virology 1996, 223, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Buck, C.B.; Pang, Y.Y.; Thompson, C.D.; Castle, P.E.; FitzGerald, P.C.; Kruger Kjaer, S.; Lowy, D.R.; Schiller, J.T. Reactivity of human sera in a sensitive, high-throughput pseudovirus-based papillomavirus neutralization assay for HPV16 and HPV18. Virology 2004, 321, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Jean, F.; Stella, K.; Thomas, L.; Liu, G.; Xiang, Y.; Reason, A.J.; Thomas, G. Alpha1-antitrypsin portland, a bioengineered serpin highly selective for furin: Application as an antipathogenic agent. Proc. Natl. Acad. Sci. USA 1998, 95, 7293–7298. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.M.; Klimpel, K.R.; Arora, N.; Henderson, M.A.; Leppla, S.H. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 1995, 63, 82–87. [Google Scholar] [PubMed]
- Pearton, D.J.; Nirunsuksiri, W.; Rehemtulla, A.; Lewis, S.P.; Presland, R.B.; Dale, B.A. Proprotein convertase expression and localization in epidermis: Evidence for multiple roles and substrates. Exp. Dermatol. 2001, 10, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yemelyanova, A.V.; Gambhira, R.; Jagu, S.; Meyers, C.; Kirnbauer, R.; Ronnett, B.M.; Gravitt, P.E.; Roden, R.B. Expression pattern and subcellular localization of human papillomavirus minor capsid protein L2. Am. J. Pathol. 2009, 174, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Gallimore, P.H. Identification of proteins encoded by the L1 and L2 open reading frames of human papillomavirus 1a. J. Virol. 1987, 61, 2793–2799. [Google Scholar] [PubMed]
- Jin, X.W.; Cowsert, L.M.; Pilacinski, W.P.; Jenson, A.B. Identification of L2 open reading frame gene products of bovine papillomavirus type 1 using monoclonal antibodies. J. General Virol. 1989, 70, 1133–1140. [Google Scholar] [CrossRef]
- Komly, C.A.; Breitburd, F.; Croissant, O.; Streeck, R.E. The L2 open reading frame of human papillomavirus type 1a encodes a minor structural protein carrying type-specific antigens. J. Virol. 1986, 60, 813–816. [Google Scholar] [PubMed]
- Rose, R.C.; Bonnez, W.; Strike, D.G.; Reichman, R.C. Expression of the full-length products of the human papillomavirus type 6b (HPV-6b) and HPV-11 L2 open reading frames by recombinant baculovirus, and antigenic comparisons with HPV-11 whole virus particles. J. General Virol. 1990, 71, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Wilson, S.; Mullikin, B.; Suzich, J.; Meyers, C. Human papillomavirus type 45 propagation, infection, and neutralization. Virology 2003, 312, 1–7. [Google Scholar] [CrossRef]
- Meyers, C.; Mayer, T.J.; Ozbun, M.A. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral dna. J. Virol. 1997, 71, 7381–7386. [Google Scholar] [PubMed]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Madison, K.C.; Sando, G.N.; Howard, E.J.; True, C.A.; Gilbert, D.; Swartzendruber, D.C.; Wertz, P.W. Lamellar granule biogenesis: A role for ceramide glucosyltransferase, lysosomal enzyme transport, and the golgi. J. Investig. Dermatol. Symp. Proc. 1998, 3, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Cullander, C.; Mauro, T.; Rassner, U.; Komuves, L.; Brown, B.E.; Menon, G.K. The secretory granular cell: The outermost granular cell as a specialized secretory cell. J. Investig. Dermatol. Symp. Proc. 1998, 3, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Madison, K.C.; Howard, E.J. Ceramides are transported through the golgi apparatus in human keratinocytes in vitro. J. Investig. Dermatol. 1996, 106, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.T.; Brown, D.R. Transmission of human papillomavirus type 11 infection by desquamated cornified cells. Virology 2001, 281, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Hitzeroth, I.I.; Rybicki, E.P. Insights into the role and function of L2, the minor capsid protein of papillomaviruses. Arch. Virol. 2009, 154, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, X.Y.; Louis, K.; Frazer, I.H. Interaction of human papillomavirus (HPV) type 16 capsid proteins with HPV DNA requires an intact L2 N-terminal sequence. J. Virol. 1994, 68, 619–625. [Google Scholar] [PubMed]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, C.; Liu, Y.; Kuhling, L.; Chai, W.; Hafezi, W.; van Kuppevelt, T.H.; Kuhn, J.E.; Feizi, T.; Schelhaas, M. Heparin increases the infectivity of human papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cell. Microbiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sapp, M.; Bienkowska-Haba, M. Viral entry mechanisms: Human papillomavirus and a long journey from extracellular matrix to the nucleus. Febs. J. 2009, 276, 7206–7216. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Kuhling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Campos, S.K.; Wandinger-Ness, A.; Ozbun, M.A. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for ph-dependent uncoating. J. Virol. 2008, 82, 9505–9512. [Google Scholar] [PubMed]
- Bousarghin, L.; Touze, A.; Sizaret, P.Y.; Coursaget, P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 2003, 77, 3846–3850. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, P.L.; Laimins, L.A. Mechanisms regulating expression of the HPV 31 L1 and L2 capsid proteins and pseudovirion entry. Virol. J. 2007, 4, 19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 2005, 119, 445–462. [Google Scholar] [PubMed]
- Leder, C.; Kleinschmidt, J.A.; Wiethe, C.; Muller, M. Enhancement of capsid gene expression: Preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J. Virol. 2001, 75, 9201–9209. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Cladel, N.M.; Balogh, K.K.; Budgeon, L.R.; Mejia, A.F.; Christensen, N.D. Papillomavirus particles assembled in 293tt cells are infectious in vivo. J. Virol. 2006, 80, 11381–11384. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, L.; Biryukov, J.; Conway, M.J.; Meyers, C. Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection. Viruses 2015, 7, 5813-5830. https://doi.org/10.3390/v7112910
Cruz L, Biryukov J, Conway MJ, Meyers C. Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection. Viruses. 2015; 7(11):5813-5830. https://doi.org/10.3390/v7112910
Chicago/Turabian StyleCruz, Linda, Jennifer Biryukov, Michael J. Conway, and Craig Meyers. 2015. "Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection" Viruses 7, no. 11: 5813-5830. https://doi.org/10.3390/v7112910
APA StyleCruz, L., Biryukov, J., Conway, M. J., & Meyers, C. (2015). Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection. Viruses, 7(11), 5813-5830. https://doi.org/10.3390/v7112910