
Cell Penetrable Human scFv Specific to Middle Domain of Matrix Protein-1 Protects Mice from Lethal Influenza


Abstract
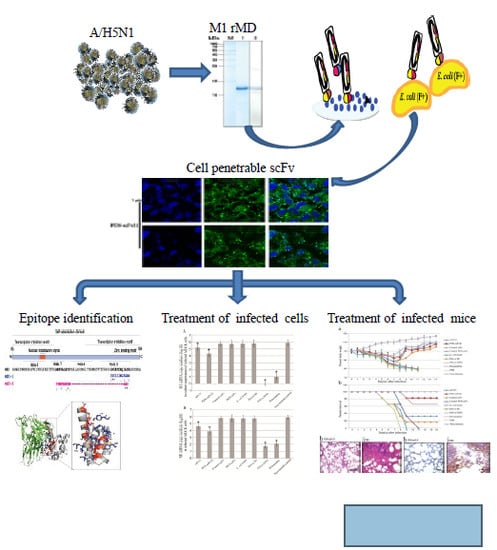
:
1. Introduction
2. Material and Methods
2.1. Influenza A Viruses
2.2. Preparation of Native M1
2.3. Recombinant MD (rMD) Preparation
2.4. Bio-Panning for Selection of Phage Clones that Bound to rMD and Production of the MD Specific-human scFv
2.5. Characterization of the Human scFvs
2.6. Large Scale Production of the Human scFv
2.7. Cell Penetrable Human scFv Specific to MD of M1
2.8. Cell Internalization of PEN-scFv
2.9. Interference of the Influenza Virus Replication by MD Specific-PEN-scFv
2.10. Therapeutic Efficacy of scFv/PEN-scFv in Influenza Virus Infected Mice
2.11. Quantitative Real-Time RT PCR (qPCR)
2.12. Viral Foci Assay
2.13. Histopathology of the Mouse Lungs
2.14. Immunohistochemical Staining of the Virus Antigen in the Mouse Lungs
2.15. A Search for Phage Mimotopic Peptides for Determining the scFv Presumptive Epitopes
2.16. Competitive ELISA for Verification of the Phage Mimotopes
2.17. Computerized Procedure for Determining the Interaction between the Human scFv and M1 MD of Influenza A Virus
2.18. Statistical Analysis
3. Results
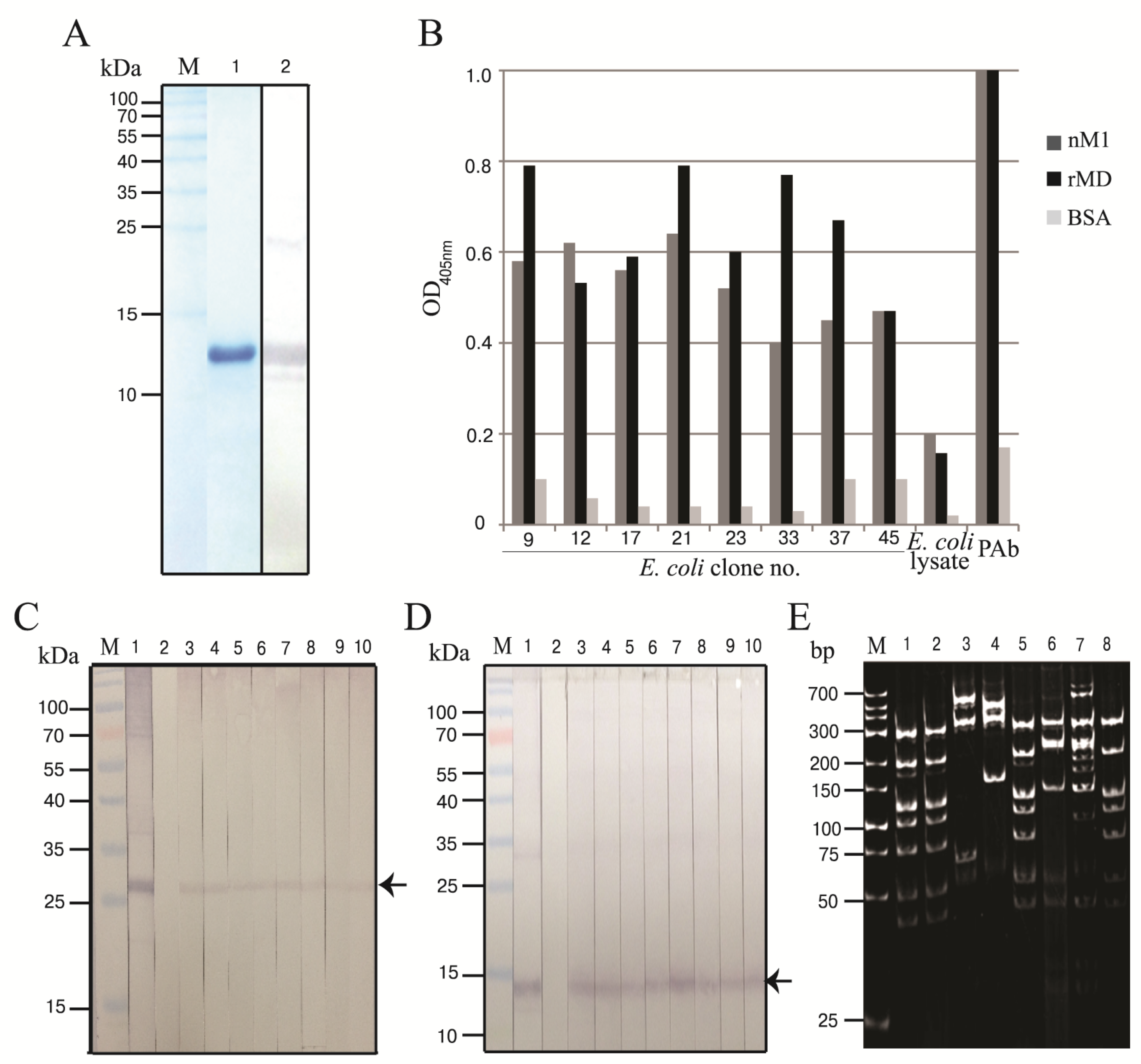
3.1. Production of Recombinant M1 Middle Domain (rMD)
3.2. Human scFvs Specific to nM1 and rMD
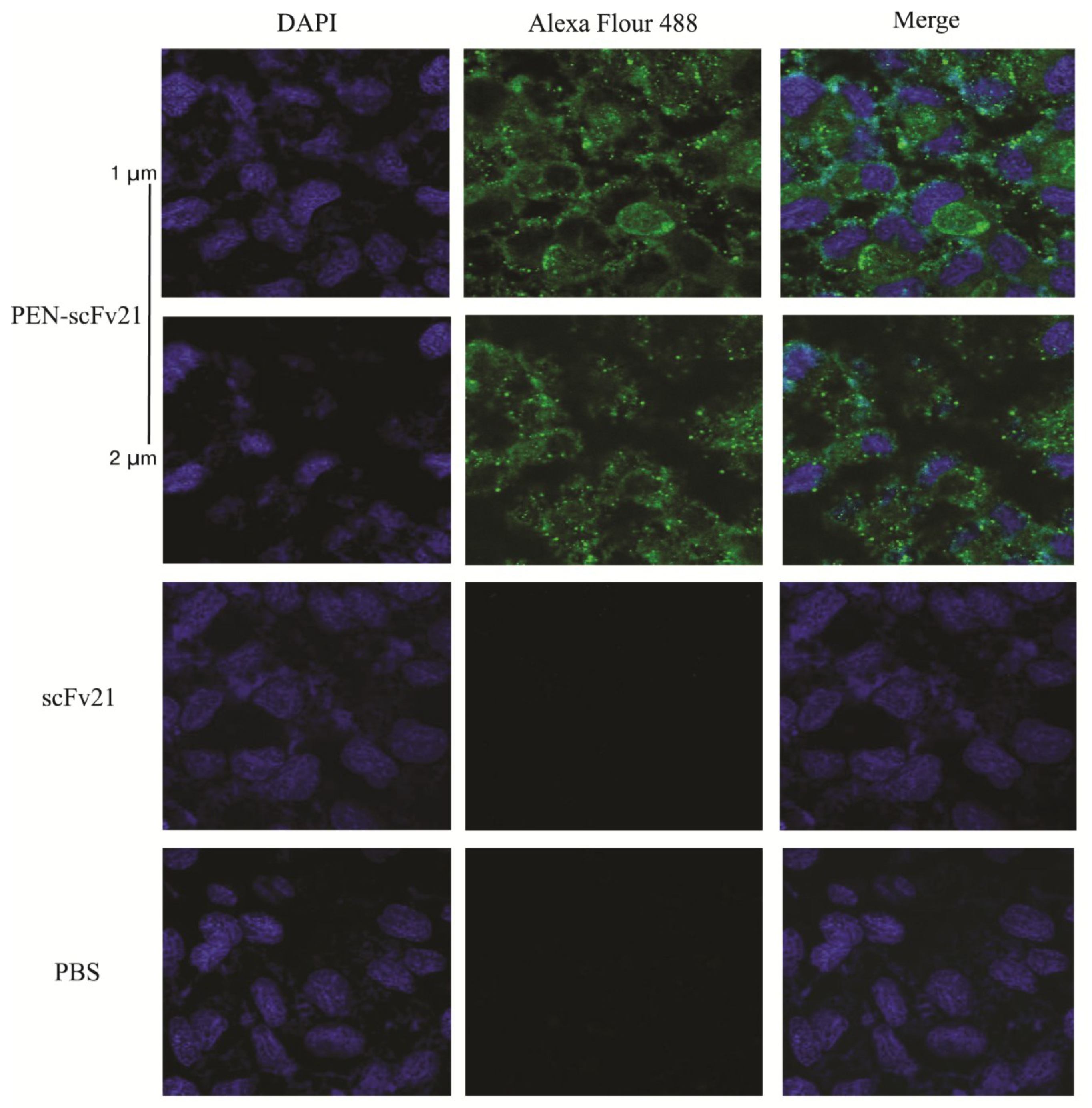
3.3. Cell Penetrable Human scFv and the Cellular Entry
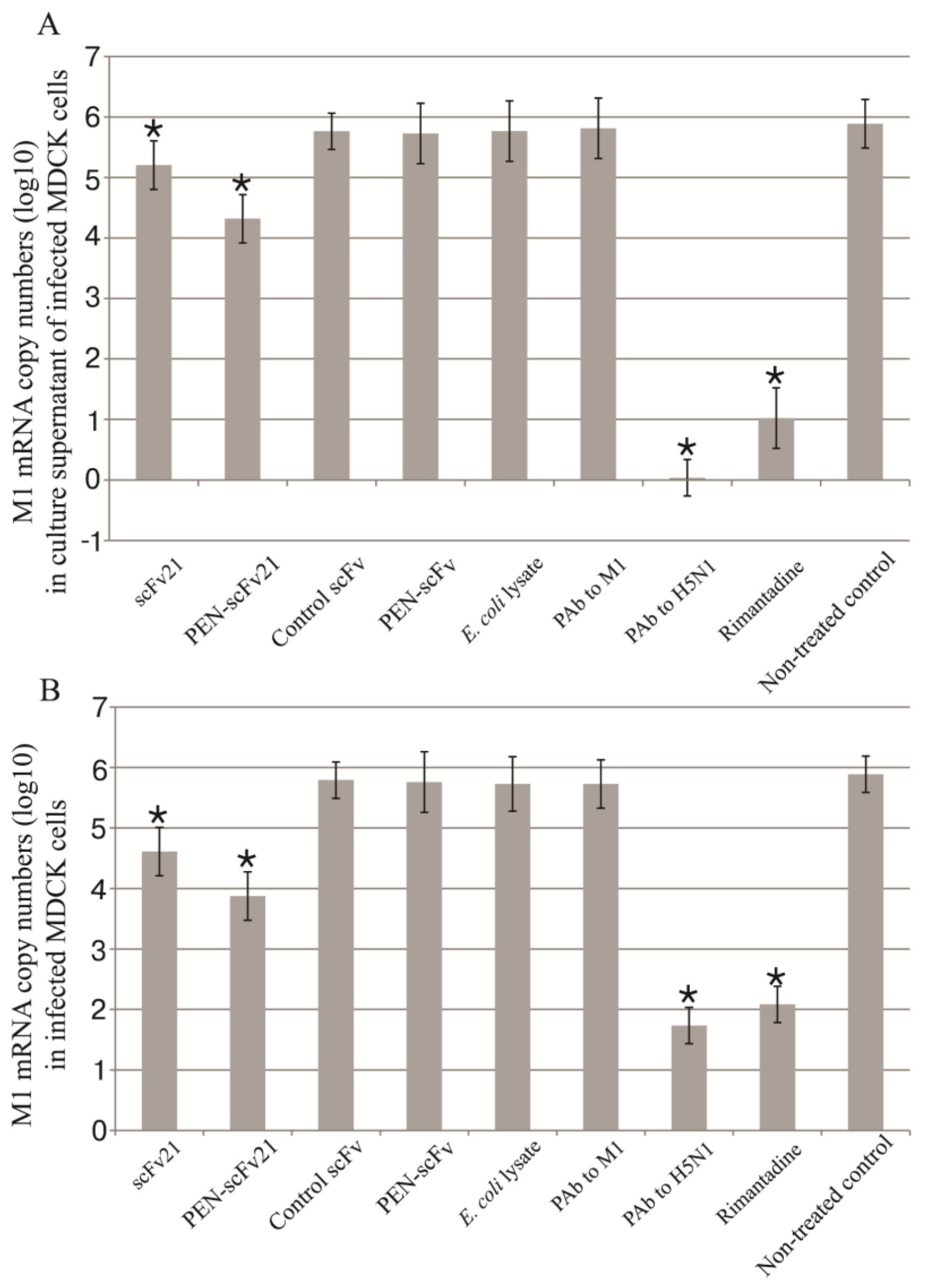
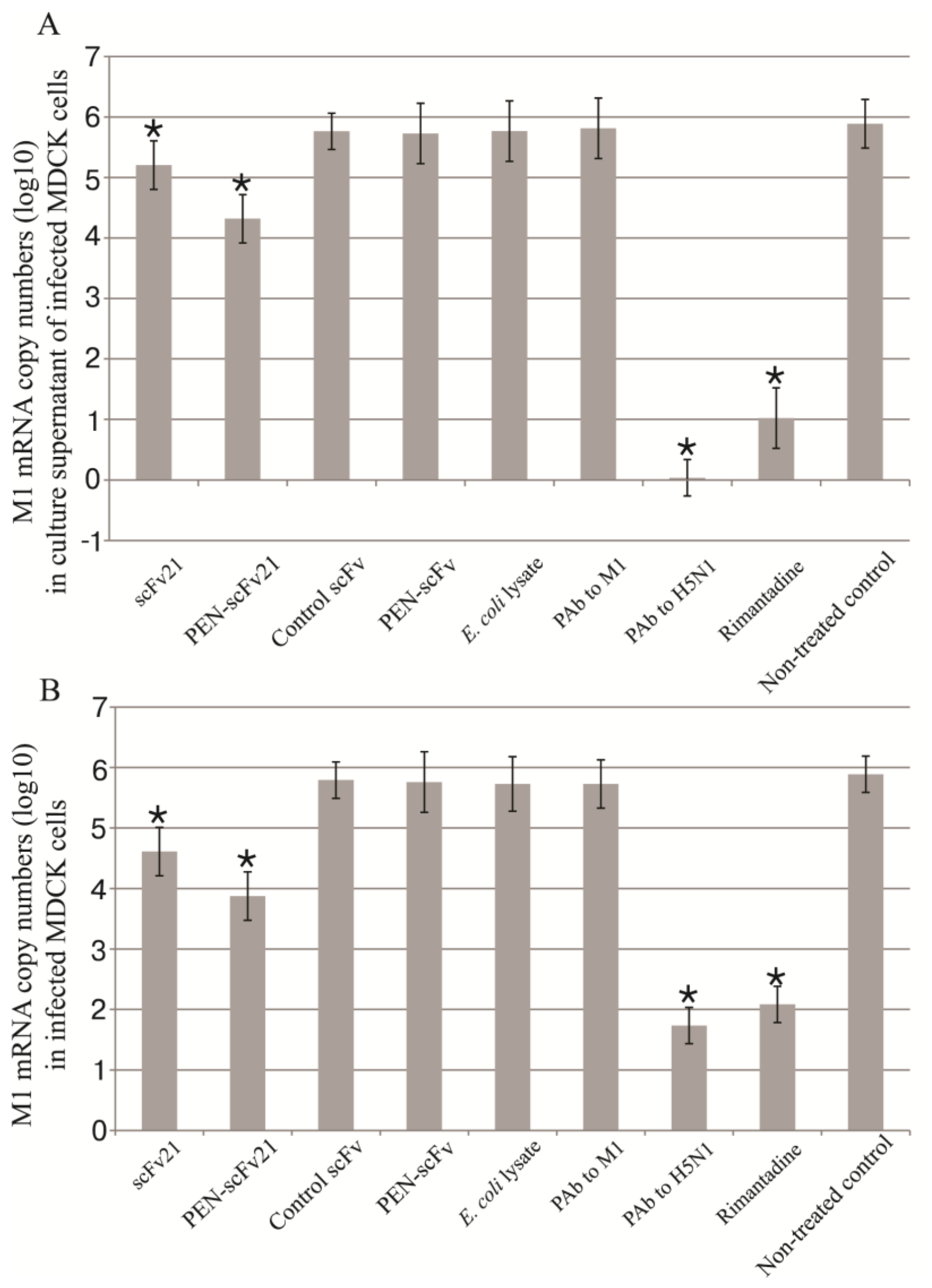
3.4. Inhibition of Influenza Virus Replication in MDCK Cells by MD Specific-scFv and PEN-scFv
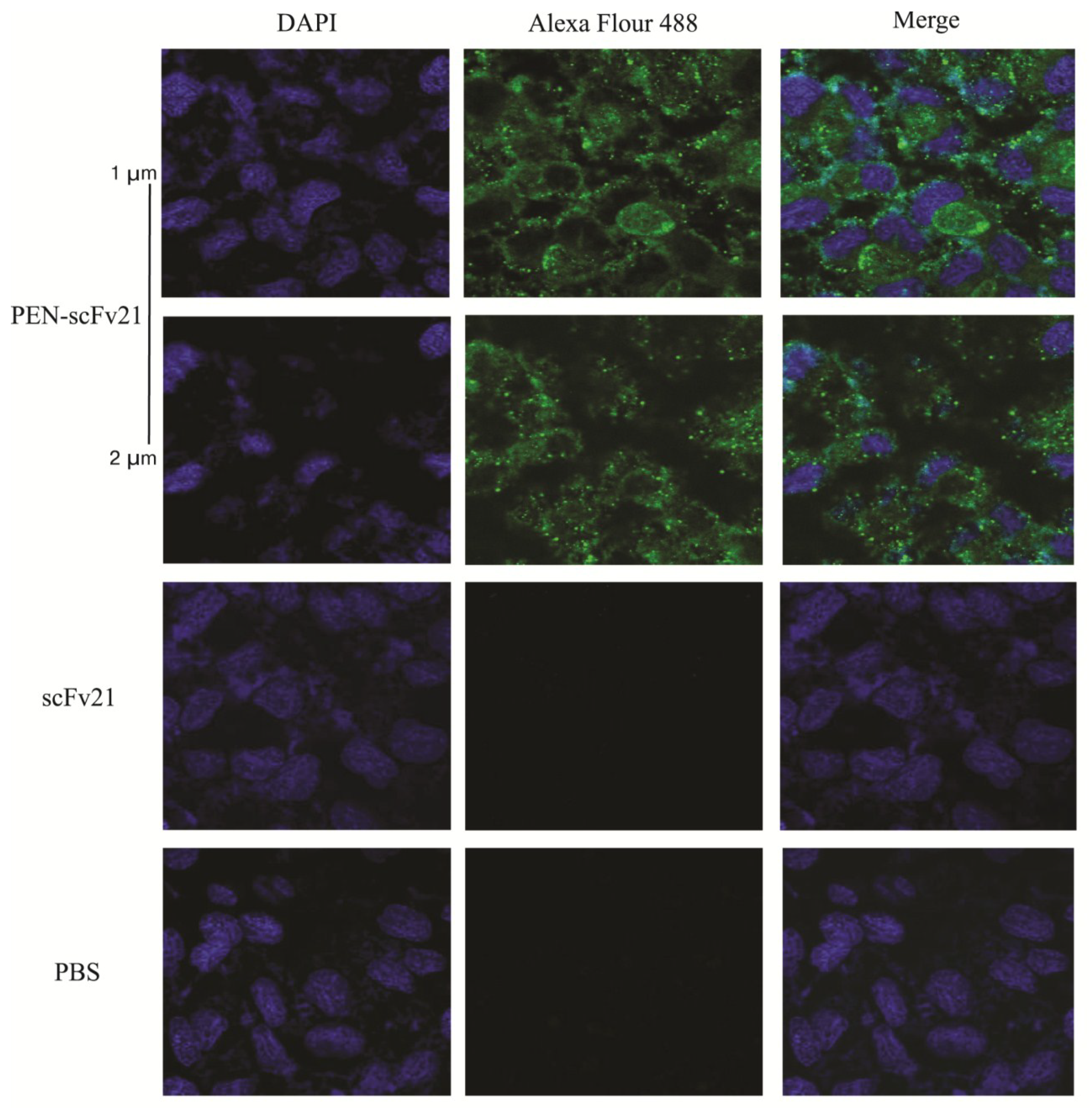
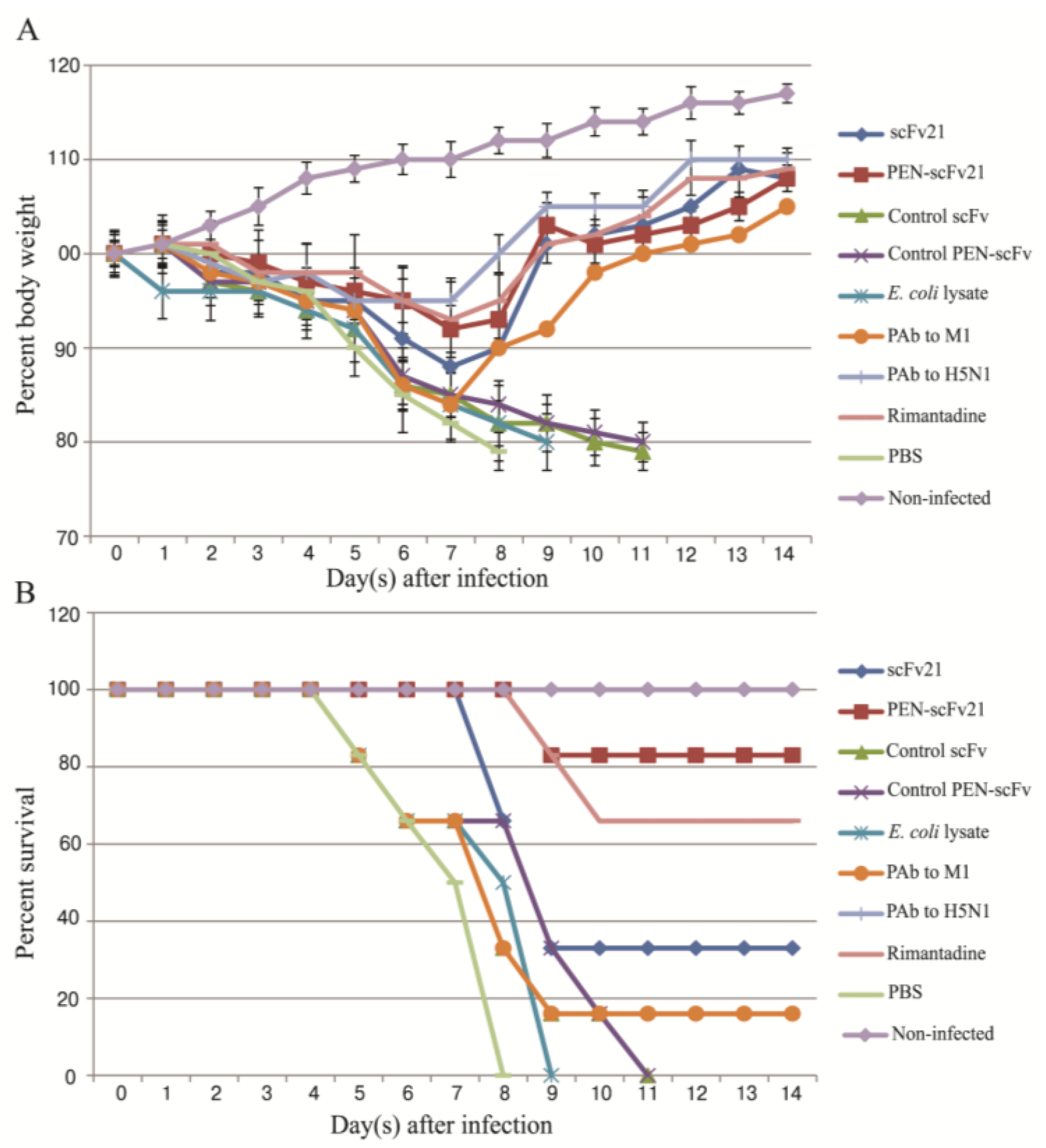
3.5. Therapeutic Efficacy of MD Specific-scFv and PEN-scFv in A/H5N1 Infected Mice
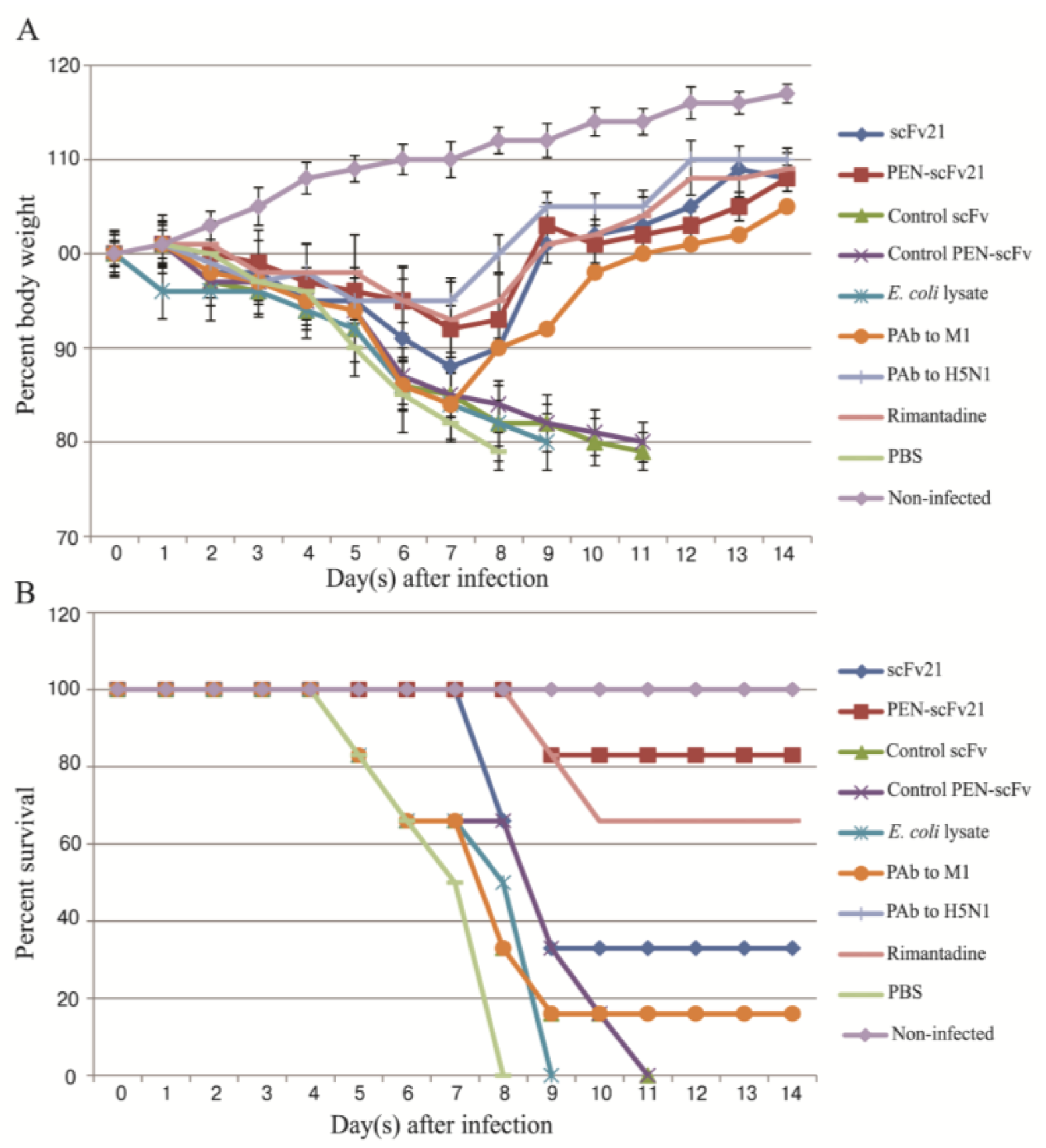
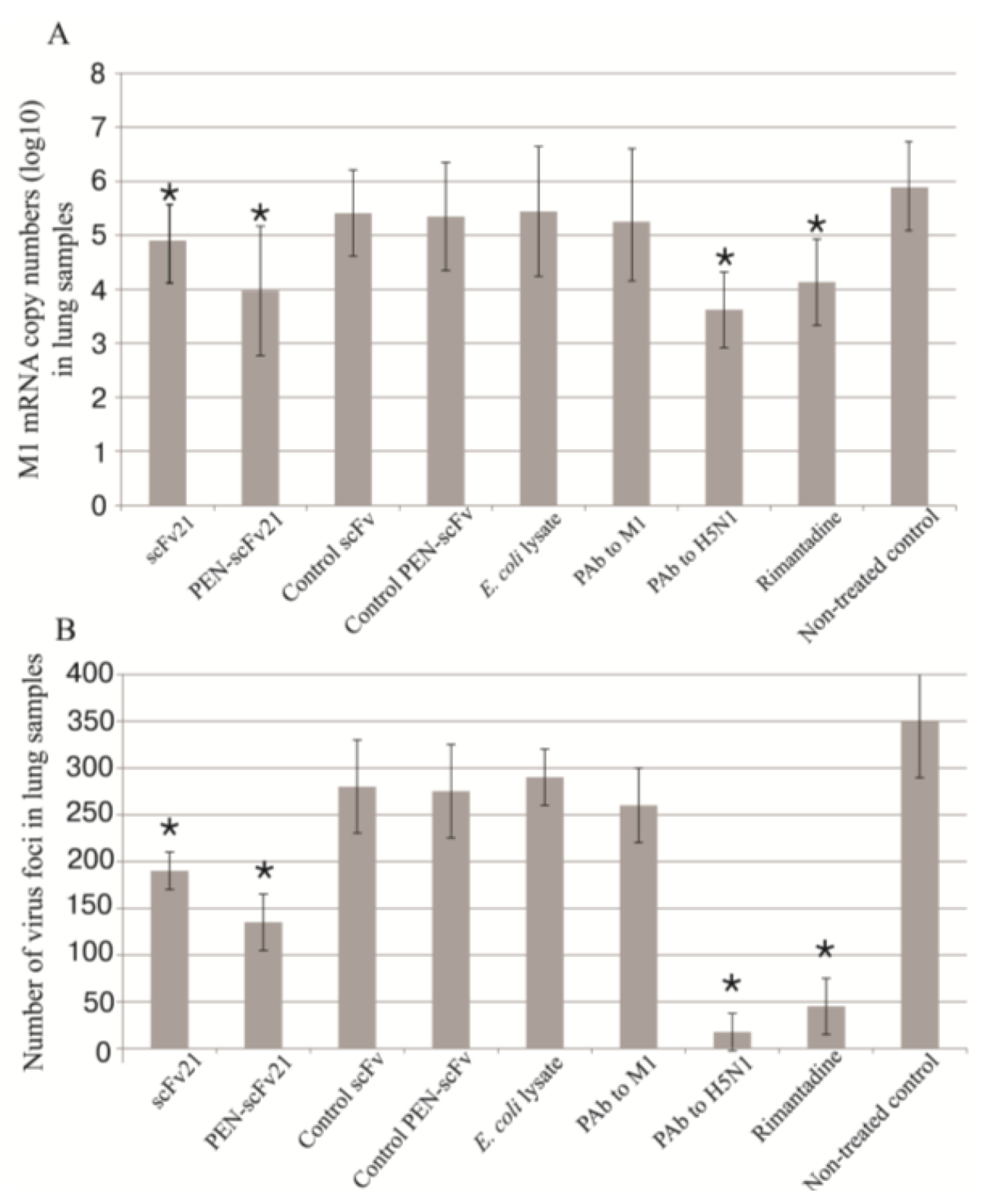
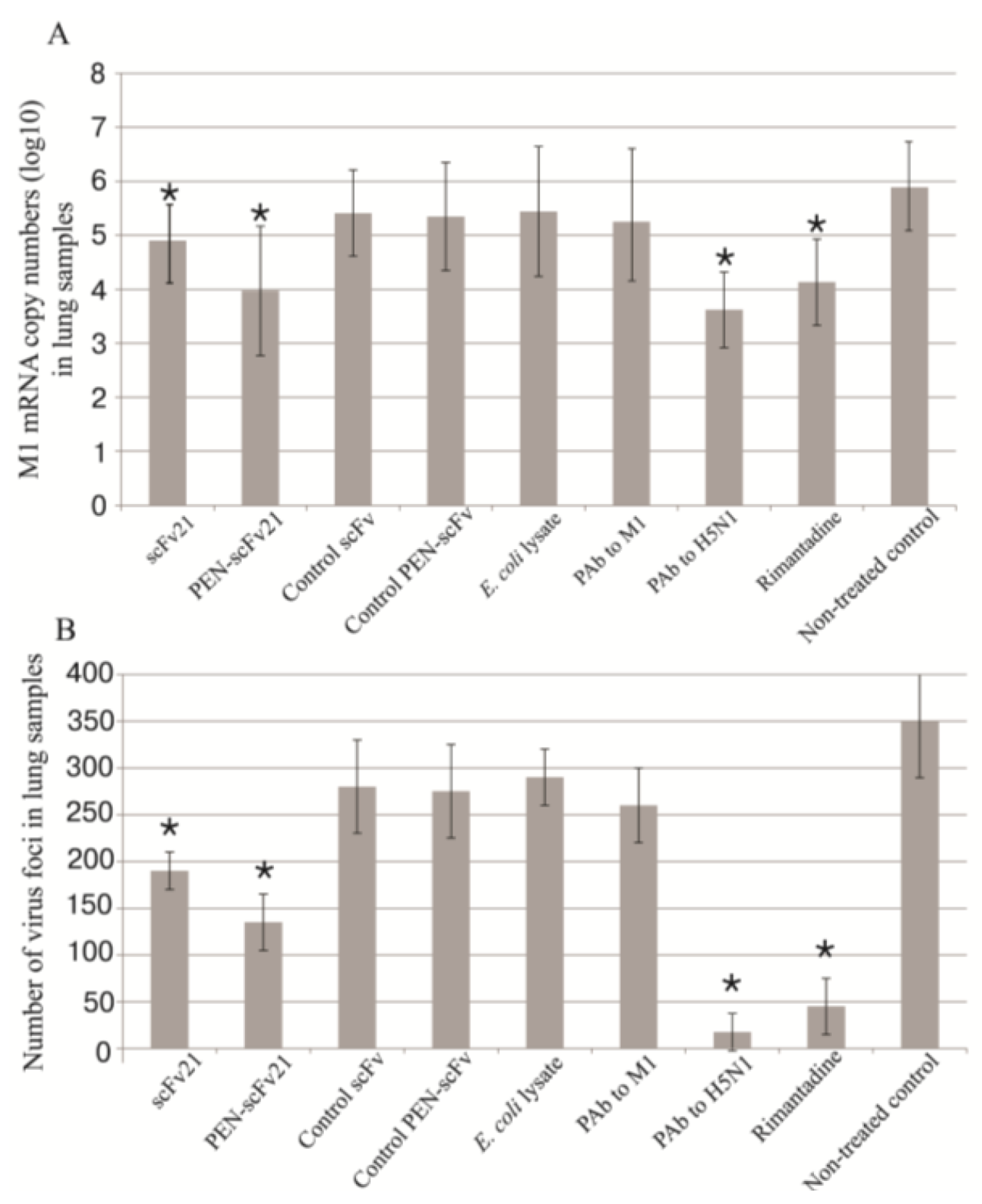
3.6. Quantification of Viral Titer in the Mouse Lungs
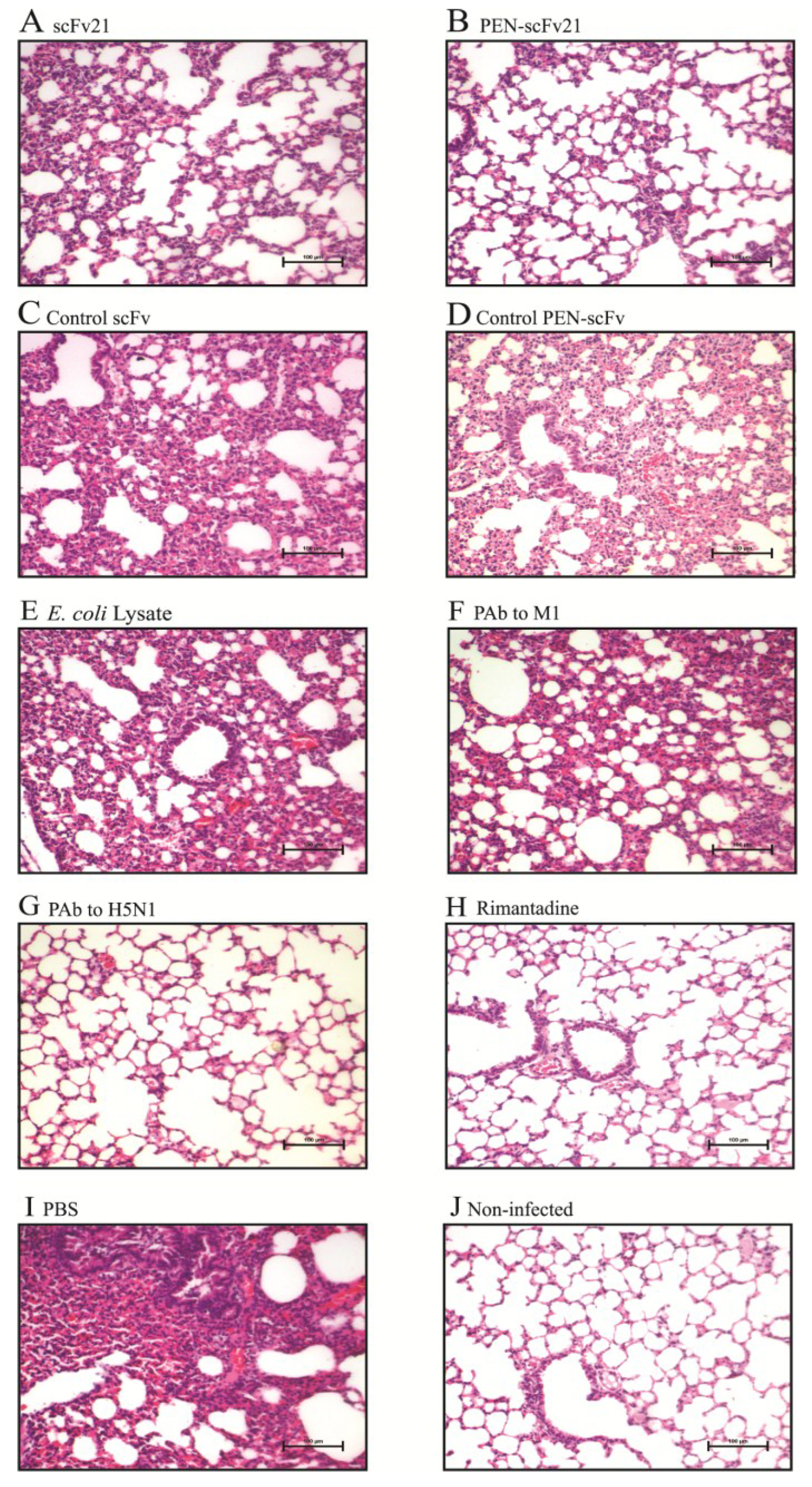
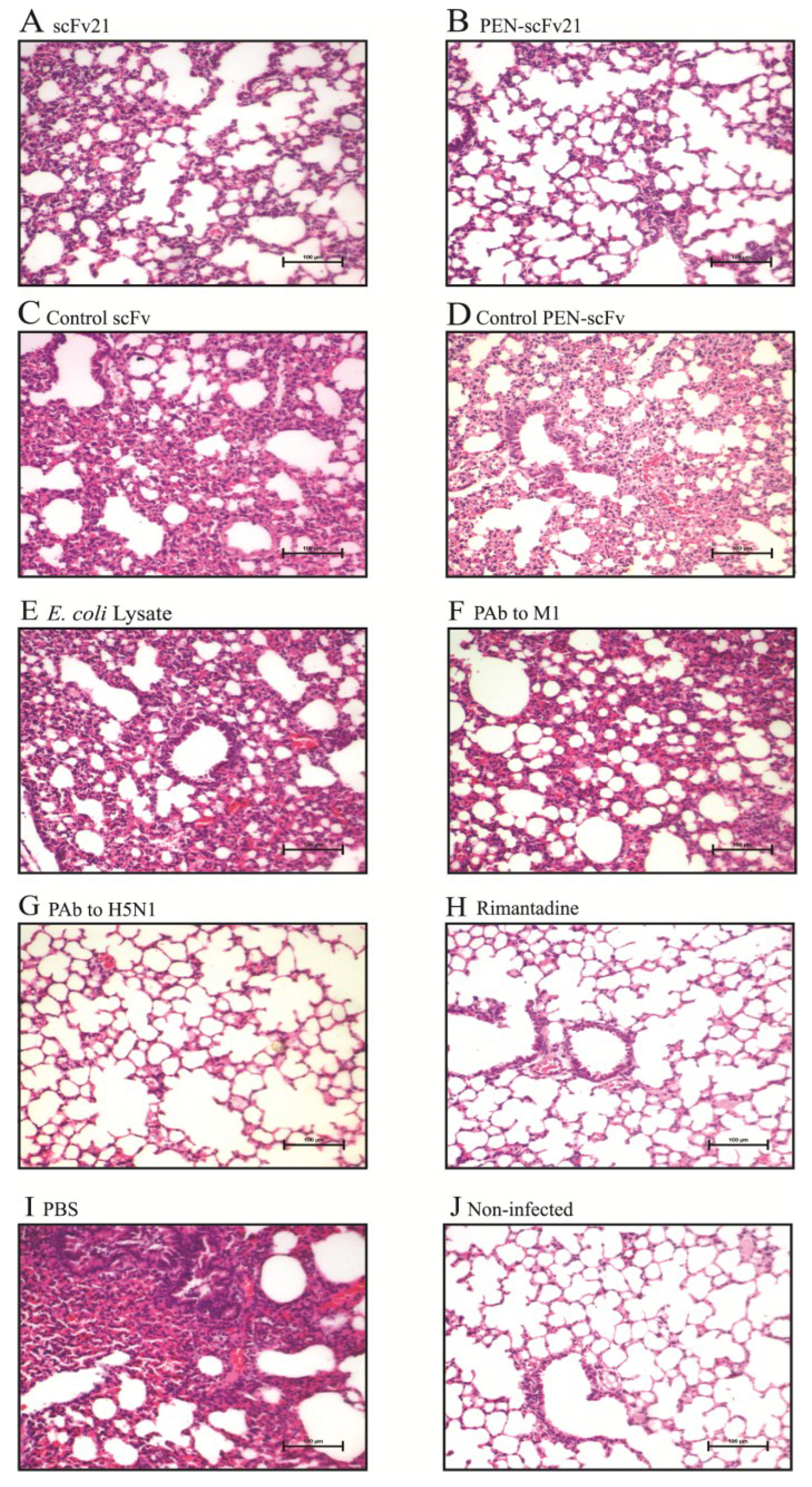
3.7. Comparative Histopathological Features
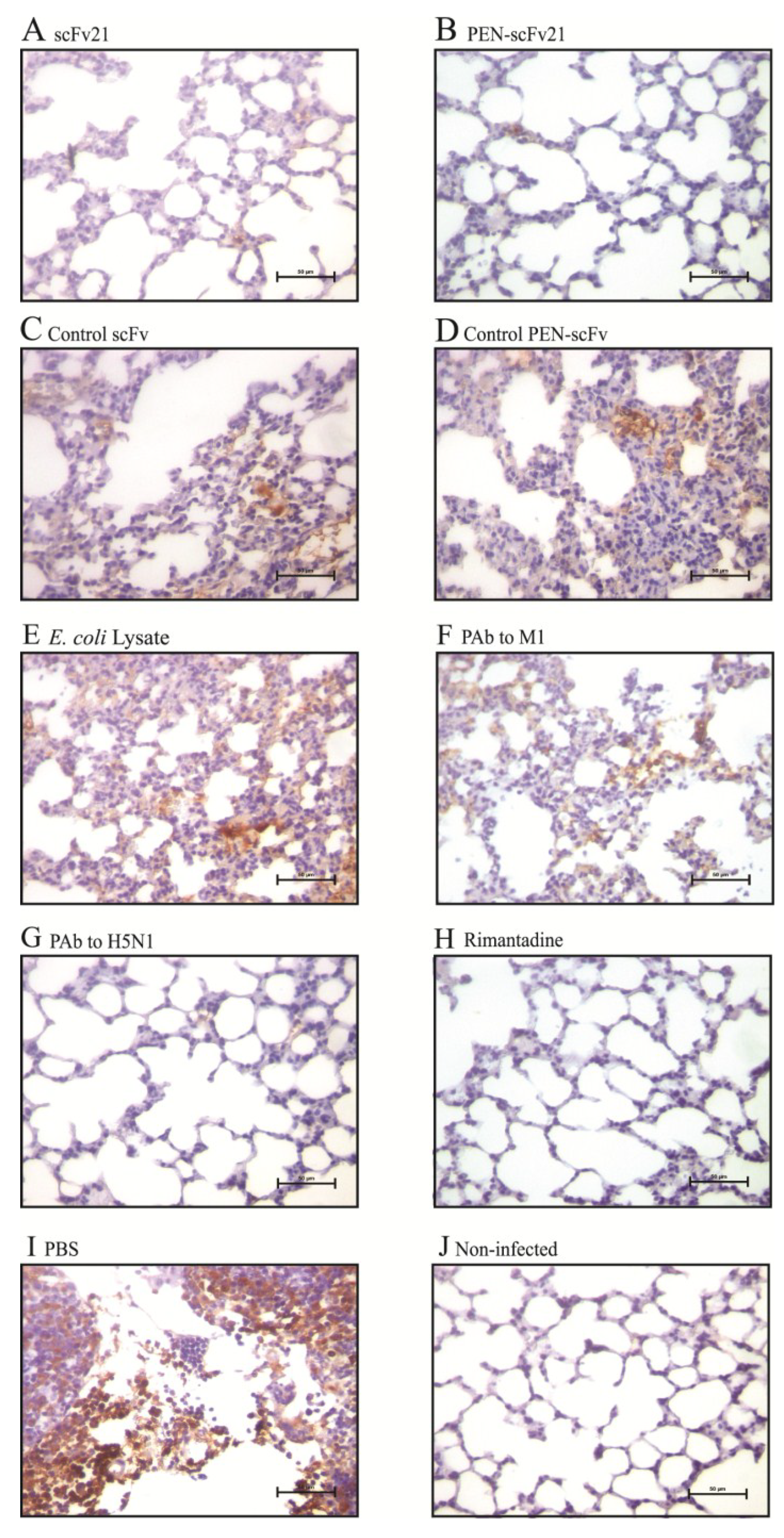
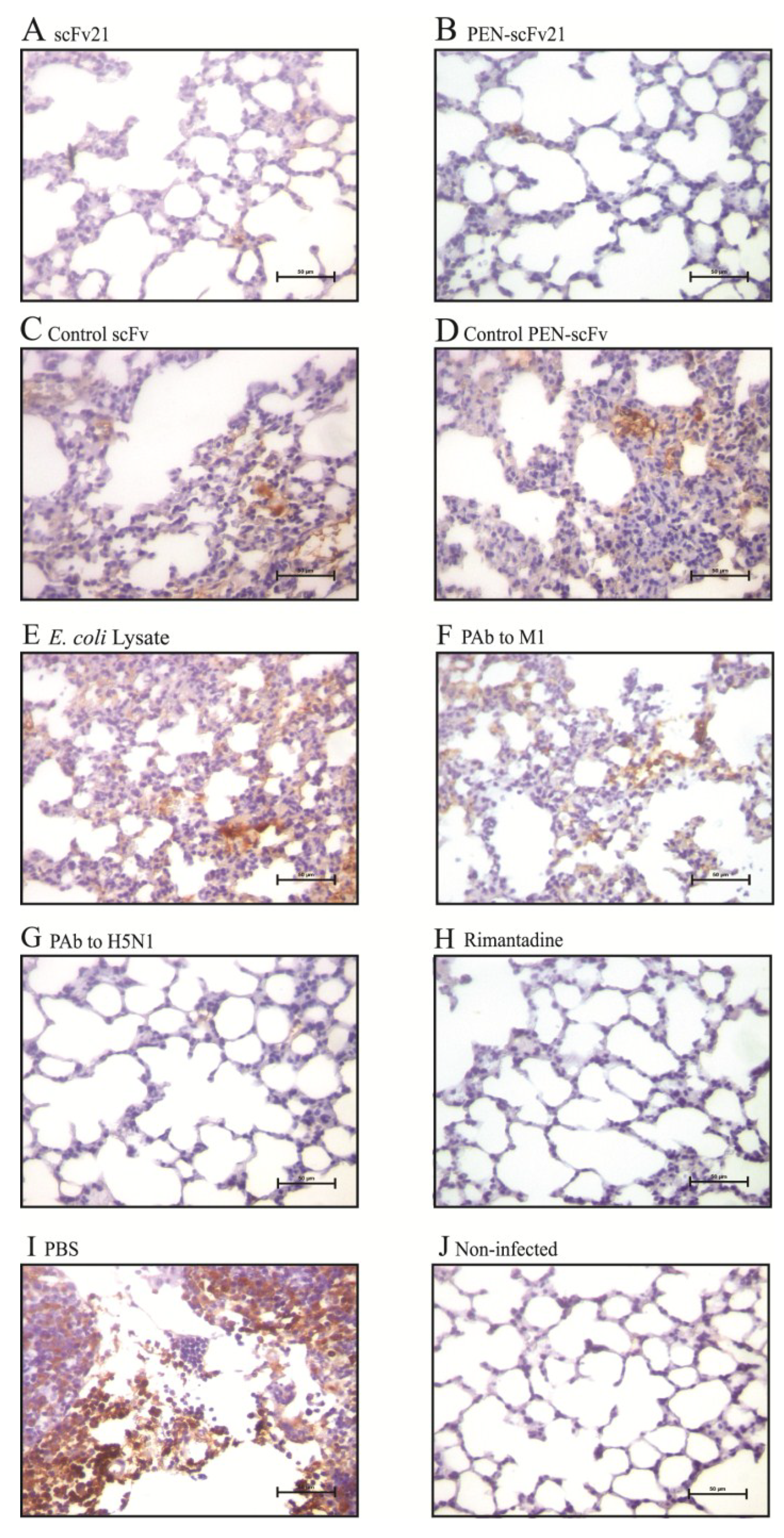
3.8. Amounts of Virus Antigen in Mouse Lungs
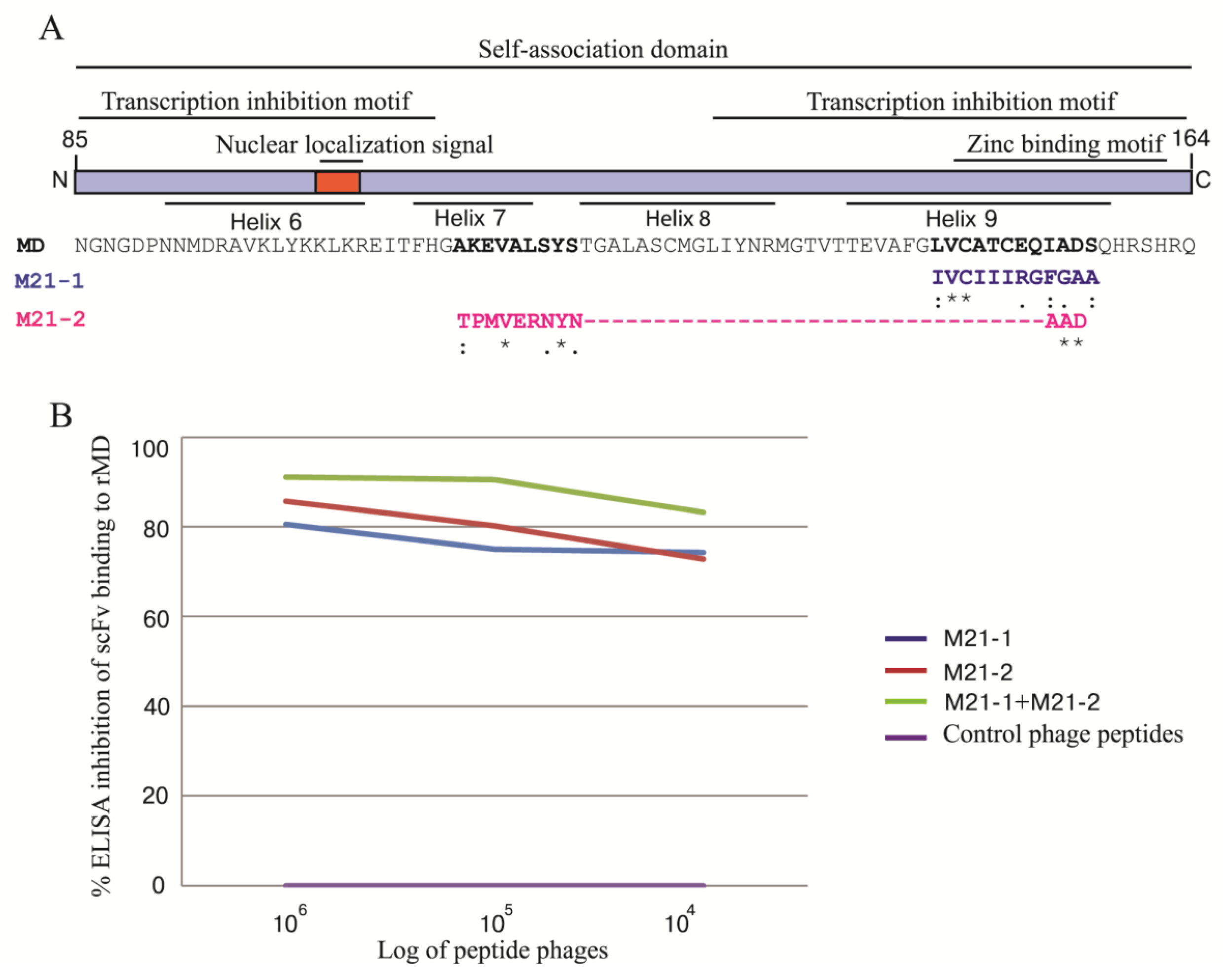
3.9. Phage Mimotopic Peptides that Bound to MD Specific-scFv and Presumptive scFv Epitope
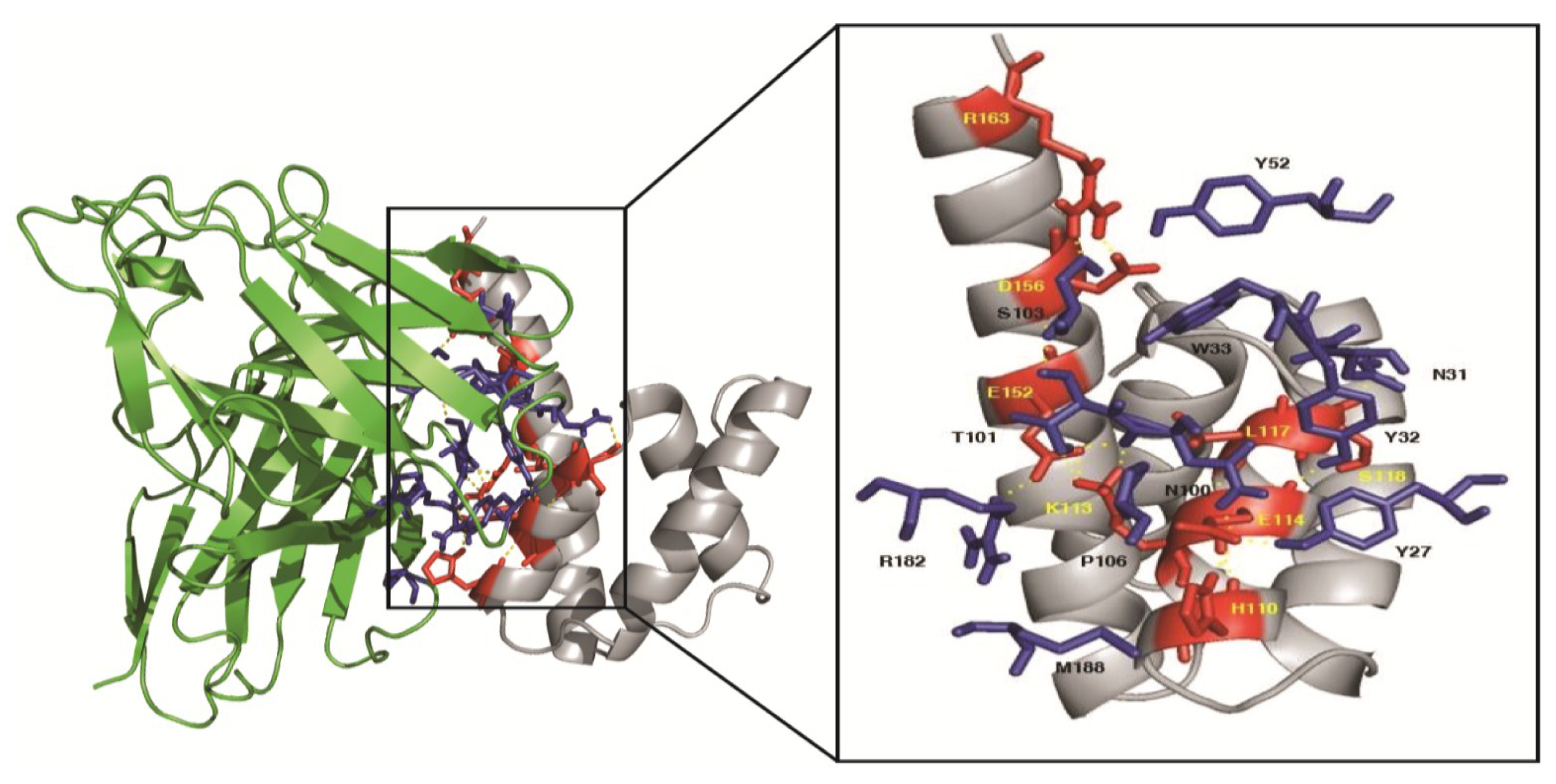
3.10. Homology Modeling and Molecular Docking

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | C-Score | TM-Score | RMSD (Å) | No. of Decoys | Cluster Density |
|---|---|---|---|---|---|
| scFv21 | 0.73 | 0.81 ± 0.09 | 4.2 ± 2.8 | 10165 | 0.5630 |
| M1 MD | 1.43 | 0.91 ± 0.06 | 1.0 ± 1.0 | 10200 | 1.1111 |
| Influenza A M1 Middle Domain | Human scFv21 | Intermolecular Bond | ||
|---|---|---|---|---|
| Amino Acid | Motif | Amino Acid | Domain | |
| H110 | Helix 7 | P106 | VH-CDR3 | Hydrophobic |
| H110 | Helix 7 | M188 | VL-FR3 | Hydrophobic |
| K113 | Helix 7 | N100 | VH-CDR3 | Hydrogen |
| K113 | Helix 7 | T101 | VH-CDR3 | Hydrogen |
| E114 | Helix 7 | Y27 | VH-CDR1 | Hydrogen |
| E114 | Helix 7 | Y32 | VH-CDR1 | Hydrogen |
| E114 | Helix 7 | N100 | VH-CDR3 | Hydrogen |
| L117 | Helix 7 | W33 | VH-CDR1 | Hydrophobic |
| S118 | Between helices 7 and 8 | N31 | VH-CDR1 | Hydrogen |
| E152 | Helix 9 | T101 | VH-CDR3 | Hydrogen |
| E152 | Helix 9 | R182 | VL-FR2 | Salt-bridge |
| D156 | Helix 9 | Y52 | VH-CDR2 | Hydrogen |
| D156 | Helix 9 | Y52 | VH-CDR2 | Hydrogen |
| R163 | Helix 9 | S103 | VH-CDR3 | Hydrogen |
4. Discussion
Acknowledgements
Author Contributions
Conflicts of Interests
References
- Lamb, R.A.; Krug, R.M. Orthomyxoviridae: The Viruses and Their Replication. In Fields’ Virology, 3rd ed.; Lippincott-Raven: Philadephia, PA, USA, 1996; pp. 1353–1395. [Google Scholar]
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yang, Y.; Qiu, Y.; Yang, Y. Annual economic impacts of seasonal influenza on US counties: Spatial heterogeneity and patterns. Int. J. Health Geogr. 2012, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, T.; Ellis, R.W. Influenza vaccine technologies and the use of the cell-culture process (cell-culture influenza vaccine). Dev. Biol. 2002, 110, 125–134. [Google Scholar]
- Reisinger, K.S.; Block, S.L.; Izu, A.; Groth, N.; Holmes, S.J. Subunit influenza vaccines produced from cell culture or in embryonated chicken eggs: Comparison of safety, reactogenicity, and immumogenicity. J. Infect. Dis. 2009, 200, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Epstein, S.L.; Tumpey, T.M.; Misplon, J.A.; Lo, C.Y.; Cooper, L.A.; Subbarao, K.; Renshaw, M.; Sambhara, S.; Katz, J.M. DNA vaccine expressing conserved influenza virus proteins protective against H5N1 challenge infection in mice. Emerg. Infect. Dis. 2002, 8, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Thueng-in, K.; Maneewatch, S.; Srimanote, P.; Songserm, T.; Tapchaisri, P.; Sookrung, N.; Tongtawe, P.; Channarong, S.; Chaicumpa, W. Heterosubtypic immunity to influenza mediated by liposome adjuvanted H5N1 recombinant protein vaccines. Vaccine 2010, 28, 6765–6777. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, A.; Abed, Y.; Boivin, G. Influenza drug resistance. Semin. Respir. Crit. Care Med. 2011, 32, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Vasin, A.V.; Temkina, O.A.; Egora, V.V.; Klotchenko, S.A.; Plotnikova, M.A.; Kiselev, O.I. Molecular mechanisms enhancing the proteome of influenza A viruses: An overview of recently discovered proteins. Virus Res. 2014, 154, 53–63. [Google Scholar] [CrossRef]
- Heiny, A.T.; Miotto, O.; Srinivasan, K.N.; Khan, A.M.; Zhang, G.L.; Brusic, V.; Tan, T.W.; August, J.T. Evolutionarily conserved protein sequences of influenza A viruses, avian and human, as vaccine targets. PLoS One 2007, 2, e1190. [Google Scholar] [CrossRef] [PubMed]
- Bui, M.; Whittaker, G.; Helenius, A. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 1996, 70, 8391–8401. [Google Scholar] [PubMed]
- Yasuda, J.; Nakada, S.; Kato, A.; Toyoda, T.; Ishihama, A. Molecular assembly of influenza virus: Association of the NS2 protein with virion matrix. Virol. 1993, 196, 249–255. [Google Scholar] [CrossRef]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Muller, C.W.; Ruigrok, R.W.H.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Takizawa, N.; Watanabe, K.; Nagata, K.; Kobayashi, N. Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP. FEBS Lett. 2011, 585, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ekstrom, M.; Garoff, H. The M1 and NP proteins of influenza A virus form homo- but nothetero-oligomeric complexes when co-expressed in BHK-21 cells. J. Gen. Virol. 1998, 79, 2435–2446. [Google Scholar] [PubMed]
- Ali, A.; Avalos, R.; Ponimaskin, E.; Nayak, D.P. Influenza virus assembly: Effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 2000, 74, 8709–8719. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puertas, P.; Albo, C.; Pérez-Pastrana, E.; Vivo, A.; Portela, A. Influenza virus matrix protein is the major driving force in virus budding. J. Virol. 2000, 74, 11538–11547. [Google Scholar] [CrossRef] [PubMed]
- Noton, S.L.; Medcalf, E.; Fisher, D.; Mullin, A.E.; Elton, D.; Digard, P. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J. Gen. Virol. 2007, 88, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, Z.; Liu, X.; Yin, C.; Basit, Z.; Xia, B.; Liu, W. Dissection of influenza A virus M1 protein: PH-dependent oligomerization of N-terminal domain and dimerization of C-terminal domain. PLoS One 2012, 7, e37786. [Google Scholar] [CrossRef] [PubMed]
- Sha, B.; Luo, M. Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat. Struct. Mol. Biol. 1997, 4, 239–244. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Yu, M.; Wang, Z.; Xu, C.; Xue, Q.; Zhang, K.; Ye, X.; Kitamura, Y.; Liu, W. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell Microbiol. 2009, 11, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Songserm, T.; Jam-on, R.; Sae-Heng, N.; Meemak, N.; Hulse-Past, D. Domestic ducks and H5N1 influenza epidemic Thailand. Emerg. Infect. Dis. 2006, 12, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungtrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia neurotoxin. J. Proteomics 2009, 72, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Poungpair, O.; Pootong, A.; Maneewatch, S.; Srimanote, P.; Tongtawe, P.; Songserm, T.; Tapchaisri, P.; Chaicumpa, W. A Human single chain transbody specifc to matrix protein (M1) interferes with the replication of influenza A virus. Bioconjug. Chem. 2011, 21, 1134–1141. [Google Scholar] [CrossRef]
- Thueng-in, K.; Thanongsaksrikul, J.; Srimanote, P.; Bangphoomi, K.; Poungpair, O.; Maneewatch, S.; Choowongkomon, K.; Chaicumpa, W. Cell penetrable humanized-VH/VHH that inhibit RNA dependent RNA polymerase (NS5B) of HCV. PLoS One 2012, 7, e49254. [Google Scholar] [CrossRef] [PubMed]
- Chulanetra, M.; Bangphoomi, K.; Sookrung, N.; Thanongsaksrikul, J.; Srimanote, P.; Sakolvarvaree, Y.; Choowongkomon, K.; Chaicumpa, W. Human ScFv that block sodium ion channel activity of tetrodotoxin. Toxicon 2012, 59, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Pissawong, T.; Maneewatch, S.; Thueng-in, K.; Srimanote, P.; Dong-din-on, F.; Thanongsaksrikul, J.; Songserm, T.; Tongtawe, P.; Bangphoomi, B.; Chaicumpa, W. Human monoclonal ScFv that bind to different functional domains of M2 and inhibit H5N1 influenza virus replication. Virol. J. 2013, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004, 32, 96–99. [Google Scholar] [CrossRef]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Hall, D.R.; Chuang, G.Y.; Comeau, S.R.; Bohnuud, T.; Beglov, D.; Schueler-Furman, O.; Vajda, S.; Kozakov, D. Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics 2012, 28, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Prot. Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.3r1 edu; Schrodinger, LLC: Portland, OR, USA, 2010.
- Haidar, J.N.; Yuan, Q.A.; Zeng, L.; Snavely, M.; Luna, X.; Zhang, H.; Zhu, W.; Ludwig, D.L.; Zhu, Z. A universal combinatorial design of antibody framework to graft distinct CDR sequences: A bioinformatics approach. Proteins 2012, 80, 896–912. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [PubMed]
- Yodsheewan, R.; Maneewatch, S.; Srimanote, P.; Thueng-In, K.; Songserm, T.; Dong-din-on, F.; Bangphoomi, K.; Sookrung, N.; Choowongkomon, K.; Chaicumpa, W. Human monoclonal ScFv specific to NS1 protein inhibits replication of influenza viruses across types and subtypes. Antivir. Res. 2013, 100, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Vancell, R.; Beaty, G.; Stefani, E.; Rodríguez-Boulan, E.E.; Cereijido, M. Changes in paracellular and cellular ionic permeability of monolayers of MDCK cells infected with influenza or vesicular stomatitis viruses. J. Membr. Biol. 1984, 81, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, R.; Morris, S.J. The influenza haemagglutinin-induced fusion cascade: Effects of target membrane permeability changes. Mol. Membr. Biol. 1999, 16, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Frolov, V.A.; Dunina-Barkovskaya, A.Y.; Samsonov, A.V.; Zimmerberg, J. Membrane permeabilitychanges at early stages of influenza hemagglutinin-mediated fusion. Biophys. J. 2003, 85, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Koff, W.C.; Knight, V. Inhibition of influenza virus uncoating by rimantadine hydrochloride. J. Virol. 1979, 31, 261–263. [Google Scholar] [PubMed]
- Bukrinskaya, A.G.; Vorkunova, N.K.; Kornilayeva, G.V.; Narmanbetova, R.A.; Vorkunova, G.K. Influenza virus uncoating in infected cells and effect of rimantadine. J. Gen. Virol. 1982, 60, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wurtzer, S.; Rameix-Welti, M.; Dwyer, D.; van der Werf, S.; Naffakh, N.; Clavel, F.; Labrosse, B. Enhancement of the influenza A hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS One 2009, 4, e8495. [Google Scholar] [CrossRef] [PubMed]
- Galabov, A.; Simeonova, L.; Gegova, G. Rimantadine and oseltamivir demonstrate synergistic combination effect in an experimental infection with type A (H3N2) influenza virus in mice. Antivir. Chem. Chemother. 2006, 17, 251–258. [Google Scholar] [PubMed]
- Zhang, J.; Li, G.; Liu, X.; Wang, Z.; Liu, W.; Ye, X. Influenza A virus M1 blocks the classical complement pathway through interacting with C1qA. J. Gen. Virol. 2009, 90, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, N.; Prabakaran, M.; Ho, H.; Velumani, S.; Qiang, J.; Goutama, M.; Kwang, J. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J. Virol. 2009, 83, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Robinson, D.; Wagner, R.R. Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J. Virol. 1995, 69, 1964–1970. [Google Scholar] [PubMed]
- Elster, C.; Larsen, J.; Gagnon, J.; Ruigrok, R.W.; Baudin, F. Influenza virus M1 protein binds to RNA through its nuclear localization signal. J. Gen. Virol. 1997, 78, 1589–1596. [Google Scholar] [PubMed]
- Ye, Z.P.; Baylor, N.W.; Wagner, R.R. Transcription-inhibition and RNA-binding domains of influenza A virus matrix protein mapped with anti-idiotypic antibodies and synthetic peptides. J. Virol. 1989, 63, 3586–3594. [Google Scholar] [PubMed]
- Elster, C.; Fourest, E.; Baudin, F.; Larsen, K.; Cusack, S.; Ruigrok, R.W. A small percentage of influenza virus M1 protein contains zinc but zinc does not influence in vitro M1-RNA interaction. J. Gen. Virol. 1994, 75, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Miura, T.; Takeuchi, H. Zinc- and pH- dependent conformational transition in a putative interdomain linker region of the influenza virus matrix protein M1. Biochem. 2003, 42, 1978–1984. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong-din-on, F.; Songserm, T.; Pissawong, T.; Srimanote, P.; Thanongsaksrikul, J.; Thueng-in, K.; Moonjit, P.; Lertwatcharasarakul, P.; Seesuay, W.; Chaicumpa, W. Cell Penetrable Human scFv Specific to Middle Domain of Matrix Protein-1 Protects Mice from Lethal Influenza. Viruses 2015, 7, 154-179. https://doi.org/10.3390/v7010154
Dong-din-on F, Songserm T, Pissawong T, Srimanote P, Thanongsaksrikul J, Thueng-in K, Moonjit P, Lertwatcharasarakul P, Seesuay W, Chaicumpa W. Cell Penetrable Human scFv Specific to Middle Domain of Matrix Protein-1 Protects Mice from Lethal Influenza. Viruses. 2015; 7(1):154-179. https://doi.org/10.3390/v7010154
Chicago/Turabian StyleDong-din-on, Fonthip, Thaweesak Songserm, Tippawan Pissawong, Potjanee Srimanote, Jeeraphong Thanongsaksrikul, Kanyarat Thueng-in, Pattra Moonjit, Preeda Lertwatcharasarakul, Watee Seesuay, and Wanpen Chaicumpa. 2015. "Cell Penetrable Human scFv Specific to Middle Domain of Matrix Protein-1 Protects Mice from Lethal Influenza" Viruses 7, no. 1: 154-179. https://doi.org/10.3390/v7010154
APA StyleDong-din-on, F., Songserm, T., Pissawong, T., Srimanote, P., Thanongsaksrikul, J., Thueng-in, K., Moonjit, P., Lertwatcharasarakul, P., Seesuay, W., & Chaicumpa, W. (2015). Cell Penetrable Human scFv Specific to Middle Domain of Matrix Protein-1 Protects Mice from Lethal Influenza. Viruses, 7(1), 154-179. https://doi.org/10.3390/v7010154