Coat as a Dagger: The Use of Capsid Proteins to Perforate Membranes during Non-Enveloped DNA Viruses Trafficking

Abstract
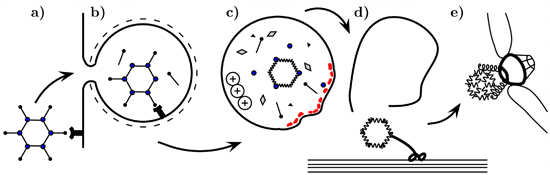
:
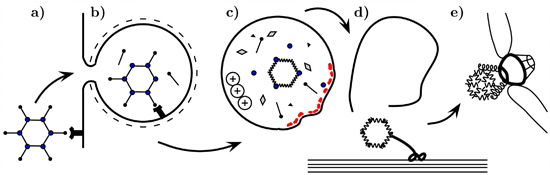
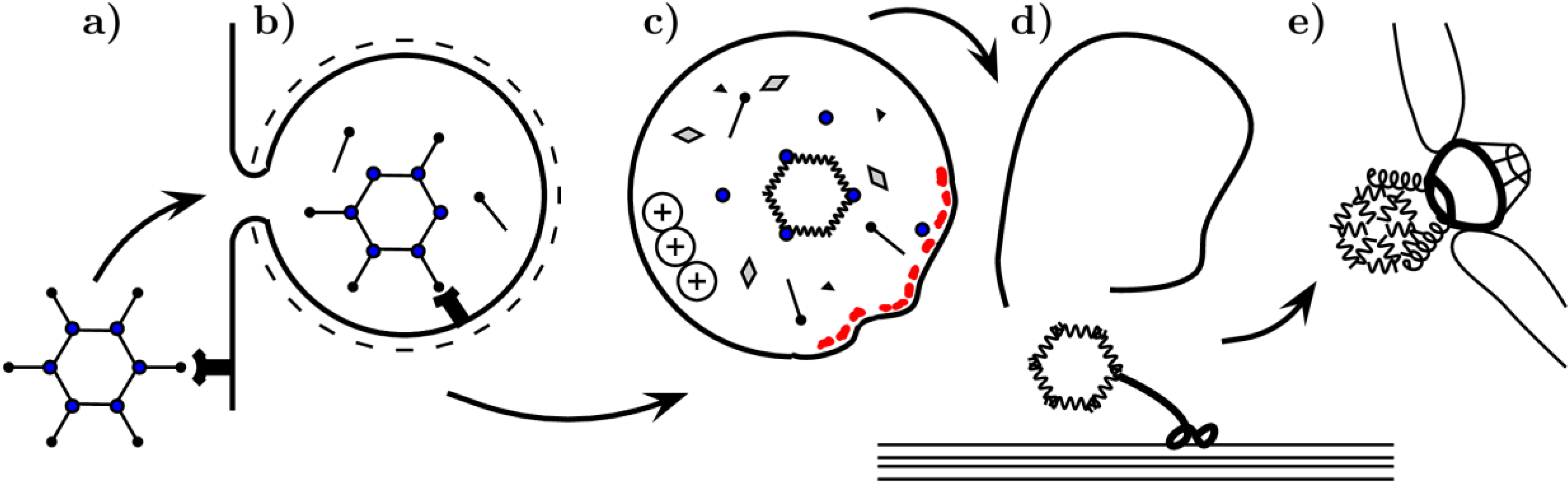
1. Introduction
2. The Role of Capsid Proteins in Membrane Penetration
2.1. Adenoviridae
2.1.1. Receptor Binding
2.1.2. Endosome Escape
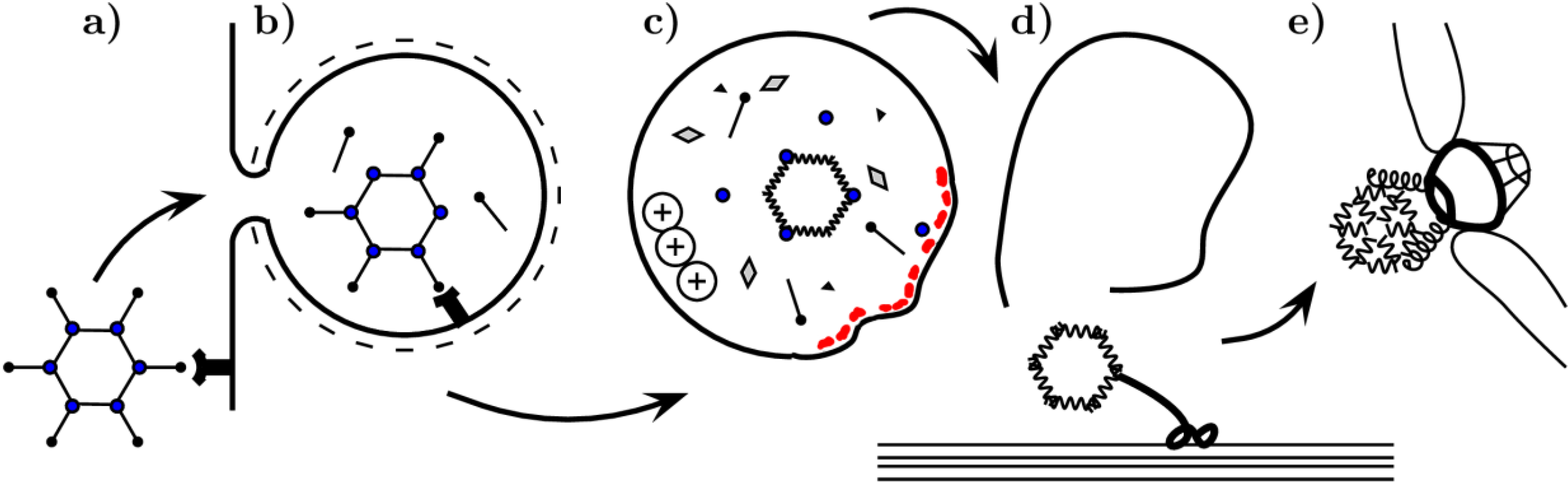
2.1.2.1. Role of Protein VI
2.1.3. Nuclear Translocation
2.2. Parvoviridae
2.2.1. Cell Entry
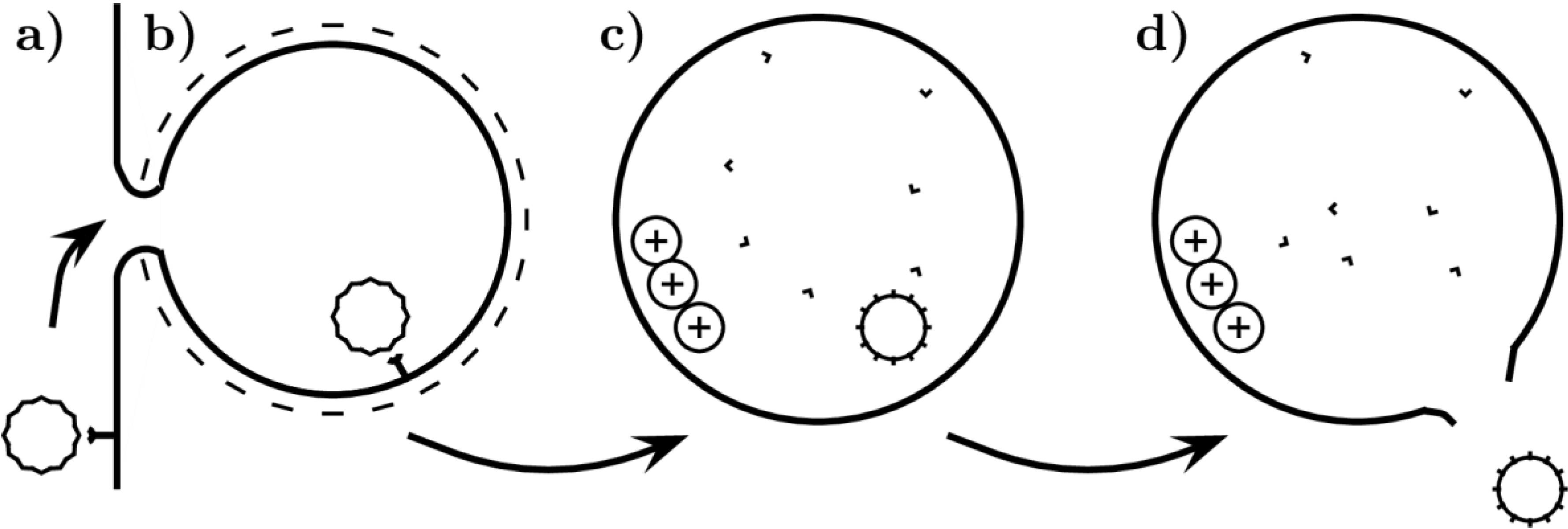
2.2.2. Endosome Escape and Capsid Disassembly
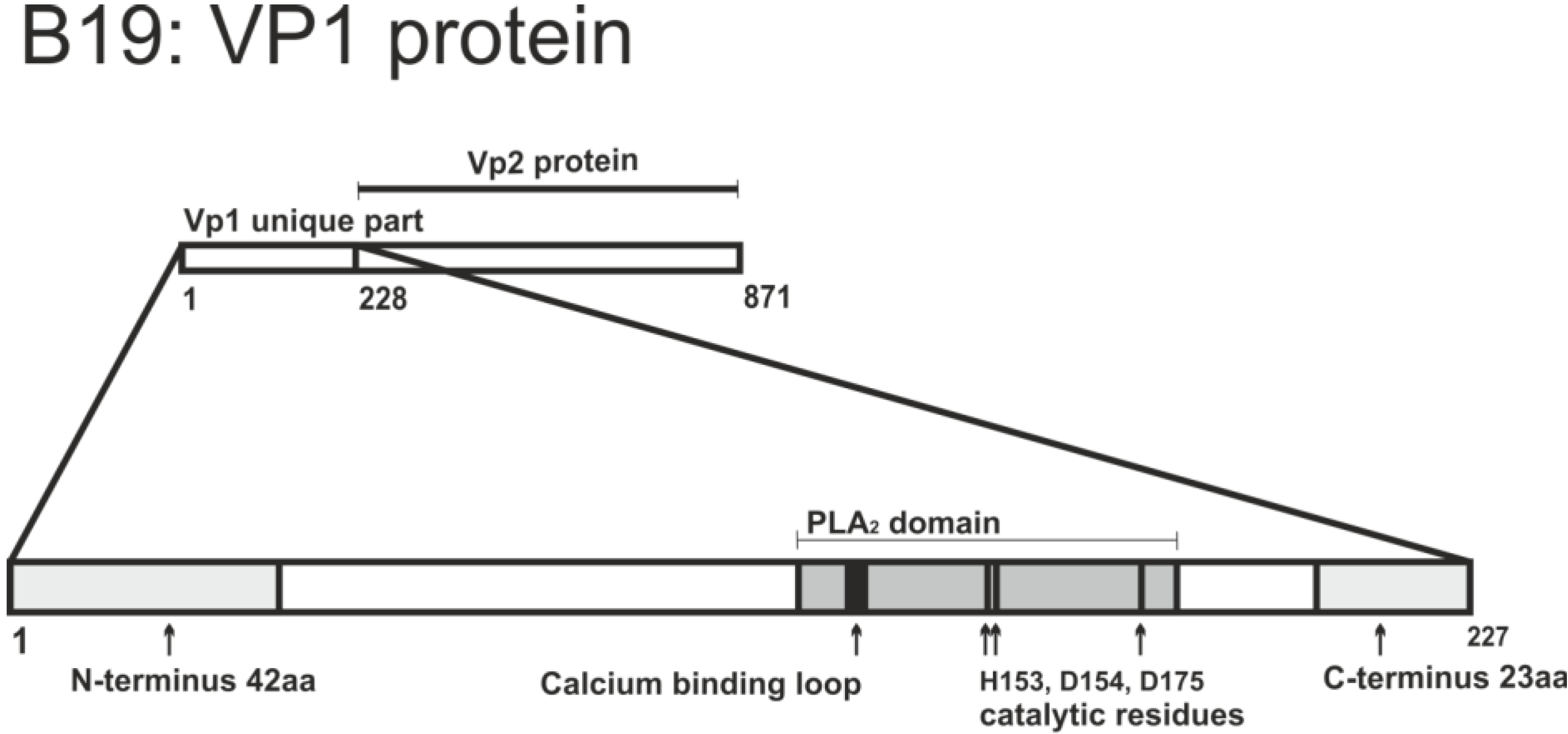
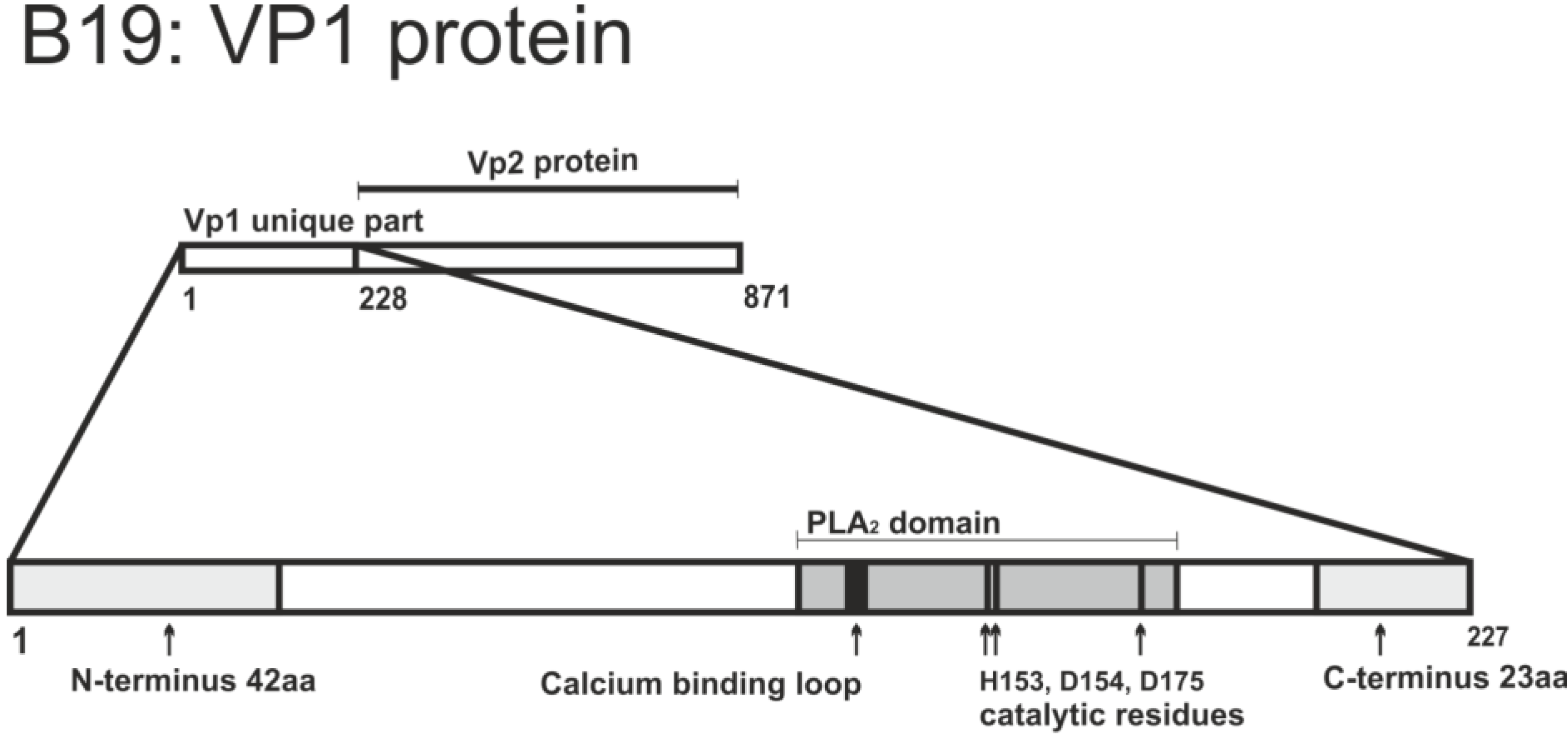
2.2.2.1. VP1 Unique Region

{kind=link}
{kind=link}
{kind=link}
{kind=link}
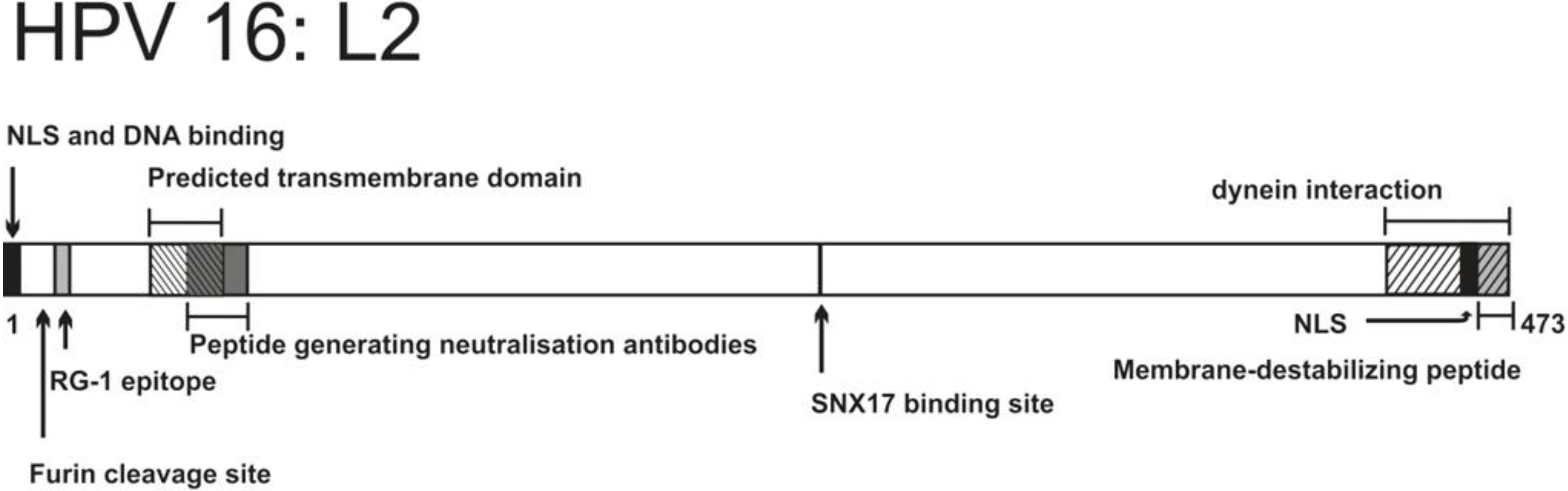{kind=link}
| Residues and motifs | Biological properties | Introduced mutation | Effects of mutation | Reference |
|---|---|---|---|---|
| 123–181 | PLA2 motif, based on sequence alignment and homology | Fragment of VP1 from 2 to 240 aa | Protein exhibits PLA2 activity | [97] |
| 131–227 | includes PLA2 motif | Deletion 131-227 | Abolished PLA2 activity of protein | [99] |
| H153 (histidine) D175 (aspartic acid) | Catalytic residues | H153A, D175A point substitution | Abolished PLA2 activity of protein | [99] |
| P133 (proline) | Calcium binding loop residue | P133R substitution | Abolished PLA2 activity of protein | [99] |
| 1–42 (N-terminus 42 aa) 205–227 (C-terminus 23 aa) | Outside PLA2 motif but required for its function | Deletion 43–227, deletion 1–204 | Severely impaired PLA2 activity of protein | [104] |
| E176 (glutamic acid) | Proximal to catalytic residues of PLA2 motif | E176D substitution | Abolished PLA2 activity of protein and infection | [103] |
2.2.3. Further Trafficking and Entry into the Nucleus
2.3. Papillomaviridae
2.3.1. Cell Entry
2.3.2. Intracellular Trafficking
2.3.3. Endosome Escape
2.3.3.1. L2 Sequence Properties Related to Endosome Escape
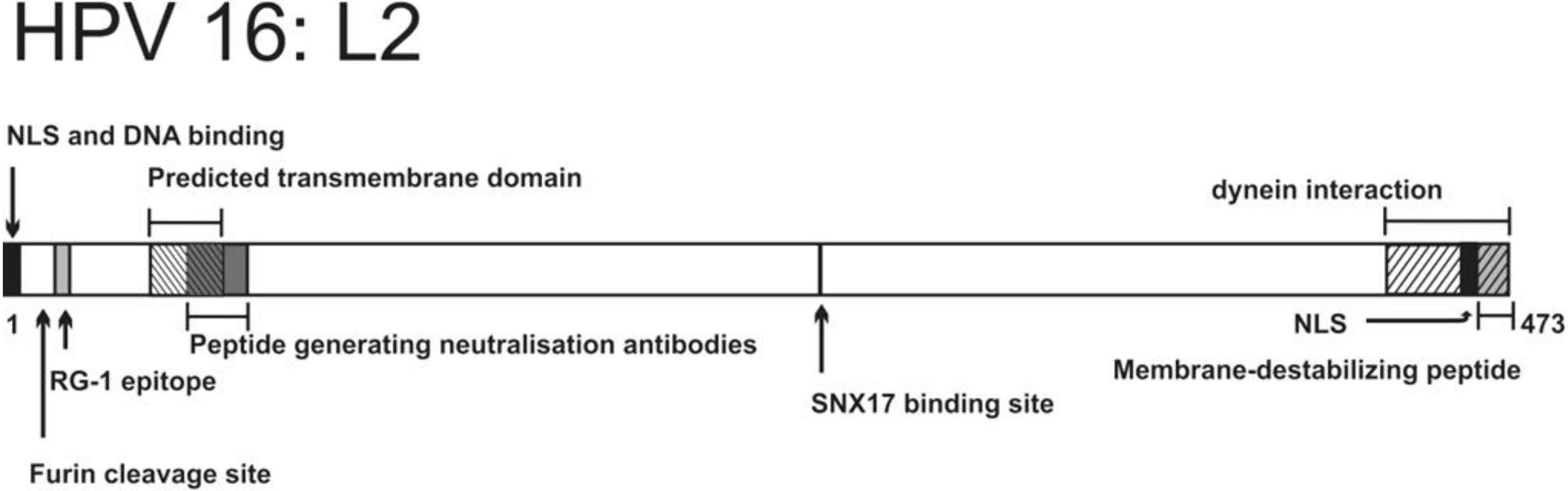
| Residues and motifs | Biological properties | Introduced mutation | Effects of mutation | Reference |
|---|---|---|---|---|
| 17–34 (RG-1 epitope) | Anti RG-1 antibodies have neutralizing properties Epitope exposed during early stages of infection | Not relevant | Not relevant | [118,119,120] |
| 9–12 (RRKR) | Furin cleavage site | R12S substitution | Impaired infectivity, virus retained in endosomal vesicles | [119] |
| C22, C28 (cysteines) | Form a disulfide bond | C22S and/or C28S substitution | Virions lack infectivity, internalization and trafficking to endolysosomes are not affected | [155,156] |
| 45–67 | Predicted transmembrane domain, adopts α-helical structure | e.g., G56V, G57V substitution, A55, A60 insertion | Non-infectious virions, viral DNA trapped in an endosomal compartment during infection | [157] |
| 56–75 | Antipeptide serum inhibits infection, mainly by blocking the viral genome transport to the nucleus | Not relevant | Not relevant | [158] |
| 254–257 | Interaction with SNX17 | N254A substitution | Abolished infectivity, capsid targeting to lysosomes increased | [159] |
| 451–473 (last 23aa at C-terminus) | Membrane-destabilizing peptide | Deletion 455–473 or 465–473 | Abolished infectivity, viral genome retained in endosomes during infection | [154] |
2.3.3.2. L2 and Viral Intracellular Targeting
| Features | Adenoviridae | Parvoviridae | Papillomaviridae | Polyomaviridae |
|---|---|---|---|---|
| MPCP | VI | VP1 | L2 | VP2, VP3 |
| Weight of MPCP (relative to the capsid) | 5% | 10% | 12% | 14% |
| MPCP exposed on the surface (>15%) | no | No | no | no |
| Site of viral genome escape from the vesicle | endosome | Endosome | Trans-Golgi network? ER? | ER |
| Conditions required for MPCP exposure | Partial capsid disassembly, release of other capsid proteins | High temperature, low pH, cleavage of other capsid proteins | Interaction of the capsid with primary receptor, extracellular enzymes | ER resident enzymes, interaction with the membrane, low pH for MPyV |
| MPCP associates with the capsid during membrane damage | no | Yes | no (but associates with the genome) | not known |
| MPCP forms oligomers | not determined | not determined | yes | yes |
2.3.4. Further Trafficking and Possible Membrane Penetration Site
2.4. Polyomaviridae
2.4.1. Cell Entry
2.4.2. Trafficking Inside the Cell
2.4.3. Roles of the Endoplasmic Reticulum
2.4.4. Coat Determinants
2.4.4.1. Membrane Insertion Properties of the Minor Capsid Proteins
| Virus families | Membrane penetration mechanism | Conformation of the protein segment required for membrane penetration |
|---|---|---|
| Parvoviridae | Enzymatic activity | α-helices [109] |
| Adenoviridae | Protein-membrane interaction | Amphipathic α-helix [33] |
| Papillomaviridae | Transmembrane segment * [157,220,221] | |
| Polyomaviridae |
2.4.4.2. Role of the Minor Structural Proteins in the Virus Escape from ER
2.4.5. Post-Vesicular Steps
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Day, P.M.; Schelhaas, M. Concepts of papillomavirus entry into host cells. Curr. Opin. Virol. 2014, 4, 24–31. [Google Scholar] [CrossRef]
- Cerqueira, C.; Schelhaas, M. Principles of polyoma- and papillomavirus uncoating. Med. Microbiol. Immunol. 2012, 201, 427–436. [Google Scholar]
- Puntener, D.; Greber, U.F. Dna-tumor virus entry-from plasma membrane to the nucleus. Semin. Cell Dev. Biol. 2009, 20, 631–642. [Google Scholar] [CrossRef]
- Benevento, M.; Di Palma, S.; Snijder, J.; Moyer, C.L.; Reddy, V.S.; Nemerow, G.R.; Heck, A.J. Adenovirus composition, proteolysis, and disassembly studied by in-depth qualitative and quantitative proteomics. J. Biol. Chem. 2014, 289, 11421–11430. [Google Scholar]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrin-alpha-v-beta-3 and integrin-alpha-v-beta-5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Mathias, P.; Wickham, T.; Moore, M.; Nemerow, G. Multiple adenovirus serotypes use alpha-v integrins for infection. J. Virol. 1994, 68, 6811–6814. [Google Scholar]
- Huang, S.A.; Endo, R.I.; Nemerow, G.R. Up-regulation of integrins alpha-v-beta-3 and alpha-v-beta-5 on human monocytes and t-lymphocytes facilitates adenovirus-mediated gene delivery. J. Virol. 1995, 69, 2257–2263. [Google Scholar]
- Davison, E.; Diaz, R.M.; Hart, I.R.; Santis, G.; Marshall, J.F. Integrin alpha 5 beta 1-mediated adenovirus infection is enhanced by the integrin-activating antibody ts2/16. J. Virol. 1997, 71, 6204–6207. [Google Scholar]
- Nemerow, G.R.; Stewart, P.L. Role of alpha(v) integrins in adenovirus cell entry and gene delivery. Microbiol. Mol. Biol. Rev. 1999, 63, 725–734. [Google Scholar]
- Wang, K.N.; Huang, S.; Kapoor-Munshi, A.; Nemerow, G. Adenovirus internalization and infection require dynamin. J. Virol. 1998, 72, 3455–3458. [Google Scholar]
- Amstutz, B.; Gastaldelli, M.; Kalin, S.; Imelli, N.; Boucke, K.; Wandeler, E.; Mercer, J.; Hemmi, S.; Greber, U.F. Subversion of ctbp1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J. 2008, 27, 956–969. [Google Scholar] [CrossRef]
- Imelli, N.; Ruzsics, Z.; Puntener, D.; Gastaldelli, M.; Greber, U.F. Genetic reconstitution of the human adenovirus type 2 temperature-sensitive 1 mutant defective in endosomal escape. Virol. J. 2009, 6, 174. [Google Scholar] [CrossRef]
- Meier, O.; Boucke, K.; Hammer, S.V.; Keller, S.; Stidwill, R.P.; Hemmi, S.; Greber, U.F. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J. Cell Biol. 2002, 158, 1119–1131. [Google Scholar] [CrossRef]
- Kaelin, S.; Amstutz, B.; Gastaldelli, M.; Wolfrum, N.; Boucke, K.; Havenga, M.; DiGennaro, F.; Liska, N.; Hemmi, S.; Greber, U.F. Macropinocytotic uptake and infection of human epithelial cells with species b2 adenovirus type 35. J. Virol. 2010, 84, 5336–5350. [Google Scholar] [CrossRef]
- Burckhardt, C.J.; Suomalainen, M.; Schoenenberger, P.; Boucke, K.; Hemmi, S.; Greber, U.F. Drifting motions of the adenovirus receptor car and immobile integrins initiate virus uncoating and membrane lytic protein exposure. Cell Host Microbe 2011, 10, 105–117. [Google Scholar] [CrossRef]
- Nakano, M.Y.; Boucke, K.; Suomalainen, M.; Stidwill, R.P.; Greber, U.F. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J. Virol. 2000, 74, 7085–7095. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Li, Z.Y.; Gaggar, A.; Gharwan, H.; Ternovoi, V.; Sandig, V.; Lieber, A. Genome size and structure determine efficiency of postinternalization steps and gene transfer of capsid-modified adenovirus vectors in a cell-type-specific manner. J. Virol. 2004, 78, 10009–10022. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Eberly, A.M.; Li, Z.Y.; Lieber, A. Deletion of penton rgd motifs affects the efficiency of both the internalization and the endosorne escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005, 79, 1053–1061. [Google Scholar] [CrossRef]
- Miyazawa, N.; Crystal, R.G.; Leopold, P.L. Adenovirus serotype 7 retention in a late endosomal compartment prior to cytosol escape is modulated by fiber protein. J. Virol. 2001, 75, 1387–1400. [Google Scholar] [CrossRef]
- Miyazawa, N.; Leopold, P.L.; Hackett, N.R.; Ferris, B.; Worgall, S.; Falck-Pedersen, E.; Crystal, R.G. Fiber swap between adenovirus subgroups b and c alters intracellular trafficking of adenovirus gene transfer vectors. J. Virol. 1999, 73, 6056–6065. [Google Scholar]
- Martin-Fernandez, M.; Longshaw, S.V.; Kirby, I.; Santis, G.; Tobin, M.J.; Clarke, D.T.; Jones, G.R. Adenovirus type-5 entry and disassembly followed in living cells by fret, fluorescence anisotropy, and flim. Biophys. J. 2004, 87, 1316–1327. [Google Scholar] [CrossRef]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus-2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef]
- Carey, B.; Staudt, M.K.; Bonaminio, D.; van der Loo, J.C.M.; Trapnell, B.C. Pu.1 redirects adenovirus to lysosomes in alveolar macrophages, uncoupling internalization from infection. J. Immunol. 2007, 178, 2440–2447. [Google Scholar]
- Engesaeter, B.O.; Tveito, S.; Bonsted, A.; Engebraaten, O.; Berg, K.; Maelandsmo, G.M. Photochemical treatment with endosomally localized photosensitizers enhances the number of adenoviruses in the nucleus. J. Gene Med. 2006, 8, 707–718. [Google Scholar] [CrossRef]
- Maier, O.; Marvin, S.A.; Wodrich, H.; Campbell, E.M.; Wiethoff, C.M. Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J. Virol. 2012, 86, 10821–10828. [Google Scholar] [CrossRef]
- Gastaldelli, M.; Imelli, N.; Boucke, K.; Amstutz, B.; Meier, O.; Greber, U.F. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 2008, 9, 2265–2278. [Google Scholar] [CrossRef]
- Morgan, C.; Rosenkra, H.S.; Mednis, B. Structure and development of viruses as observed in electron microscope. 10. Entry and uncoating of adenovirus. J. Virol. 1969, 4, 777–796. [Google Scholar]
- Seth, P. Adenovirus-dependent release of choline from plasma-membrane vesicles at an acidic ph is mediated by the penton base protein. J. Virol. 1994, 68, 1204–1206. [Google Scholar]
- Prchla, E.; Plank, C.; Wagner, E.; Blaas, D.; Fuchs, R. Virus-mediated release of endosomal content in-vitro—Different behavior of adenovirus and rhinovirus serotype-2. J. Cell Biol. 1995, 131, 111–123. [Google Scholar] [CrossRef]
- Suomalainen, M.; Luisoni, S.; Boucke, K.; Bianchi, S.; Engel, D.A.; Greber, U.F. A direct and versatile assay measuring membrane penetration of adenovirus in single cells. J. Virol. 2013, 87, 12367–12379. [Google Scholar] [CrossRef]
- Farr, G.A.; Zhang, L.G.; Tattersall, P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. USA 2005, 102, 17148–17153. [Google Scholar]
- Kato, S.E.; Chahal, J.S.; Flint, S.J. Reduced infectivity of adenovirus type 5 particles and degradation of entering viral genomes associated with incomplete processing of the preterminal protein. J. Virol. 2012, 86, 13554–13565. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein vi mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef]
- Greber, U.F.; Webster, P.; Weber, J.; Helenius, A. The role of the adenovirus protease in virus entry into cells. EMBO J. 1996, 15, 1766–1777. [Google Scholar]
- Wodrich, H.; Guan, T.L.; Cingolani, G.; Von Seggern, D.; Nemerow, G.; Gerace, L. Switch from capsid protein import to adenovirus assembly by cleavage of nuclear transport signals. EMBO J. 2003, 22, 6245–6255. [Google Scholar] [CrossRef]
- Van Oostrum, J.; Burnett, R.M. The structure of the adenovirus capsid. Biophys. J. 1985, 47, 394A–394A. [Google Scholar]
- Lehmberg, E.; Traina, J.A.; Chakel, J.A.; Chang, R.J.; Parkman, M.; McCaman, M.T.; Murakami, P.K.; Lahidji, V.; Nelson, J.W.; Hancock, W.S.; et al. Reversed-phase high-performance liquid chromatographic assay for the adenovirus type 5 proteome. J. Chrom. B Biomed. Sci. Appl. 1999, 732, 411–423. [Google Scholar] [CrossRef]
- Saban, S.D.; Nepomuceno, R.R.; Gritton, L.D.; Nemerow, G.R.; Stewart, P.L. Cryoem structure at 9 angstrom resolution of an adenovirus vector targeted to hematopoietic cells. J. Mol. Biol. 2005, 349, 526–537. [Google Scholar] [CrossRef]
- Silvestry, M.; Lindert, S.; Smith, J.G.; Maier, O.; Wiethoff, C.M.; Nemerow, G.R.; Stewart, P.L. Cryo-electron microscopy structure of adenovirus type 2 temperature-sensitive mutant 1 reveals insight into the cell entry defect. J. Virol. 2009, 83, 7375–7383. [Google Scholar] [CrossRef]
- Liu, H.R.; Jin, L.; Koh, S.B.S.; Atanasov, I.; Schein, S.; Wu, L.; Zhou, Z.H. Atomic structure of human adenovirus by cryo-em reveals interactions among protein networks. Science 2010, 329, 1038–1043. [Google Scholar] [CrossRef]
- Mangel, W.F.; McGrath, W.J.; Toledo, D.L.; Anderson, C.W. Viral-dna and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 1993, 361, 274–275. [Google Scholar] [CrossRef]
- Wodrich, H.; Henaff, D.; Jammart, B.; Segura-Morales, C.; Seelmeir, S.; Coux, O.; Ruzsics, Z.; Wiethoff, C.M.; Kremer, E.J. A capsid-encoded ppxy-motif facilitates adenovirus entry. PLoS Pathog. 2010, 6, e1000808. [Google Scholar] [CrossRef]
- Moyer, C.L.; Wiethoff, C.M.; Maier, O.; Smith, J.G.; Nemerow, G.R. Functional genetic and biophysical analyses of membrane disruption by human adenovirus. J. Virol. 2011, 85, 2631–2641. [Google Scholar] [CrossRef]
- Moyer, C.L.; Nemerow, G.R. Disulfide-bond formation by a single cysteine mutation in adenovirus protein vi impairs capsid release and membrane lysis. Virology 2012, 428, 41–47. [Google Scholar] [CrossRef]
- Maier, O.; Galan, D.L.; Wodrich, H.; Wiethoff, C.M. An n-terminal domain of adenovirus protein vi fragments membranes by inducing positive membrane curvature. Virology 2010, 402, 11–19. [Google Scholar] [CrossRef]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef]
- Nguyen, E.K.; Nemerow, G.R.; Smith, J.G. Direct evidence from single-cell analysis that human alpha-defensins block adenovirus uncoating to neutralize infection. J. Virol. 2010, 84, 4041–4049. [Google Scholar] [CrossRef]
- Smith, J.G.; Silvestry, M.; Lindert, S.; Lu, W.Y.; Nemerow, G.R.; Stewart, P.L. Insight into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog. 2010, 6, e1000959. [Google Scholar] [CrossRef]
- Flatt, J.W.; Kim, R.; Smith, J.G.; Nemerow, G.R.; Stewart, P.L. An intrinsically disordered region of the adenovirus capsid is implicated in neutralization by human alpha defensin 5. PLoS One 2013, 8, e61571. [Google Scholar]
- Snijder, J.; Reddy, V.S.; May, E.R.; Roos, W.H.; Nemerow, G.R.; Wuite, G.J.L. Integrin and defensin modulate the mechanical properties of adenovirus. J. Virol. 2013, 87, 2756–2766. [Google Scholar]
- Bremner, K.H.; Scherer, J.; Yi, J.L.; Vershinin, M.; Gross, S.P.; Vallee, R.B. Adenovirus transport via direct interaction of cytoplasmic dynein with the viral capsid hexon subunit. Cell Host Microbe 2009, 6, 523–535. [Google Scholar] [CrossRef]
- Suomalainen, M.; Nakano, M.Y.; Boucke, K.; Keller, S.; Greber, U.F. Adenovirus-activated pka and p38/mapk pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 2001, 20, 1310–1319. [Google Scholar] [CrossRef]
- Saphire, A.C.S.; Guan, T.L.; Schirmer, E.C.; Nemerow, G.R.; Gerace, L. Nuclear import adenovirus dna in vitro involves the nuclear protein import pathway and hsc70. J. Biol. Chem. 2000, 275, 4298–4304. [Google Scholar]
- Trotman, L.C.; Mosberger, N.; Fornerod, M.; Stidwill, R.P.; Greber, U.F. Import of adenovirus dna involves the nuclear pore complex receptor can/nup214 and histone h1. Nat. Cell Biol. 2001, 3, 1092–1100. [Google Scholar] [CrossRef]
- Hindley, C.E.; Lawrence, F.J.; Matthews, D.A. A role for transportin in the nuclear import of adenovirus core proteins and dna. Traffic 2007, 8, 1313–1322. [Google Scholar] [CrossRef]
- Strunze, S.; Trotman, L.C.; Boucke, K.; Greber, U.F. Nuclear targeting of adenovirus type 2 requires crm1-mediated nuclear export. Mol. Biol. Cell 2005, 16, 2999–3009. [Google Scholar] [CrossRef]
- Ozawa, K.; Young, N. Characterization of capsid and noncapsid proteins of b19 parvovirus propagated in human erythroid bone-marrow cell-cultures. J. Virol. 1987, 61, 2627–2630. [Google Scholar]
- Tattersall, P.; Cawte, P.J.; Shatkin, A.J.; Ward, D.C. 3 structural polypeptides coded for by minute virus of mice, a parvovirus. J. Virol. 1976, 20, 273–289. [Google Scholar]
- Cotmore, S.F.; Tattersall, P. The autonomously replicating parvoviruses of vertebrates. Adv. Virus Res. 1987, 33, 91–174. [Google Scholar] [CrossRef]
- Paradiso, P.R.; Rhode, S.L.; Singer, II. Canine parvovirus—A biochemical and ultrastructural characterization. J. Gen. Virol. 1982, 62, 113–125. [Google Scholar]
- Rose, J.A.; Maizel, J.V.; Inman, J.K.; Shatkin, A.J. Structural proteins of adenovirus-associated viruses. J. Virol. 1971, 8, 766–770. [Google Scholar]
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol. 2000, 74, 2777–2785. [Google Scholar] [CrossRef]
- Summerford, C.; Samulski, R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar]
- Parker, J.S.L.; Murphy, W.J.; Wang, D.; O'Brien, S.J.; Parrish, C.R. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 2001, 75, 3896–3902. [Google Scholar] [CrossRef]
- Hueffer, K.; Parker, J.S.L.; Weichert, W.S.; Geisel, R.E.; Sgro, J.Y.; Parrish, C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef]
- Johnson, F.B.; Fenn, L.B.; Owens, T.J.; Faucheux, L.J.; Blackburn, S.D. Attachment of bovine parvovirus to sialic acids on bovine cell membranes. J. Gen. Virol. 2004, 85, 2199–2207. [Google Scholar] [CrossRef]
- Brown, K.E.; Anderson, S.M.; Young, N.S. Erythrocyte-p antigen—Cellular receptor for b19 parvovirus. Science 1993, 262, 114–117. [Google Scholar]
- Munakata, Y.; Saito-Ito, T.; Kumura-Ishii, K.; Huang, J.; Kodera, T.; Ishii, T.; Hirabayashi, Y.; Koyanagi, Y.; Sasaki, T. Ku80 autoantigen as a cellular coreceptor for human parvovirus b19 infection. Blood 2005, 106, 3449–3456. [Google Scholar] [CrossRef]
- Weigel-Kelley, K.A.; Yoder, M.C.; Srivastava, A. Alpha 5 beta 1 integrin as a cellular coreceptor for human parvovirus b19: Requirement of functional activation of beta 1 integrin for viral entry. Blood 2003, 102, 3927–3933. [Google Scholar] [CrossRef]
- Duan, D.S.; Li, Q.; Kao, A.W.; Yue, Y.P.; Pessin, J.E.; Engelhardt, J.F. Dynamin is required for recombinant adeno-associated virus type 2 infection. J. Virol. 1999, 73, 10371–10376. [Google Scholar]
- Parker, J.S.L.; Parrish, C.R. Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J. Virol. 2000, 74, 1919–1930. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Hub, B.; Kartenbeck, J. Endocytosis of adeno-associated virus type 5 leads to accumulation of virus particles in the golgi compartment. J. Virol. 2002, 76, 2340–2349. [Google Scholar] [CrossRef]
- Dudleenamjil, E.; Lin, C.-Y.; Dredge, D.; Murray, B.K.; Robison, R.A.; Johnson, F.B. Bovine parvovirus uses clathrin-mediated endocytosis for cell entry. J. Gen. Virol. 2010, 91, 3032–3041. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Braspenning-Wesch, I.; Kartenbeck, J. Adeno-associated virus type 5 exploits two different entry pathways in human embryo fibroblasts. J. Gen. Virol. 2009, 90, 317–322. [Google Scholar] [CrossRef]
- Boisvert, M.; Fernandes, S.; Tijssen, P. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 2010, 84, 7782–7792. [Google Scholar] [CrossRef]
- Suikkanen, S.; Saarjarvi, K.; Hirsimaki, J.; Valilehto, O.; Reunanen, H.; Vihinen-Ranta, M.; Vuento, M. Role of recycling endosomes and lysosomes in dynein-dependent entry of canine parvovirus. J. Virol. 2002, 76, 4401–4411. [Google Scholar] [CrossRef]
- VihinenRanta, M.; Kalela, A.; Makinen, P.; Kakkola, L.; Marjomaki, V.; Vuento, M. Intracellular route of canine parvovirus entry. J. Virol. 1998, 72, 802–806. [Google Scholar]
- Vihinen-Ranta, M.; Yuan, W.; Parrish, C.R. Cytoplasmic trafficking of the canine parvovirus capsid and its role in infection and nuclear transport. J. Virol. 2000, 74, 4853–4859. [Google Scholar] [CrossRef]
- Vendeville, A.; Ravallec, M.; Jousset, F.X.; Devise, M.; Mutuel, D.; Lopez-Ferber, M.; Fournier, P.; Dupressoir, T.; Ogliastro, M. Densovirus infectious pathway requires clathrin-mediated endocytosis followed by trafficking to the nucleus. J. Virol. 2009, 83, 4678–4689. [Google Scholar] [CrossRef]
- Sanlioglu, S.; Benson, P.K.; Yang, J.S.; Atkinson, E.M.; Reynolds, T.; Engelhardt, J.F. Endocytosis and nuclear trafficking of adeno-associated virus type 2 are controlled by rac1 and phosphatidylinositol-3 kinase activation. J. Virol. 2000, 74, 9184–9196. [Google Scholar]
- Pakkanen, K.; Karttunen, J.; Virtanen, S.; Vuento, M. Sphingomyelin induces structural alteration in canine parvovirus capsid. Virus Res. 2008, 132, 187–191. [Google Scholar] [CrossRef]
- Mani, B.; Baltzer, C.; Valle, N.; Almendral, J.M.; Kempf, C.; Ros, C. Low ph-dependent endosomal processing of the incoming parvovirus minute virus of mice virion leads to externalization of the vp1n-terminal sequence (n-vp1), n-vp2 cleavage, and uncoating of the full-length genome. J. Virol. 2006, 80, 1015–1024. [Google Scholar] [CrossRef]
- Ros, C.; Baltzer, C.; Mani, B.; Kempf, C. Parvovirus uncoating in vitro reveals a mechanism of dna release without capsid disassembly and striking differences in encapsidated dna stability. Virology 2006, 345, 137–147. [Google Scholar] [CrossRef]
- Suikkanen, S.; Antila, M.; Jaatinen, A.; Vihinen-Ranta, M.; Vuento, M. Release of canine parvovirus from endocytic vesicles. Virology 2003, 316, 267–280. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-associated virus type 2 capsids with externalized vp1/vp2 trafficking domains are generated prior to passage through the cytoplasm and are maintained until uncoating occurs in the nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef]
- Farr, G.A.; Cotmore, S.F.; Tattersall, P. Vp2 cleavage and the leucine ring at the base of the fivefold cylinder control ph-dpendent externalization of both the vp1 n terminus and the genome of minute virus of mice. J. Virol. 2006, 80, 161–171. [Google Scholar] [CrossRef]
- Weichert, W.S.; Parker, J.S.L.; Wahid, A.T.M.; Chang, S.F.; Meier, E.; Parrish, C.R. Assaying for structural variation in the parvovirus capsid and its role in infection. Virology 1998, 250, 106–117. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Hafenstein, S.; Tattersall, P. Depletion of virion-associated divalent cations induces parvovirus minute virus of mice to eject its genome in a 3'-to-5' direction from an otherwise intact viral particle. J. Virol. 2010, 84, 1945–1956. [Google Scholar]
- Vihinen-Ranta, M.; Yuan, W.; Weichert, W.; Vuento, M.; Parrish, C.R. The vp1n-terminal sequence of canine parvovirus is important for efficient cell infection. Mol. Biol. Cell 2000, 11, 291A–291A. [Google Scholar]
- Vihinen-Ranta, M.; Wang, D.; Weichert, W.S.; Parrish, C.R. The vp1n-terminal sequence of canine parvovirus affects nuclear transport of capsids and efficient cell infection. J. Virol. 2002, 76, 1884–1891. [Google Scholar] [CrossRef]
- Farr, G.A.; Tattersall, P. A conserved leucine that constricts the pore through the capsid fivefold cylinder plays a central role in parvoviral infection. Virology 2004, 323, 243–256. [Google Scholar] [CrossRef]
- Cotmore, S.F.; D'Abramo, A.M.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the mvm virion expose the vp1 n-terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef]
- Akache, B.; Grimm, D.; Shen, X.; Fuess, S.; Yant, S.R.; Glazer, D.S.; Park, J.; Kay, M.A. A two-hybrid screen identifies cathepsins b and l as uncoating factors for adeno-associated virus 2 and 8. Mol. Ther. 2007, 15, 330–339. [Google Scholar] [CrossRef]
- Dorsch, S.; Liebisch, G.; Kaufmann, B.; von Landenberg, P.; Hoffmann, J.H.; Drobnik, W.; Modrow, S. The vp1 unique region of parvovirus b19 and its constituent phospholipase a2-like activity. J. Virol. 2002, 76, 2014–2018. [Google Scholar] [CrossRef]
- Girod, A.; Wobus, C.E.; Zadori, Z.; Ried, M.; Leike, K.; Tijssen, P.; Kleinschmidt, J.A.; Hallek, M. The vp1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase a2 domain required for virus infectivity. J. Gen. Virol. 2002, 83, 973–978. [Google Scholar]
- VihinenRanta, M.; Kakkola, L.; Kalela, A.; Vilja, P.; Vuento, M. Characterization of a nuclear localization signal of canine parvovirus capsid proteins. Eur. J. Biochem. 1997, 250, 389–394. [Google Scholar]
- Zadori, Z.; Szelei, J.; Lacoste, M.C.; Li, Y.; Gariepy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase a(2) is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Canaan, S.; Zadori, Z.; Ghomashchi, F.; Bollinger, J.; Sadilek, M.; Moreau, M.E.; Tijssen, P.; Gelb, M.H. Interfacial enzymology of parvovirus phospholipases a(2). J. Biol. Chem. 2004, 279, 14502–14508. [Google Scholar] [CrossRef]
- Lu, J.; Zhi, N.; Wong, S.; Brown, K.E. Activation of synoviocytes by the secreted phospholipase a(2) motif in the vp1-unique region of parvovirus b19 minor capsid protein. J. Infect. Dis. 2006, 193, 582–590. [Google Scholar] [CrossRef]
- Murakami, M.; Kudo, I. Recent advances in molecular biology and physiology of the prostaglandin e-2-biosynthetic pathway. Prog. Lipid Res. 2004, 43, 3–35. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Pelletier, J.P.; Fahmi, H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin. Arthritis Rheum. 2003, 33, 155–167. [Google Scholar] [CrossRef]
- Stahnke, S.; Lux, K.; Uhrig, S.; Kreppel, F.; Hosel, M.; Coutelle, O.; Ogris, M.; Hallek, M.; Buning, H. Intrinsic phospholipase a2 activity of adeno-associated virus is involved in endosomal escape of incoming particles. Virology 2011, 409, 77–83. [Google Scholar] [CrossRef]
- Filippone, C.; Zhi, N.; Wong, S.; Lu, J.; Kajigaya, S.; Gallinella, G.; Kakkola, L.; Soderlund-Venenno, M.; Young, N.S.; Brown, K.E. Vp1u phospholipase activity is critical for infectivity of full-length parvovirus b19 genomic clones. Virology 2008, 374, 444–452. [Google Scholar] [CrossRef]
- Deng, X.F.; Dong, Y.M.; Yi, Q.H.; Huang, Y.; Zhao, D.; Yang, Y.B.; Tijssen, P.; Qiu, J.M.; Liu, K.Y.; Li, Y. The determinants for the enzyme activity of human parvovirus b19 phospholipase a2 (pla2) and its influence on cultured cells. PLoS One 2013, 8, e61440. [Google Scholar]
- Pakkanen, K.; Kirjavainen, S.; Makela, A.R.; Rintanen, N.; Oker-Blom, C.; Jalonen, T.O.; Vuento, M. Parvovirus capsid disorders cholesterol-rich membranes. Biochem. Biophys. Res. Comm. 2009, 379, 562–566. [Google Scholar] [CrossRef]
- Bonsch, C.; Kempf, C.; Ros, C. Interaction of parvovirus b19 with human erythrocytes alters virus structure and cell membrane integrity. J. Virol. 2008, 82, 11784–11791. [Google Scholar] [CrossRef]
- Lupescu, A.; Bock, C.T.; Lang, P.A.; Aberle, S.; Kaiser, H.; Kandolf, R.; Lang, F. Phospholipase a2 activity-dependent stimulation of ca2+ entry by human parvovirus b19 capsid protein vp1. J. Virol. 2006, 80, 11370–11380. [Google Scholar] [CrossRef]
- Almilaji, A.; Szteyn, K.; Fein, E.; Pakladok, T.; Munoz, C.; Elvira, B.; Towhid, S.T.; Alesutan, I.; Shumilina, E.; Bock, C.T.; et al. Down-regulation of na+/k+ atpase activity by human parvovirus b19 capsid protein vp1. Cell. Physiol. Biochem. 2013, 31, 638–648. [Google Scholar] [CrossRef]
- Venkatakrishnan, B.; Yarbrough, J.; Domsic, J.; Bennett, A.; Bothner, B.; Kozyreva, O.G.; Samulski, R.J.; Muzyczka, N.; McKenna, R.; Agbandje-McKenna, M. Structure and dynamics of adeno-associated virus serotype 1 vp1-unique n-terminal domain and its role in capsid trafficking. J. Virol. 2013, 87, 4974–4984. [Google Scholar] [CrossRef]
- Lux, K.; Goerlitz, N.; Schlemminger, S.; Perabo, L.; Goldnau, D.; Endell, J.; Leike, K.; Kofler, D.M.; Finke, S.; Hallek, M.; et al. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 2005, 79, 11776–11787. [Google Scholar] [CrossRef]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Pante, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef]
- Cohen, S.; Marr, A.K.; Garcin, P.; Pante, N. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 2011, 85, 4863–4874. [Google Scholar] [CrossRef]
- Hansen, J.; Qing, K.; Srivastava, A. Infection of purified nuclei by adeno-associated virus 2. Mol. Ther. 2001, 4, 289–296. [Google Scholar] [CrossRef]
- Nicolson, S.C.; Samulski, R.J. Adeno-associated virus utilizes host cell nuclear import machinery to enter the nucleus. Mol. Ther. 2013, 21, S30–S30. [Google Scholar]
- Baker, T.S.; Newcomb, W.W.; Olson, N.H.; Cowsert, L.M.; Olson, C.; Brown, J.C. Structures of bovine and human papillomaviruses—Analysis by cryoelectron microscopy and 3-dimensional image-reconstruction. Biophys. J. 1991, 60, 1445–1456. [Google Scholar] [CrossRef]
- Giroglou, T.; Florin, L.; Schafer, F.; Streeck, R.E.; Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef]
- Joyce, J.G.; Tung, J.S.; Przysiecki, C.T.; Cook, J.C.; Lehman, E.D.; Sands, J.A.; Jansens, K.U.; Keller, P.M. The l1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 1999, 274, 5810–5822. [Google Scholar]
- Gambhira, R.; Karanam, B.; Jagu, S.; Roberts, J.N.; Buck, C.B.; Bossis, I.; Alphs, H.; Culp, T.; Christensen, N.D.; Roden, R.B.S. A protective and broadly cross-neutralizing epitope of human papillomavirus l2. J. Virol. 2007, 81, 13927–13931. [Google Scholar] [CrossRef]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, l2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef]
- Chen, X.; Liu, H.; Zhang, T.; Liu, Y.; Xie, X.; Wang, Z.; Xu, X. A vaccine of l2 epitope repeats fused with a modified igg1 fc induced cross-neutralizing antibodies and protective immunity against divergent human papillomavirus types. PLoS One 2014, 9, e95448. [Google Scholar]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef]
- Horvath, C.A.J.; Boulet, G.A.V.; Renoux, V.M.; Delvenne, P.O.; Bogers, J.P.J. Mechanisms of cell entry by human papillomaviruses: An overview. Virol. J. 2010, 7, 11. [Google Scholar] [CrossRef]
- Laniosz, V.; Holthusen, K.A.; Meneses, P.I. Bovine papillomavirus type 1: From clathrin to caveolin. J. Virol. 2008, 82, 6288–6298. [Google Scholar] [CrossRef]
- Smith, J.L.; Campos, S.K.; Ozbun, M.A. Human papillomavirus type 31 uses a caveolin 1-and dynamin 2-mediated entry pathway for infection of human keratinocytes. J. Virol. 2007, 81, 9922–9931. [Google Scholar] [CrossRef]
- Bousarghin, L.; Touze, A.; Sizaret, P.Y.; Coursaget, P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 2003, 77, 3846–3850. [Google Scholar]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 2003, 307, 1–11. [Google Scholar] [CrossRef]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and caveolin-independent entry of human papillomavirus type 16-involvement of tetraspanin-enriched microdomains (tems). PLoS One 2008, 3, e3313. [Google Scholar] [CrossRef]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef]
- Spoden, G.; Kuhling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.A.; Poffenberger, A.C.; Nelson, C.D.S.; Atwood, W.J.; et al. Genome-wide sirna screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef]
- Bossis, L.; Roden, R.B.S.; Gambhira, R.; Yang, R.; Tagaya, M.; Howley, P.A.; Meneses, P.I. Interaction of tsnare syntaxin 18 with the papillomavirus minor capsid protein mediates infection. J. Virol. 2005, 79, 6723–6731. [Google Scholar] [CrossRef]
- Laniosz, V.; Dabydeen, S.A.; Havens, M.A.; Meneses, P.I. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. J. Virol. 2009, 83, 8221–8232. [Google Scholar] [CrossRef]
- Karanam, B.; Peng, S.W.; Li, T.; Buck, C.; Day, P.M.; Roden, R.B.S. Papillomavirus infection requires gamma secretase. J. Virol. 2010, 84, 10661–10670. [Google Scholar] [CrossRef]
- Campos, S.K.; Chapman, J.A.; Deymier, M.J.; Bronnimann, M.P.; Ozbun, M.A. Opposing effects of bacitracin on human papillomavirus type 16 infection: Enhancement of binding and entry and inhibition of endosomal penetration. J. Virol. 2012, 86, 4169–4181. [Google Scholar] [CrossRef]
- Selinka, H.C.; Giroglou, T.; Sapp, M. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 2002, 299, 279–287. [Google Scholar] [CrossRef]
- Dabydeen, S.A.; Meneses, P.I. The role of nh(4)cl and cysteine proteases in human papillomavirus type 16 infection. Virol. J. 2009, 6, 109. [Google Scholar] [CrossRef]
- Muller, K.H.; Spoden, G.A.; Scheffer, K.D.; Brunnhofer, R.; De Brabander, J.K.; Maier, M.E.; Florin, L.; Muller, C.P. Inhibition by cellular vacuolar atpase impairs human papillomavirus uncoating and infection. Antimicrob. Agents Chemother. 2014, 58, 2905–2911. [Google Scholar] [CrossRef]
- Selinka, H.C.; Giroglou, T.; Nowak, T.; Christensen, N.D.; Sapp, M. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 2003, 77, 12961–12967. [Google Scholar] [CrossRef]
- Finnen, R.L.; Erickson, K.D.; Chen, X.J.S.; Garcea, R.L. Interactions between papillomavirus l1 and l2 capsid proteins. J. Virol. 2003, 77, 4818–4826. [Google Scholar] [CrossRef]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of l2 within the papillomavirus capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef]
- Trus, B.L.; Roden, R.B.S.; Greenstone, H.L.; Vrhel, M.; Schiller, J.T.; Booy, F.P. Novel structural features of bovine papillomavirus capsid revealed by a three-dimensional reconstruction to 9 angstrom resolution. Nat. Struct. Biol. 1997, 4, 413–420. [Google Scholar] [CrossRef]
- Liu, W.J.; Gissmann, L.; Sun, X.Y.; Kanjanahaluethai, A.; Muller, M.; Doorbar, J.; Zhou, J. Sequence close to the n-terminus of l2 protein is displayed on the surface of bovine papillomavirus type 1 virions. Virology 1997, 227, 474–483. [Google Scholar] [CrossRef]
- Kondo, K.; Ishii, Y.; Ochi, H.; Matsumoto, T.; Yoshikawa, H.; Kanda, T. Neutralization of hpv16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the hpv 16 minor capsid protein l2 surface region. Virology 2007, 358, 266–272. [Google Scholar] [CrossRef]
- Mamoor, S.; Onder, Z.; Karanam, B.; Kwak, K.; Bordeaux, J.; Crosby, L.; Roden, R.B.S.; Moroianu, J. The high risk hpv16 l2 minor capsid protein has multiple transport signals that mediate its nucleocytoplasmic traffic. Virology 2012, 422, 413–424. [Google Scholar] [CrossRef]
- Darshan, M.S.; Lucchi, J.; Harding, E.; Moroianu, J. The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J. Virol. 2004, 78, 12179–12188. [Google Scholar] [CrossRef]
- Fay, A.; Yutzy, W.H.; Roden, R.B.S.; Moroianu, J. The positively charged termini of l2 minor capsid protein required for bovine papillomavirus infection function separately in nuclear import and dna binding. J. Virol. 2004, 78, 13447–13454. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, X.Y.; Louis, K.; Frazer, I.H. Interaction of human papillomavirus (hpv) type-16 capsid proteins with hpv dna requires an intact l2 n-terminal sequence. J. Virol. 1994, 68, 619–625. [Google Scholar]
- Kawana, Y.; Kawana, K.; Yoshikawa, H.; Taketani, Y.; Yoshiike, K.; Kanda, T. Human papillomavirus type 16 minor capsid protein l2 n-terminal region containing a common neutralization epitope binds to the cell surface and enters the cytoplasm. J. Virol. 2001, 75, 2331–2336. [Google Scholar] [CrossRef]
- Woodham, A.W.; Da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The s100a10 subunit of the annexin a2 heterotetramer facilitates l2-mediated human papillomavirus infection. PLoS One 2012, 7, e43519. [Google Scholar] [CrossRef]
- Yang, R.C.; Day, P.M.; Yutzy, W.H.; Lin, K.Y.; Hung, C.F.; Roden, R.B.S. Cell surface-binding motifs of l2 that facilitate papillomavirus infection. J. Virol. 2003, 77, 3531–3541. [Google Scholar] [CrossRef]
- Holmgren, S.C.; Patterson, N.A.; Ozbun, M.A.; Lambert, P.F. The minor capsid protein l2 contributes to two steps in the human papillomavirus type 31 life cycle. J. Virol. 2005, 79, 3938–3948. [Google Scholar] [CrossRef]
- Roden, R.B.S.; Day, P.M.; Bronzo, B.K.; Yutzy, W.H.; Yang, Y.Q.; Lowy, D.R.; Schiller, J.T. Positively charged termini of the l2 minor capsid protein are necessary for papillomavirus infection. J. Virol. 2001, 75, 10493–10497. [Google Scholar] [CrossRef]
- Kamper, N.; Day, P.M.; Nowak, T.; Selinka, H.C.; Florin, L.; Bolscher, J.; Hilbig, L.; Schiller, J.T.; Sapp, M. A membrane-destabilizing peptide in capsid protein l2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 2006, 80, 759–768. [Google Scholar] [CrossRef]
- Campos, S.K.; Ozbun, M.A. Two highly conserved cysteine residues in hpv16 l2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes. PLoS One 2009, 4, e4463. [Google Scholar] [CrossRef]
- Gambhira, R.; Jagu, S.; Karanam, B.; Day, P.M.; Roden, R. Role of l2 cysteines in papillomavirus infection and neutralization. Virol. J. 2009, 6, 176. [Google Scholar] [CrossRef]
- Bronnimann, M.P.; Chapman, J.A.; Park, C.K.; Campos, S.K. A transmembrane domain and gxxxg motifs within l2 are essential for papillomavirus infection. J. Virol. 2013, 87, 464–473. [Google Scholar] [CrossRef]
- Ishii, Y.; Tanaka, K.; Kondo, K.; Takeuchi, T.; Mori, S.; Kanda, T. Inhibition of nuclear entry of hpv16 pseudovirus-packaged dna by an anti-hpv16 l2 neutralizing antibody. Virology 2010, 406, 181–188. [Google Scholar] [CrossRef]
- Marusic, M.B.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human papillomavirus l2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef]
- Bergant, M.; Banks, L. Snx17 facilitates infection with diverse papillomavirus types. J. Virol. 2013, 87, 1270–1273. [Google Scholar]
- Laniosz, V.; Nguyen, K.C.; Meneses, P.I. Bovine papillomavirus type 1 infection is mediated by snare syntaxin 18. J. Virol. 2007, 81, 7435–7448. [Google Scholar] [CrossRef]
- Ishii, Y.; Nakahara, T.; Kataoka, M.; Kusumoto-Matsuo, R.; Mori, S.; Takeuchi, T.; Kukimoto, I. Identification of trappc8 as a host factor required for human papillomavirus cell entry. PLoS One 2013, 8, e80297. [Google Scholar]
- Liu, W.J.; Qi, Y.M.; Zhao, K.N.; Liu, Y.H.; Liu, X.S.; Frazer, I.H. Association of bovine papillomavirus type 1 with microtubules. Virology 2001, 282, 237–244. [Google Scholar] [CrossRef]
- Zhou, J.; Gissmann, L.; Zentgraf, H.; Muller, H.; Picken, M.; Muller, M. Early phase in the infection of cultured-cells with papillomavirus virions. Virology 1995, 214, 167–176. [Google Scholar] [CrossRef]
- Florin, L.; Becker, K.A.; Lambert, C.; Nowak, T.; Sapp, C.; Strand, D.; Streeck, R.E.; Sapp, M. Identification of a dynein interacting domain in the papillomavirus minor capsid protein l2. J. Virol. 2006, 80, 6691–6696. [Google Scholar] [CrossRef]
- Schneider, M.A.; Spoden, G.A.; Florin, L.; Lambert, C. Identification of the dynein light chains required for human papillomavirus infection. Cell. Microbiol. 2011, 13, 32–46. [Google Scholar] [CrossRef]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (pml) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein l1 from the l2/dna complex following virus entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef]
- Lee, J.E.; Lim, H.J. Ldp12, a novel cell-permeable peptide derived from l1 capsid protein of the human papillomavirus. Mol. Biol. Rep. 2012, 39, 1079–1086. [Google Scholar] [CrossRef]
- Liddington, R.C.; Yan, Y.; Moulai, J.; Sahli, R.; Benjamin, T.L.; Harrison, S.C. Structure of simian virus-40 at 3.8-a resolution. Nature 1991, 354, 278–284. [Google Scholar] [CrossRef]
- Barouch, D.H.; Harrison, S.C. Interactions among the major and minor coat proteins of polyomavirus. J. Virol. 1994, 68, 3982–3989. [Google Scholar]
- Chen, X.J.S.; Stehle, T.; Harrison, S.C. Interaction of polyomavirus internal protein vp2 with the major capsid protein vp1 and implications for participation of vp2 in viral entry. EMBO J. 1998, 17, 3233–3240. [Google Scholar] [CrossRef]
- Streuli, C.H.; Griffin, B.E. Myristic acid is coupled to a structural protein of polyoma-virus and sv40. Nature 1987, 326, 619–622. [Google Scholar] [CrossRef]
- Gross, L. A filterable agent, recovered from ak leukemic extracts, causing salivary gland carcinomas in c3h mice. Proc. Soc. Exp. Biol. Med. 1953, 83, 414–421. [Google Scholar] [CrossRef]
- Dalianis, T.; Hirsch, H.H. Human polyomaviruses in disease and cancer. Virology 2013, 437, 63–72. [Google Scholar] [CrossRef]
- Feng, H.C.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Tsai, B.; Gilbert, J.M.; Stehle, T.; Lencer, W.; Benjamin, T.L.; Rapoport, T.A. Gangliosides are receptors for murine polyoma virus and sv40. EMBO J. 2003, 22, 4346–4355. [Google Scholar] [CrossRef]
- Qian, M.D.; Tsai, B. Lipids and proteins act in opposing manners to regulate polyomavirus infection. J. Virol. 2010, 84, 9840–9852. [Google Scholar] [CrossRef]
- Low, J.A.; Magnuson, B.; Tsai, B.; Imperiale, M.J. Identification of gangliosides gd1b and gt1b as receptors for bk virus. J. Virol. 2006, 80, 1361–1366. [Google Scholar] [CrossRef]
- Ewers, H.; Romer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.G.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. Gm1 structure determines sv40-induced membrane invagination and infection. Nat. Cell Biol. 2010, 12, U11–U36. [Google Scholar] [CrossRef]
- Cavaldesi, M.; Caruso, M.; Sthandier, O.; Amati, P.; Garcia, M.I. Conformational changes of murine polyomavirus capsid proteins induced by sialic acid binding. J. Biol. Chem. 2004, 279, 41573–41579. [Google Scholar]
- Qian, M.D.; Cai, D.W.; Verhey, K.J.; Tsai, B. A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog. 2009, 5, e1000465. [Google Scholar] [CrossRef]
- Pho, M.T.; Ashok, A.; Atwood, W.J. Jc virus enters human glial cells by clathrin-dependent receptor-mediated endocytosis. J. Virol. 2000, 74, 2288–2292. [Google Scholar] [CrossRef]
- Anderson, H.A.; Chen, Y.Z.; Norkin, L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 1996, 7, 1825–1834. [Google Scholar] [CrossRef]
- Richterova, Z.; Liebl, D.; Horak, M.; Palkova, Z.; Stokrova, J.; Hozak, P.; Korb, J.; Forstova, J. Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial vp1 pseudocapsids toward cell nuclei. J. Virol. 2001, 75, 10880–10891. [Google Scholar] [CrossRef]
- Eash, S.; Querbes, W.; Atwood, W.J. Infection of vero cells by bk virus is dependent on caveolae. J. Virol. 2004, 78, 11583–11590. [Google Scholar] [CrossRef]
- Gilbert, J.M.; Goldberg, I.G.; Benjamin, T.L. Cell penetration and trafficking of polyomavirus. J. Virol. 2003, 77, 2615–2622. [Google Scholar] [CrossRef]
- Ashok, A.; Atwood, W.J. Contrasting roles of endosomal ph and the cytoskeleton in infection of human glial cells by jc virus and simian virus 40. J. Virol. 2003, 77, 1347–1356. [Google Scholar] [CrossRef]
- Moriyama, T.; Sorokin, A. Intracellular trafficking pathway of bk virus in human renal proximal tubular epithelial cells. Virology 2008, 371, 336–349. [Google Scholar] [CrossRef]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the er. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef]
- Damm, E.M.; Pelkmans, L.; Kartenbeck, J.; Mezzacasa, A.; Kurzckalia, T.; Helenius, A. Clathrin- and caveolin-1-independent endocytosis: Entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 2005, 168, 477–488. [Google Scholar] [CrossRef]
- Liebl, D.; Difato, F.; Hornikova, L.; Mannova, P.; Strokrova, J.; Forstova, J. Mouse polyomavirus enters early endosomes, requires their acidic ph for productive infection, and meets transferrin cargo in rab11-positive endosomes. J. Virol. 2006, 80, 4610–4622. [Google Scholar] [CrossRef]
- Mannova, P.; Forstova, J. Mouse polyomavirus utilizes recycling endosomes for a traffic pathway independent of copi vesicle transport. J. Virol. 2003, 77, 1672–1681. [Google Scholar] [CrossRef]
- Gilbert, J.; Benjamin, T. Uptake pathway of polyomavirus via ganglioside gd1a. J. Virol. 2004, 78, 12259–12267. [Google Scholar] [CrossRef]
- Richards, A.A.; Stang, E.; Pepperkok, R.; Parton, R.G. Inhibitors of cop-mediated transport and cholera toxin action inhibit simian virus 40 infection. Mol. Biol. Cell 2002, 13, 1750–1764. [Google Scholar] [CrossRef]
- Norkin, L.C.; Anderson, H.A.; Wolfrom, S.A.; Oppenheim, A. Caveolar endocytosis of simian virus 40 is followed by brefeldin a-sensitive transport to the endoplasmic reticulum, where the virus disassembles. J. Virol. 2002, 76, 5156–5166. [Google Scholar]
- Norkin, L.C.; Kuksin, D. The caveolae-mediated sv40 entry pathway bypasses the golgi complex en route to the endoplasmic reticulum. Virol. J. 2005, 2, 38. [Google Scholar] [CrossRef]
- Zila, V.; Difato, F.; Klimova, L.; Huerfano, S.; Forstova, J. Involvement of microtubular network and its motors in productive endocytic trafficking of mouse polyomavirus. PLoS One 2014, 9, e96922. [Google Scholar]
- Walczak, C.P.; Tsai, B. A pdi family network acts distinctly and coordinately with erp29 to facilitate polyomavirus infection. J. Virol. 2011, 85, 2386–2396. [Google Scholar] [CrossRef]
- Schelhaas, M.; Malmstrom, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grunewald, K.; Helenius, A. Simian virus 40 depends on er protein folding and quality control factors for entry into host cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef]
- Gilbert, J.; Ou, W.; Silver, J.; Benjamin, T. Downregulation of protein disulfide isomerase inhibits infection by the mouse polyomavirus. J. Virol. 2006, 80, 10868–10870. [Google Scholar] [CrossRef]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. Bap31 and bip are essential for dislocation of sv40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, U1305–U1360. [Google Scholar] [CrossRef]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.B.; Van Goor, K.E.Y.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. Bip and multiple dnaj molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. Mbio 2011, 2, e00101-11. [Google Scholar]
- Lilley, B.N.; Gilbert, J.M.; Ploegh, H.L.; Benjamin, T.L. Murine polyomavirus requires the endoplasmic reticulum protein derlin-2 to initiate infection. J. Virol. 2006, 80, 8739–8744. [Google Scholar] [CrossRef]
- Bennett, S.M.; Jiang, M.; Imperiale, M.J. Role of cell-type-specific endoplasmic reticulum-associated degradation in polyomavirus trafficking. J. Virol. 2013, 87, 8843–8852. [Google Scholar] [CrossRef]
- Chen, P.L.; Wang, M.L.; Ou, W.C.; Lii, C.K.; Chen, L.S.; Chang, D.C. Disulfide bonds stabilize jc virus capsid-like structure by protecting calcium ions from chelation. FEBS Lett. 2001, 500, 109–113. [Google Scholar] [CrossRef]
- Stehle, T.; Harrison, S.C. High-resolution structure of a polyomavirus vp1-oligosaccharide complex: Implications for assembly and receptor binding. EMBO J. 1997, 16, 5139–5148. [Google Scholar] [CrossRef]
- Gharakhanian, E.; Fasching, C.L.; Orlando, S.J.; Perez, A.R. Cys(9), cys(104) and cys(207) of simian virus 40 vp1 are essential for infectious virion formation in cv-1 celss. J. Gen. Virol. 2001, 82, 1935–1939. [Google Scholar]
- Salunke, D.M.; Caspar, D.L.D.; Garcea, R.L. Self-assembly of purified polyomavirus capsid protein-vp1. Cell 1986, 46, 895–904. [Google Scholar] [CrossRef]
- Daniels, R.; Rusan, N.M.; Wadsworth, P.; Hebert, D.N. Sv40vp2 and vp3 insertion into er membranes is controlled by the capsid protein vp1: Implications for dna translocation out of the er. Mol. Cell 2006, 24, 955–966. [Google Scholar] [CrossRef]
- Gharakhanian, E.; Munoz, L.; Mayorca, L. The simian virus 40 minor structural protein vp3, but not vp2, is essential for infectious virion formation. J. Gen. Virol. 2003, 84, 2111–2116. [Google Scholar] [CrossRef]
- Enomoto, T.; Kukimoto, I.; Kawano, M.A.; Yamaguchi, Y.; Berk, A.J.; Handa, H. In vitro reconstitution of sv40 particles that are composed of vp1/2/3 capsid proteins and nucleosomal dna and direct efficient gene transfer. Virology 2011, 420, 1–9. [Google Scholar] [CrossRef]
- Nakanishi, A.; Itoh, N.; Li, P.P.; Handa, H.; Liddington, R.C.; Kasamatsu, H. Minor capsid proteins of simian virus 40 are dispensable for nucleocapsid assembly and cell entry but are required for nuclear entry of the viral genome. J. Virol. 2007, 81, 3778–3785. [Google Scholar] [CrossRef]
- Stehle, T.; Harrison, S.C. Crystal structures of murine polyomavirus in complex with straight-chain and branched-chain sialyloligosaccharide receptor fragments. Structure 1996, 4, 183–194. [Google Scholar] [CrossRef]
- Neu, U.; Woellner, K.; Gauglitz, G.; Stehle, T. Structural basis of gm1 ganglioside recognition by simian virus 40. Proc. Natl. Acad. Sci. USA 2008, 105, 5219–5224. [Google Scholar]
- Inoue, T.; Tsai, B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011, 7, e1002037. [Google Scholar] [CrossRef]
- Forstova, J.; Krauzewicz, N.; Wallace, S.; Street, A.J.; Dilworth, S.M.; Beard, S.; Griffin, B.E. Cooperation of structural proteins during late events in the life-cycle of polyomavirus. J. Virol. 1993, 67, 1405–1413. [Google Scholar]
- Delos, S.E.; Montross, L.; Moreland, R.B.; Garcea, R.L. Expression of the polyomavirus vp2 and vp3 proteins in insect cells—Coexpression with the major capsid protein vp1 alters vp2/vp3 subcellular-localization. Virology 1993, 194, 393–398. [Google Scholar] [CrossRef]
- Huerfano, S.; Zila, V.; Boura, E.; Spanielova, H.; Stokrova, J.; Forstova, J. Minor capsid proteins of mouse polyomavirus are inducers of apoptosis when produced individually but are only moderate contributors to cell death during the late phase of viral infection. FEBS J. 2010, 277, 1270–1283. [Google Scholar] [CrossRef]
- Giorda, K.M.; Raghava, S.; Zhang, M.W.; Hebert, D.N. The viroporin activity of the minor structural proteins vp2 and vp3 is required for sv40 propagation. J. Biol. Chem. 2013, 288, 2510–2520. [Google Scholar]
- Raghava, S.; Giorda, K.M.; Romano, F.B.; Heuck, A.P.; Hebert, D.N. Sv40 late protein vp4 forms toroidal pores to disrupt membranes for viral release. Biochemistry 2013, 52, 3939–3948. [Google Scholar]
- Daniels, R.; Sadowicz, D.; Hebert, D.N. A very late viral protein triggers the lytic release of sv40. PLoS Pathog. 2007, 3, 928–938. [Google Scholar]
- Fiser, R.; Forstova, J.; Department of Genetics and Microbiology, Charles University, Prague, Czech Republic. Unpublished work. 2008.
- Cai, X.Y.; Chang, D.C.; Rottinghaus, S.; Consigli, R.A. Expression and purification of recombinant polyomavirus vp2 protein and its interactions with polyomavirus proteins. J. Virol. 1994, 68, 7609–7613. [Google Scholar]
- Hornikova, L.; Forstova, J.; Department of Genetics and Microbiology, Charles University, Prague, Czech Republic. Unpublished work. 2012.
- Schowalter, R.M.; Buck, C.B. The merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef]
- Magnuson, B.; Rainey, E.K.; Benjamin, T.; Baryshev, M.; Mkrtchian, S.; Tsai, B. Erp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 2005, 20, 289–300. [Google Scholar] [CrossRef]
- Attar, N.; Cullen, P.J. The retromer complex. Adv. Enzym. Regul. 2010, 50, 216–236. [Google Scholar] [CrossRef]
- Walczak, C.P.; Ravindran, M.S.; Inoue, T.; Tsai, B. A cytosolic chaperone complexes with dynamic membrane j-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog 2014, 10, e1004007. [Google Scholar] [CrossRef]
- Nakanishi, A.; Shum, D.; Morioka, H.; Otsuka, E.; Kasamatsu, H. Interaction of the vp3 nuclear localization signal with the importin alpha-2/beta heterodimer directs nuclear entry of infecting simian virus 40. J. Virol. 2002, 76, 9368–9377. [Google Scholar] [CrossRef]
- Nakanishi, A.; Li, P.P.; Qu, Q.M.; Jafri, Q.H.; Kasamatsu, H. Molecular dissection of nuclear entry-competent sv40 during infection. Virus Res. 2007, 124, 226–230. [Google Scholar]
- Kuksin, D.; Norkin, L.C. Disassociation of the sv40 genome from capsid proteins prior to nuclear entry. Virol. J. 2012, 9, 158. [Google Scholar] [CrossRef]
- Hummeler, K.; Tomassin, N.; Sokol, F. Morphological aspects of uptake of simian-virus-40 by permissive cells. J. Virol. 1970, 6, 87–93. [Google Scholar]
- Mackay, R.L.; Consigli, R.A. Early events in polyoma-virus infection - attachment, penetration, and nuclear entry. J. Virol. 1976, 19, 620–636. [Google Scholar]
- Butin-Israeli, V.; Ben-nun-Shaul, O.; Kopatz, I.; Adam, S.A.; Shimi, T.; Goldman, R.D.; Oppenheim, A. Simian virus 40 induces lamin a/c fluctuations and nuclear envelope deformation during cell entry. Nucleus-Austin 2011, 2, 320–330. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bilkova, E.; Forstova, J.; Abrahamyan, L. Coat as a Dagger: The Use of Capsid Proteins to Perforate Membranes during Non-Enveloped DNA Viruses Trafficking. Viruses 2014, 6, 2899-2937. https://doi.org/10.3390/v6072899
Bilkova E, Forstova J, Abrahamyan L. Coat as a Dagger: The Use of Capsid Proteins to Perforate Membranes during Non-Enveloped DNA Viruses Trafficking. Viruses. 2014; 6(7):2899-2937. https://doi.org/10.3390/v6072899
Chicago/Turabian StyleBilkova, Eva, Jitka Forstova, and Levon Abrahamyan. 2014. "Coat as a Dagger: The Use of Capsid Proteins to Perforate Membranes during Non-Enveloped DNA Viruses Trafficking" Viruses 6, no. 7: 2899-2937. https://doi.org/10.3390/v6072899
APA StyleBilkova, E., Forstova, J., & Abrahamyan, L. (2014). Coat as a Dagger: The Use of Capsid Proteins to Perforate Membranes during Non-Enveloped DNA Viruses Trafficking. Viruses, 6(7), 2899-2937. https://doi.org/10.3390/v6072899