Abstract
In this review, we summarize the current knowledge about the membranous replication factories of members of plus-strand (+) RNA viruses. We discuss primarily the architecture of these complex membrane rearrangements, because this topic emerged in the last few years as electron tomography has become more widely available. A general denominator is that two “morphotypes” of membrane alterations can be found that are exemplified by flaviviruses and hepaciviruses: membrane invaginations towards the lumen of the endoplasmatic reticulum (ER) and double membrane vesicles, representing extrusions also originating from the ER, respectively. We hypothesize that either morphotype might reflect common pathways and principles that are used by these viruses to form their membranous replication compartments.
1. The Family Flaviviridae
Members of the family Flaviviridae are enveloped viruses with a single stranded RNA genome of positive polarity. This family contains four different genera: Hepacivirus (from the Greek hepar, hepatos, “liver”), Flavivirus (from the Latin flavus, “yellow”), Pestivirus (from the Latin pestis, “plague”) and the recently included genus Pegivirus [1,2] (Figure 1).
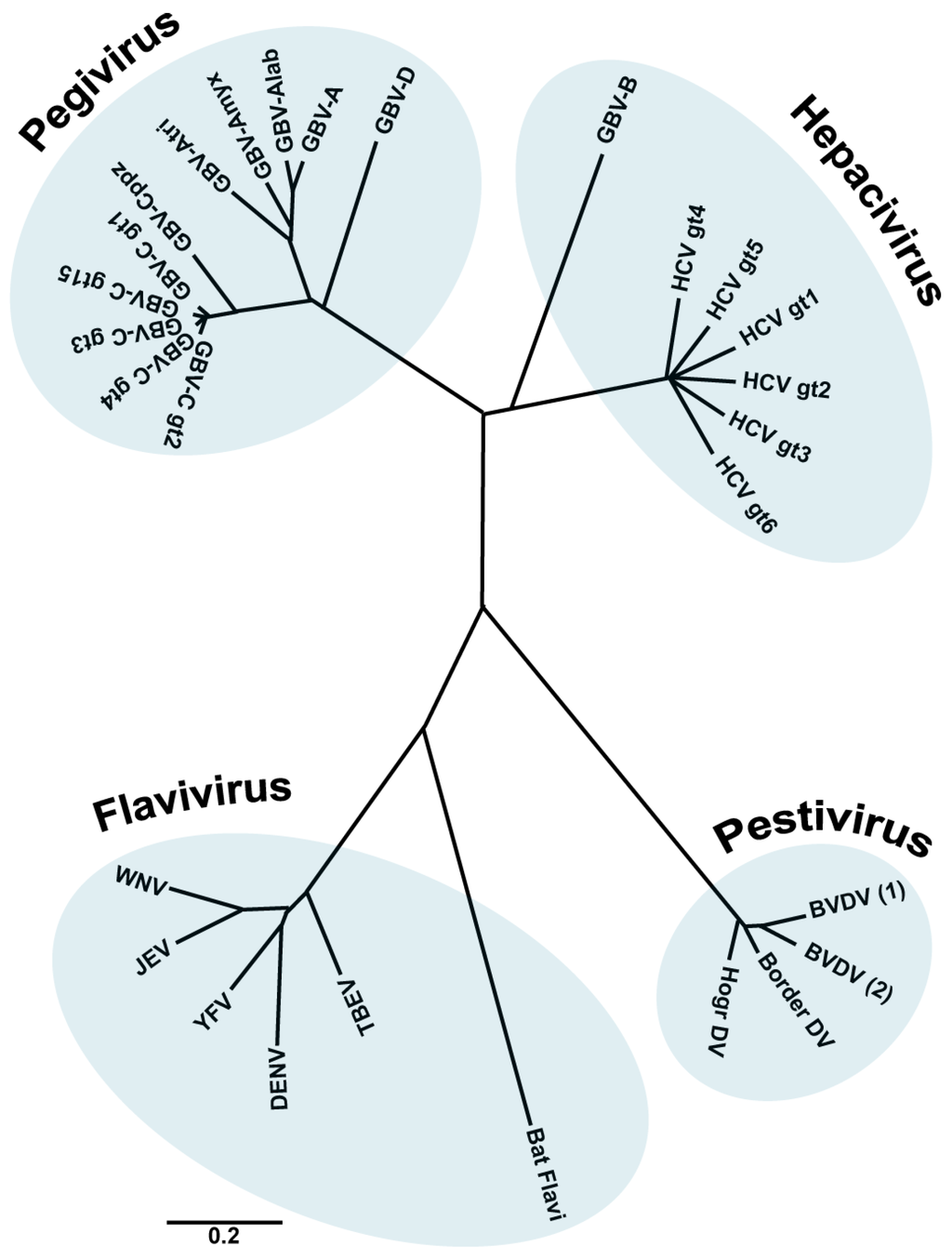
Figure 1.
The family Flaviviridae. Phylogenetic tree based on the analysis (neighbour joining) of aligned conserved motifs of the RNA dependent RNA polymerase (RdRp). Shown are selected members of the family. A distance scale corresponding to amino acid substitutions per position is shown. (Figure adapted from [1], with permission).
Hepatitis C Virus (HCV) is the prototype species of the genus Hepacivirus. It was first discovered by Choo et al. [3] in the serum and tissues of a chimpanzee experimentally inoculated with serum from an individual with chronic, non-A, non-B hepatitis. This virus was associated with a mild form of chronic hepatitis frequently observed in recipients of blood transfusions [4] and was called HCV. A second species within the genus Hepacivirus is GBV-B which was first identified in tamarins that developed hepatitis following inoculation with the serum from a surgeon (initials G.B.) with acute hepatitis. Additional GB-like viruses were discovered later on and have been assigned to the new genus Pegivirus (an acronym derived from pe, persistent; g, GB or G) within the family Flaviviridae [1].
The genus Flavivirus comprises 53 species and represents the largest genus within the family Flaviviridae. Many of these viruses are transmitted by arthropods and therefore called ARBO viruses (arthropod-borne). Flaviviruses such as Dengue Virus (DENV), Japanese Encephalitis Virus (JEV), West Nile Virus (WNV) or Yellow Fever Virus (YFV) cause a variety of diseases, including fever, encephalitis and hemorrhage [5,6].
Pestiviruses are important animal pathogens causing major losses in stock farming. The genus comprises the species Bovine Viral Diarrhea Virus 1 and 2 (BVDV-1/-2), Border Disease Virus (BDV), Classical Swine Fever Virus (CSFV), the tentative virus species “Giraffe”, as well as several unassigned viruses [7]. Within the family Flaviviridae, pestiviruses show greater similarity in genome structure and mechanism of translation initiation to the hepaciviruses than to flaviviruses. Pestivirus infections can be subclinical or induce a range of clinical conditions, including acute diarrhea, acute hemorrhagic syndrome, acute fatal disease, and a wasting disease [5].
These viruses have in common a single strand RNA genome containing one long open reading frame flanked by 5’- and 3’-untranslated regions (UTRs) [5]. Viral proteins are produced by cleavage of a single polyprotein that has a length of about 3000 amino acid residues. The N-terminal half of the polyprotein contains the structural proteins forming the virus particle, while the C-terminal part contains the nonstructural proteins (NS) involved in synthesis of the viral RNA genome. The NS proteins comprise the enzymatic components of the RNA replicase, including a RNA helicase and a RNA-dependent RNA polymerase (RdRp) [8].
Binding and uptake of viruses belonging to the Flaviviridae family into their host cells occur by receptor-mediated endocytosis involving interactions between cellular receptors and viral envelope proteins. Upon uncoating, the (+) RNA genome is released into the cytoplasm, where it serves as messenger RNA (mRNA) for translation of all viral proteins, as template for RNA replication to produce new (−) RNA copies and as genetic material packaged into new virus particles. During RNA replication, the (+) RNA genome is converted into a (−) RNA copy that serves as template for production of new (+) RNA genomes. Most of these steps occur in close association with intracellular membranes that are rearranged upon viral infection giving rise to complex membranous replication factories. These rearrangements correspond to invaginations or protrusions of membranes that are frequently derived from the endoplasmic reticulum (ER). In this review, we summarize our current knowledge about the 3D architecture of these complex membrane rearrangements, focusing on members of the family Flaviviridae that generate either morphotype of replication compartment.
2. Architecture and Properties of the Replication Factories of Members of the Flaviviridae Family
2.1. Flaviviruses
The first publications on DENV-induced membrane modifications in mammalian cells (marked cytoplasmic vacuolization and accumulation of virions in crystalloid arrays surrounded by membranes) date from the seventies [9,10]. Several years later, studies on DENV-infected mosquito cells were published in which the visualization of virions was reported [11,12]. In 1997, Grief and coworkers described DENV-induced membrane alterations of various morphologies, including convoluted membranes (CMs) and vesicle packets (VPs) [13]. Subsequent immuno-EM studies indicated that VPs and CMs may represent the site of DENV replication and RNA translation/polyprotein processing, respectively [14]. The most complete characterization of DENV-induced intracellular membrane rearrangements elucidated their 3D architecture as well as their spatial connection with viral assembly sites [15]. TEM of resin-embedded infected cells revealed a complex collection of convoluted and vesicular structures, including CMs that were usually surrounded by multiple vesicles, often appearing as longitudinal vesicle arrays. By using electron tomography (ET), the latter were found to correspond to ER tubules containing 80–90 nm single-membrane vesicles (Ve) that result from the invagination of the ER membrane into the ER lumen. By conventional EM, these vesicles appeared as double membrane vesicles, likely corresponding to the VPs described earlier [13]. Immuno-EM confirmed that the vesicles visible in resin-embedded cells were induced by DENV infection and contained all NS proteins. However, only NS3 was detectable within the CMs, which could be due to lower affinity of the antibodies or poor accessibility of the other NS proteins in the CMs. Double-stranded RNA (dsRNA) detected by immunostaining appeared as discrete electron-dense structures inside or on the cytosolic surface of a subset of vesicles, suggesting that dsRNA might be present only in some of the vesicles at a given time point. Furthermore, the vesicles contain rather uniform pores of ~10 nm diameter towards the cytosol (Figure 2A). Thus, both the topology of the vesicles and the immunolabeling results support the idea that the vesicles might be the site of RNA replication. Moreover, these results showed that replication factories are a continuous membrane network that provides a platform for the transport of viral proteins and genomes between sites of RNA replication, ribosome-containing compartments (RNA translation) and virus assembly sites. In fact, virus budding sites were found in close proximity to the pores of the replication vesicles. This topological link may ensure efficient production and delivery of viral RNA for the assembly of infectious virus progeny. Consistent with these findings, a very recent publication using ET showed that these virally modified structures were also observed in DENV-infected mosquito cells, with one exception: CMs were absent from DENV-infected C6/36 mosquito cells [16]. In addition, after multiple rounds of virus replication, tubular structures were also observed in the vicinity of VPs. These structures might represent a hallmark of chronically infected insect cells, since these structures are also induced by TBEV in tick cells (see below).
The first reports on WNV-infected cells described the visualization of virions [17]. An extensive characterization of Kunjin Virus—the Australian variant of WNV (WNVKUN)—infected cells has been carried out more recently [18,19,20]. Three well-defined structures were found, corresponding to large CMs, paracrystalline arrays (PCs) and VPs that appeared as membrane sacs containing small vesicles (Ve) [18,21]. Based on immunolocalization studies, a distinct redistribution of the trans-Golgi network (TGN) and colocalization of TGN markers with dsRNA has been observed, suggesting that the replication factories of WNVKUN were derived from the TGN [22]. Three-dimensional reconstructions of the WNVKUN replication sites revealed an intimate association of the rough ER (rER) with the bounding membrane of the VPs [20] (Figure 2B), resembling the vesicles observed in DENV-infected cells. These results argue for an additional role of the rER in the formation of the WNVKUN replication factories. Similar to DENV, individual necks were observed in the vesicles as well as the majority of the viral RNA, as detected by immunolabeling with a dsRNA-specific antibody, resided within these vesicles [18,20,22,23,24]. In most cases, viral RNA spanned the breadth of the vesicles and was juxtaposed to the necks open to the cytoplasm [20].
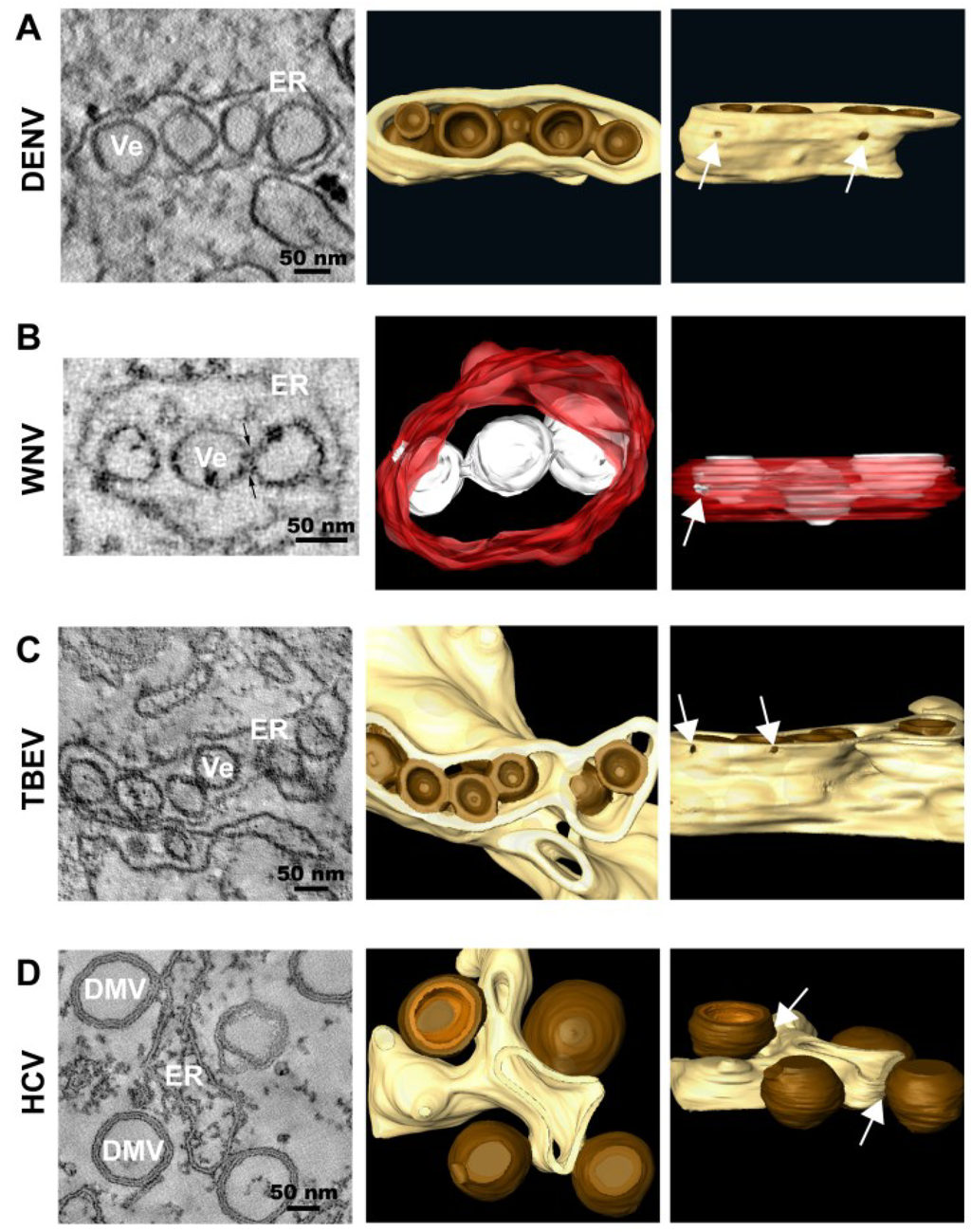
Figure 2.
Representative images of membrane rearrangements induced by different members of the family Flaviviridae. (A) Dengue Virus (DENV); (B) West Nile Virus (WNV); (C) Tick-borne Encephalitis Virus (TBEV); (D) Hepatitis C Virus (HCV). Slices through tomograms of infected cells (on the left) and 3D top and lateral (90° rotation) views of the same tomograms (on the right) are depicted, showing the characteristic virus-induced structures. The replication vesicles (Ve) of DENV, WNV and TBEV (genus Flavivirus) correspond to invaginations of ER membranes that remain connected to the cytosol via 10 nm-pores (highlighted with white arrows in the 3D lateral views), forming vesicle packets (VPs). The replication factory of HCV (genus Hepacivirus) is primarily composed of double membrane vesicles (DMVs) that seem to be formed asER protusions connected to ER membranes via neck-like structures (highlighted with white arrows in the 3D lateral view). The ER is shown in yellow (DENV, TBEV and HCV) or in red (WNV) and the replication organelles in brown (DENV, TBEV and HCV) or in white (WNV). The outer and inner membranes of DMVs are depicted in different shades of brown (outer membrane in dark brown and inner membrane in light brown). Figure 2B is reproduced with permission from [20].
In cells infected with TBEV, one of the most important tick-transmitted viruses in Europe and Asia, virus particles and membrane-connected vesicles were also observed inside the ER [25], similar to what was described for DENV and WNVKUN. The viral dsRNA was only detected inside the vesicular structures within rER, suggesting that TBEV rearranges internal cell membranes to generate a compartment that protects viral RNA from detection by cytoplasmic pathogen recognition receptors (PRRs) [26,27,28]. This localization of dsRNA might suffice to delay the onset of the IFN response [25]. For TBEV [25] and WNVKUN [19] it was shown that treatment with brefeldin A (BFA), a drug which disrupts the Golgi apparatus, did not interfere with viral replication. However, this treatment rendered WNVKUN sensitive to the antiviral action of the IFN-induced protein MxA, indicating that BFA might have disrupted the membranous WNVKUN replication compartments, thus leading to exposure of dsRNA and its detection by PRRs. In contrast, treatment of TBEV-infected cells with BFA neither affected viral replication, nor the level of IFN production. These findings indicate that TBEV dsRNA might be stored inside BFA-resistant membrane vesicles that robustly protect the viral RNA from recognition by cellular sensors.
Vector-borne flaviviruses like DENV and TBEV must replicate in both mammalian and arthropod cells. A few comparative studies have been published describing virus-induced structures such as cytoplasmic membrane proliferations and vesicle formation, also in insect cells [11,16,21,29,30,]. A detailed comparative ultrastructural analysis of TBEV-induced modifications revealed that the extent of membrane expansion and the abundance of vesicles were lower in insect cells [31]. Single-membrane vesicles, ranging in diameter from 60–100 nm were frequently found within proliferated ER areas, often occurring in large groups contained within ER cisternae. Pore-like openings connected these vesicles to the cytoplasm and to other vesicles. Apart from these vesicles, in tick-infected cells elongated vesicles or tubules were found that were much more prevalent in persistently than in acutely infected cells. These tubules were only occasionally noted in infected mammalian cells, similar to what was found with DENV-infected cells [15]. The tubular structures had a cross-sectional diameter of 60–100 nm, similar to the one of vesicles, reached up to 800 nm in length, were closed at the ends and often arranged in fascicle-like bundles, shrouded with the ER membrane. However, no pores between the tubules or towards the cytoplasm were observed [31]. The function of these tubules is unclear and it is not known whether they represent bona fide features of replication factories, aberrant structures as a result of incorrect membrane remodeling, or the result of a cellular process to restrict infection [31]. In any case, the tubules might be a feature of persistent infection, eventually linked to the high number of defective virus particles, because the lack of pores could prevent proper replication or packaging of the viral genome [32,33,34]. Further studies are required to shed light on the biogenesis and biological significance of these membranous tubular structures.
A recent study identified 80 nm-diameter vesicles within the ER lumen of TBEV-infected BHK-21 cells and in cells transfected with a TBEV replicon [35]. ET revealed that these vesicles are invaginations of the ER within a highly organized network of interconnected membranes with half of vesicles containing pore-like connections to the cytoplasm (Figure 2C). However, no pore-like openings were observed between adjacent/neighboring vesicles, in contrast to what has been described for cells infected with Langat Virus (LGTV), a naturally attenuated tick-borne flavivirus [31] or in WNVKUN-infected cells [20]. Interestingly, in TBEV replicon cells, the number of pore-containing vesicles was slightly larger (~75%) and they were found in much more fragmented ER tubules as compared to TBEV-infected cells. However, despite more extensive ER rearrangements in replicon cells, they contained fewer vesicles, consistent with the lower level of viral replication [36,37,38].
Conventional EM analysis of neurons infected with Murray Valley Encephalitis Virus (MVEV) revealed several ultrastructural features, including proliferation of ER and Golgi complex membranes as well as the appearance of membrane-bound spherical vesicles (75–145 nm diameter) [39], similar to those observed for the related flaviviruses Japanese Encephalitis Virus (JEV) [40,41] and St. Louis Encephalitis Virus (SLEV) [42]. In the latter case, cylindrical membranous structures (or tubules) were also observed [43]. The presence of vesicles was also detected in monkey liver cells infected with Yellow Fever Virus (YFV) [44]. These findings indicate that all members of the genus Flavivirus utilize the ER as a source of membranes for the formation of their replication factories, whereas assembly of new virions seems to occur at ER sacs in close proximity to the replication sites [15,35], thus creating an optimized membranous environment to support efficient viral replication and assembly. Maturation of the newly synthesized virions takes place in the Golgi apparatus, where flavirirus virions are often observed [15].
2.2. Hepaciviruses
In stark contrast to flaviviruses, HCV, the prototype of the genus Hepacivirus, provokes an alternative rearrangement of intracellular membranes, originally designated “membranous web” (MW). This term referred to compact vesicle accumulations embedded into a membranous matrix [45] as detected in cells inducibly expressing the HCV polyprotein. By using different EM methods, we and others have recently found that the MW is primarily composed of double membrane vesicles (DMVs) [46,47,48]. The fact that the kinetics of their appearance correlates with HCV replication suggests that these structures play an important role for viral RNA amplification [48]. Indeed, immunolabeling of purified DMVs revealed an enrichment for viral proteins as well as dsRNA [46,49]. Importantly, DMVs contain enzymatically active viral replicase [49] and they originate from ER membranes, similar to what has been found for other members of the family Flaviviridae. ET analysis showed that most of the DMVs remain connected to the ER via their outer membrane [48] (Figure 2D). Although DMVs are primarily closed structures, ~10% of them have an opening towards the cytosol. Late in infection, multi-membrane vesicles (MMVs) with an average diameter of 390 nm are generated, likely originating from DMVs by secondary enwrapping events [48].
By using Huh7.5 cells infected with the highly replicative HCV strain JFH-1, Ferraris and coworkers observed three different types of membrane alterations: vesicles in clusters (ViCs), contiguous vesicles (CVs) and DMVs [47]. The ViCs were small single-membrane vesicles of variable size (100–200 nm), grouped together in well-delimited areas. Most of them had an internal invagination. The CVs were small single-membrane vesicles, present in large numbers and widely distributed throughout the cytoplasm, with a more homogeneous size (around 100 nm). They were tightly associated to each other and tended to form a collar around lipid droplets (LDs). DMVs were heterogeneous in size (150–1000 nm) and had a thick, electron-dense membrane consisting of two closely apposed membranes. The increase of CVs’ number correlated with an increase of intracellular HCV RNA levels, arguing for a possible role of CVs in the early stages of viral replication. The presence of NS5A in CVs, as demonstrated by immunogold staining, is consistent with this hypothesis. Alternatively, CVs might constitute the membranous platform for viral assembly. In fact, the core protein is present in these structures (16%) as well as on the LD surface (81%). However, so far visualization of virus particles in infected cells has not been possible, making this hypothesis difficult to prove. While most of the dsRNA signal was located within DMVs or at DMV membranes, ViCs were free of viral components and RNA and these structures as well as CVs were very rarely observed in cells with a subgenomic JFH-1 replicon [46] or absent in cells infected with a JFH-1 variant designated Jc1 [48]. The first 3D reconstruction of a complete HCV-infected cell revealed that all these three membrane structures were tightly connected and closely associated with LD clusters [47].
Taken together, these findings indicate a fundamental role of DMVs in HCV replication. An in-depth comparison of the study by Ferraris and coworkers [47] and our publication [48] suggests that CVs might be also DMVs for several reasons: first, CVs have electron dense tightly apposed membranes; second, by using correlative light and electron microscopy, we also detected DMV accumulations around LDs, reminiscent of the CVs described by Ferraris and coworkers [47]; third, taking into consideration the density of content and morphology, some of the structures described as DMVs by Ferraris and colleagues might correspond to MMVs according to our nomenclature. This might account for the differences in size between the DMVs reported in both studies (up to 1000 nm versus 150 nm, respectively). Alternatively, the difference might be due to the use of distinct virus strains (JFH-1 and Jc1) that differ in their capacity to produce infectious virus particles by ~3 orders of magnitude [50], which might also explain the presence of ViCs only in JFH-1 infected cells.
2.3. Pestiviruses
Much less about membranous replication factories is known for pestiviruses. TEM-based studies from the times in which the genus Pestivirus was still belonging to the family Togaviridae reported that pestivirus-infected cells exhibited ultrastructural modifications of rER and contained small numbers of virus-like particles (VLPs) [51,52]. Gray and Nettleton (1987) reported that Border Disease Virus (BDV)-infected cells contained several profiles of ER and many dense lamellar bodies, which when transversely sectioned appeared as multiple rows of tubules, 33 nm in diameter [52]. These lamellae were often found in association with rER and in one occasion VLPs appeared to be budding within them. Bovine Viral Diarrhea Virus (BVDV)-infected cells contained rER modified into tubules, in which electron-dense VLPs were present. More recent studies on BVDV-infected cells revealed cytoplasmic vacuolization and VLPs in dilated ER cisternae [53,54]. In addition, membrane structures consisting of vesicles of various sizes enclosed in much larger vesicles have been reported [55]. These structures that morphologically resemble multivesicular bodies (MVBs) are distinct from the HCV-induced membranous web and more reminiscent of the flavivirus-induced VPs.
Studies on the morphogenesis of pestiviral particles were hampered by a low rate of virion production. In a recent study, Schmeiser and colleagues have overcome this problem by using high multiplicity of infection in MDBK cells with a distinct virus strain, the Giraffe-1 strain [56]. Obtained results define the ER as the site of pestivirus particle assembly, where budding of virions was observed. Virus particles were also found inside the lumen of the Golgi and in vesicles associated with the Golgi compartment, suggesting that virus egress occurs via the conventional secretory pathway. Interestingly, replication kinetics of pestiviral RNA did not correlate with distinct membrane rearrangements and only slight dilatation of the ER lumen was noticed. The absence of significant membrane rearrangements argues for a major difference between pestiviruses and other members of the Flaviviridae family. Interestingly, the authors detected the capsid protein and dsRNA, the marker for viral replication intermediates, mainly in MVBs, indicating that pestiviruses are either using this compartment for replication or that viral RNA and proteins are transferred to this compartment for degradation. Similar assumptions have been made for HIV [57] and Marburg virus, a member of the Filoviridae family [58,59]. Alternatively, pestiviral RNA and protein in MVBs might be intermediates of the entry process, prior to fusion of the envelope with the endosomal membrane. Indeed, particles inside MVBs matching the morphological criteria of pestivirus virions were detected [56]. However, MVBs of non-infected cells also contain vesicles for lysosomal degradation termed intraluminal vesicles (ILVs) that display a very similar morphology to pestiviral virions. Thus, unambiguous discrimination between ILVs and pestivirus particles will require detailed immunolabeling approaches.
3. Architecture and Properties of the Replication Factories of Other (+) Strand RNA Viruses
3.1. Nodaviruses
The first visualization of the 3D architecture of a (+) strand RNA virus replication factory was reported for Flock House Virus (FHV), a member of the family Nodaviridae [60]. This insect nodavirus induces the formation of invaginations at the outer mitochondrial membrane (OMM) with an average diameter of ~50 nm [60,61] (Figure 3A). The interior of these vesicles (called spherules) is connected to the cytoplasm by a necked channel of ~10 nm diameter, which is wide enough to allow import of ribonucleotides and export of synthesized RNA (diameter < 2 nm) [60]. Furthermore, metabolically labeled FHV RNA localized between inner and outer mitochondrial membranes inside these spherules, thus validating the spherules as bona fide FHV-induced compartments for viral RNA synthesis [60].
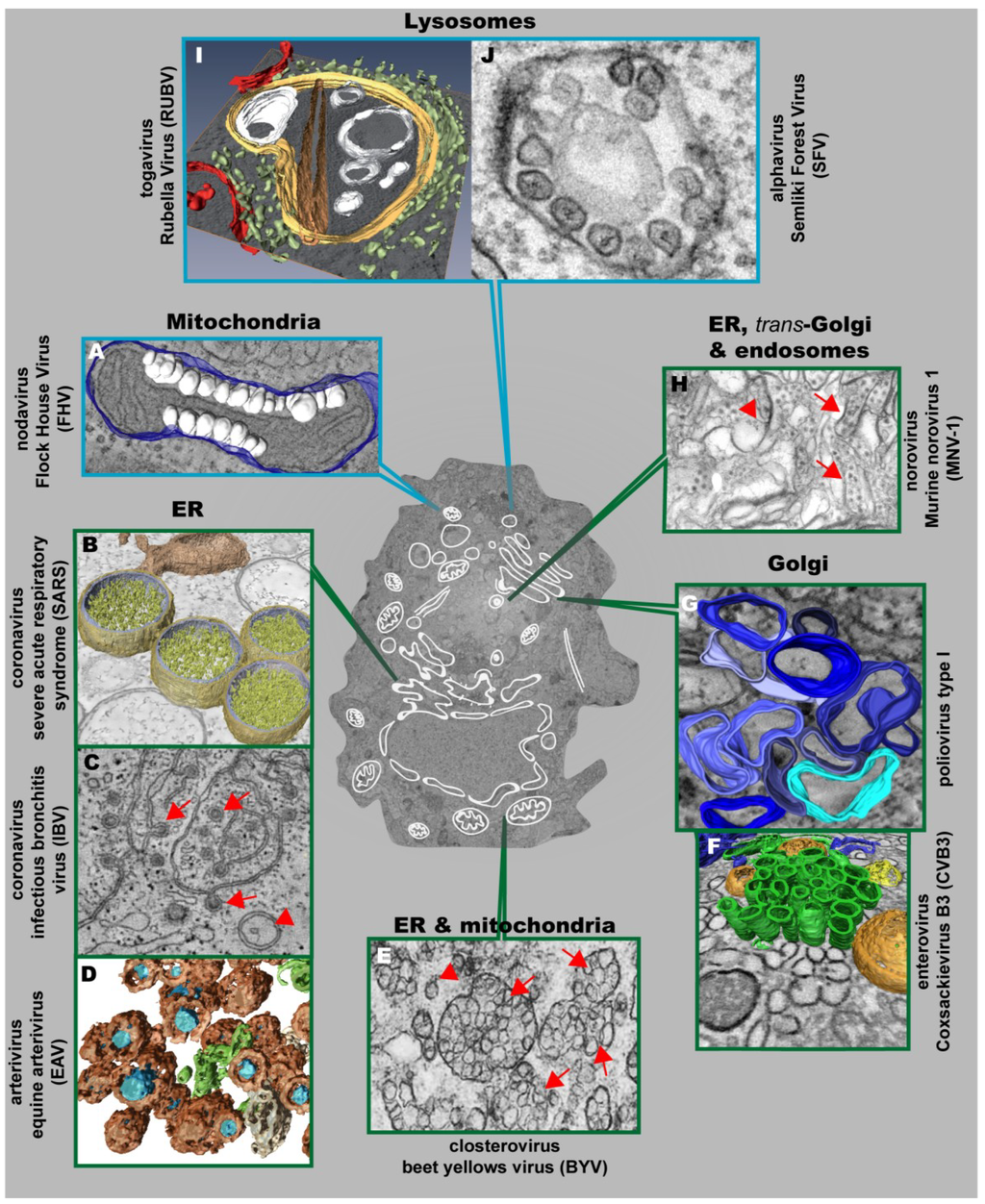
Figure 3.
Positive strand RNA viruses usurp and modify cell membranes of different origins to replicate their genomes. (A) Mitochondrial membranes are targeted by Flock House Virus (FHV), which induces the formation of vesicles (white) at the outer mitochondrial membrane (blue); (B) 3D surface-rendered model of SARS-CoV–infected Vero E6 cells containing large double membrane vesicles (DMVs) (outer membrane, gold; inner membrane, silver) that remain connected to their source, the ER (in bronze); (C) Zippered ER, found in cells infected with the gammacoronavirus Infectious Bronchitis Virus (IBV), is connected to spherules (red arrows). DMVs (red arrowheads) are also found, but to a lesser extent; (D) Equine Arteriviruses (EAV) infection of HeLa cells results in the formation of DMVs (brown), depicting a core (blue) that is associated with the ER (beige) and close to ER tubules (green); (E) Closterovirus rearranges ER and mitochondrial membranes to form DMVs (red arrowheads) and vesicles packets (VPs) (red arrows); (F) Coxsackie B3 Virus (CVB3) usurps donor membranes most likely derived from the Golgi, appearing early in infection as single-membrane tubules (green), open (orange) and closed (yellow) DMVs and ER (blue); (G) 3D architecture of poliovirus membranous replication factories at intermediate stages of development, originating from cis-Golgi membranes. Single-membrane structures are depicted in different shades of blue. Note that these single membrane vesicles undergo secondary invaginations giving rise to DMVs at the late stages of infection; (H) Murine Norovirus 1 (MNV-1) infection results in the formation of vesiculated areas (VAs, red arrowheads) within aggregates of MNV-1 particles (red arrows). These VAs seem to originate from ER, trans-Golgi and endosomes; (I) 3D model of a cytopathic vacuole (CPV) found in Rubella Virus-infected cells. This CPV (yellow) is surrounded by the rER (light green) and contains a number of vacuoles, vesicles (white) and a rigid straight sheet (brown) that is connected with the periphery of the CPV. Mitochondria are depicted in red; (J) Typical cytoplasmic vacuoles induced by Semliki Forest Virus (SFV) in baby hamster kidney cells. Color code is as follows: blue-framed images depict single-membrane vesicle inducers; green-framed images correspond to double-membrane vesicle inducers. (The different parts were reproduced with permission: see acknowledgement section)
3.4. Togaviruses
Rubella Virus (RUBV) is an important human teratogenic virus and the only member of the genus Rubivirus (family Togaviridae) [82]. RUBV anchors its RNA synthesis machinery to membranes of a cell organelle known as “cytopathic vacuoles” (CPVs) that is derived from modified endosomes or lysosomes and has an average diameter of 600–2000 nm [83,84,85]. Freeze-fracture and ET analysis of RUBV-infected cells revealed a high complexity of CPVs that are composed of stacked membranes, rigid sheets, small vesicles and large vacuoles (Figure 3I) [86]. The CPVs are interconnected and linked to the endocytic pathway, as deduced from labeling experiments with endocytosed BSA-gold. Furthermore, rER cisternae, mitochondria and Golgi stacks are recruited around CPVs to build up RUBV factories. CPVs have several contacts with cellular organelles: they are coupled to the rER through protein bridges of ~10–15 nm and closely apposed membranes and they are attached to Golgi vesicles, whereas contacts with mitochondria were not detected [86]. It has been proposed that RNA synthesis occurs on vesicular membranes within the CPVs, which are linked to the cytosol and that the viral replicase molecules are associated with vesicles that transform with time into large vacuoles and straight elements [86]. This is supported by immunogold labeling revealing replicase components and dsRNA within the CPVs [83,84,87].
The modification of late endosomes and lysosomes is a feature that RUBV shares with alphaviruses, the other genus of the family Togaviridae [88,89]. Cells infected with alphaviruses like Semliki Forest Virus (SFV), Sindbis Virus and Western Equine Encephalitis Virus (WEEV) contain large CPVs with a diameter ranging between 600 and 2000 nm. The inner surface of these CPVs is covered with small invaginations or spherules that originate at the plasma membrane [90,91,92] (Figure 3J). These spherules are comprised of a single membrane forming a vesicle with a diameter of ~50 nm. In addition, the spherules were shown to be the site of viral RNA synthesis as deduced from metabolic labeling and detection by EM [89,91,92]. Importantly, the inside of the spherule is connected to the cytoplasm by a pore with a diameter of 5–10 nm. The spherules are formed at the plasma membrane by the concerted action of the viral nonstructural proteins (nsP1–nsP4) and genomic viral RNA [89]. Furthermore, Froshauer et al. [88] showed that the CPVs that contain the spherules possess endosomal and lysosomal markers.
Time course studies revealed that the spherules of SFV undergo an unprecedented large-scale movement between cellular compartments [93]. The spherules first form as blebs (exvaginations) at the plasma membrane. Then, they are internalized by an endocytic process requiring a functional actin-myosin network. The spherules therefore represent an unusual type of endocytic cargo. After endocytosis, spherule-containing vesicles, namely CPVs-I fuse with acidic endosomes and move along microtubules. This leads to the formation of a very stable compartment, where the spherules accumulate as invaginations on the outer surface of unusually large, acidic vacuoles localized in the pericentriolar region [93].
3.5. Caliciviruses
Members of the genus Norovirus (NoVs, family Caliciviridae) are major agents of acute gastroenteritis [94]. Ultrastructural examination of Murine Norovirus 1 (MNV-1)-infected cells revealed a striking change in their overall morphology and intracellular organization [95]. Structures resembling virus particles were observed within or next to single- or double-membrane vesicles in the cytoplasm. The vesiculated areas increase in size with time and by 24 hpi, large numbers of these vesicles and viral particles occupy most of the cytoplasm and displace the nucleus (Figure 3H). In addition, a complete rearrangement of the ER and loss of intact Golgi apparatus was observed. Both dsRNA and MNV-1 nonstructural protein 7, the RNA dependent RNA polymerase, localize to the limiting membrane of individual vesicle clusters by immuno-EM [96]. Immunofluorescence-based double-labeling showed that MNV-1 appears to recruit membranes derived from multiple cellular organelles and/or compartments: the ER, trans-Golgi apparatus and endosomes. However, despite extensive efforts, human norovirus cannot be grown in cultured cells [97]. Thus, detailed studies have not been possible, but it is assumed that replication structures are similar to those of MNV-1.
Feline Calicivirus (FCV), a member of the genus Vesivirus within this family, is a major agent of respiratory disease in cats, which replication originates also membranous rearrangements and vesicles [98].
3.6. Plant Viruses
Brome Mosaic Virus (BMV, family Bromoviridae) generates its replication factory by hijacking ER membranes, similar to what has been described for other plant viruses like Tobacco Mosaic Virus (TMV, family Virgaviridae) [99], Tobacco Echt Virus (TEV, family Potyviridae) [100] and Red Clover Necrosis Mosaic Virus (family Tombusviridae) [101]. However, other plant viruses such as Alfalfa Mosaic Virus (AMV) [102] and Cucumber Mosaic Virus (CMV), both belonging to the family Bromoviridae, and Turnip Yellow Mosaic Virus (TYMV, family Tymoviridae) [103] anchor their replication sites on chloroplasts. Cucumber Necrosis Virus (CNV), family Tombusviridae, utilizes peroxisomal membranes as replication platforms [104], while other plant viruses replicate on the surface of mitochondria [105].
Although the 3D architecture of these membranous replication sites remains largely unknown, their characteristics are strikingly similar to those for FHV (family Nodaviridae). Best studied is BMV that induces spherules, of similar size as the insect nodavirus FHV, in the ER close to the nucleus, where viral RNA synthesis and viral replication proteins are localized [106,107,108]. In the case of Beet Yellows Closterovirus (BYV, family Closteroviridae), TEM of infected plant cells revealed the formation of ~100 nm-diameter DMVs and multivesicular complexes (single-membrane vesicles surrounded by a common membrane) (Figure 3E) [109]. These multivesicular complexes often reside next to stacks of aligned filamentous BYV particles [110,111] and resemble the DMVs and VPs produced by nidoviruses and flaviviruses. Several BYV replication-associated proteins (L-PCP, MTR and HEL) colocalize with DMV and VP membranes, supporting the role of these structures as replications platforms [112,113]. The membranes in closterovirus DMVs and VPs are likely to be derived from ER for members of the genus Crinivirus [114] or mitochondria in case of Ampelovirus [115,116]. Whether these structures are “closed” or “necked” remain unknown.
4. Similarities and Differences between the Replication Factories of Flaviviruses and other (+) Strand RNA Viruses
Based on amino acid sequence homologies of their RNA-dependent RNA polymerases, (+) RNA viruses have been classified into three large supergroups [113,117,118]: supergroup I (Picornavirata or picorna-like group), including picorna-, corona-, arteri- and nodaviruses; supergroup II (Flavivirata or the flavi-like group), including tombus-, diantho-, pesti-, hepaci- and flaviviruses as well as single-strand RNA bacteriophages; supergroup III (Rubivirata or the alpha-like group), including tobamo-, hordei-, alpha- and rubiviruses as well as hepatitis E virus (HEV). These higher-order taxonomic units encompass diverse viruses infecting different hosts from almost all kingdoms of life. As discussed earlier [119], amongst these viruses two main architectures of remodeled membranes (morphotypes) can be found that may reflect two alternative strategies to induce the membranous microenvironments required to allow virus replication (summarized in Table 1).
The first morphotype involves the formation of negatively curved membranes, initiated by invaginations of the pre-existing membrane bilayer and giving rise to spherules, vesicles or vacuoles towards the lumen of the targeted cell organelle. These structures have been identified in a broad range of mammalian, plant and insect cells infected with viruses belonging to supergroups II and III. The second strategy involves the formation of membranes with positive curvature, giving rise to double-membrane structures that are the predominant characteristic of the replication factories of the picorna-like virus supergroup. The conservation of these two sorts of morphotypes in distantly related viruses supports the assumption of an evolutionary conserved mechanism. A striking finding in this regard was the observation that HCV, despite belonging to the flavi-like supergroup, induces DMVs whereas flaviviruses induce the formation of negatively curved membranes. To our current knowledge, HCV is the only member of the family Flaviviridae inducing the formation of membrane structures with positive curvature, suggesting that HCV might share common host cell pathways to induce membranous replication compartments with distantly related viruses such as corona-, arteri-, picorna-, calivi- or closteroviruses (Table 1). However, it still remains to be elucidated whether other members of the family Flaviviridae, belonging to the genera Pestivirus and Pegivirus, also utilize a picorna-like membrane remodeling strategy.

Table 1.
Main structures induced by different (+) strand RNA viruses.
| Morphotype | Virus Group and Representative Member | Name of Membrane Alteration | Size/Diameter | Membrane Source | |
|---|---|---|---|---|---|
| Single-membrane vesicles | Nodaviruses | FHV | Spherules | ~50 nm | Mitochondria |
| Bromoviruses | BMV | ~60 nm | ER (adjacent to the nucleus) | ||
| Togaviruses | RUBV | Cytoplasmic vacuoles (CPVs) | 600–2000 nm | Lysosomes | |
| SFV | CPVs with spherules | CPVs: 600–2000 nm Spherules: 50 nm | |||
| Flaviviruses | DENV | Invaginated vesicles | 80–120 nm | ER | |
| WNVKUN | 50–100 nm | ||||
| TBEV | 60–100 nm | ||||
| MVEV, SLEV and JEV | 75–145 nm | ||||
| YFV | unknown | ||||
| Double-membrane vesicles | Hepaciviruses | HCV | ~150 nm | ER | |
| Coronaviruses | SARS-CoV | 200–300 nm | |||
| MHV | 200–350 nm | ||||
| MERS-CoV | 150–320 nm | ||||
| IBV | ~200 nm | ||||
| Arteriviruses | EAV | ~95 nm | |||
| Picornaviruses | Poliovirus | 100–300 nm | Golgi | ||
| CV3B | ~160 nm | ||||
| FMDV | ~100 nm | unknown (probably ER) | |||
| Calicivirus | MNV-1 | ~200 nm | ER/Golgi | ||
| Closterovirus | BYV | ~100 nm | ER/Mitochondria | ||
Abbreviations: FHV, Flock House Virus; BMV, Brome Mosaic Virus; RUBV, Rubella Virus; SFV, Semliki Forest Virus; DENV, Dengue Virus; WNVKUN, West Nile Virus (Kunjin strain); TBEV, Tick Borne Encephalitis Virus; MVEV, Murray Valley Encephalitis Virus; SLEV, St. Louis Encephalitis Virus; JEV, Japanese Encephalitis Virus; YFV, Yellow Fever Virus; HCV, Hepatitis C Virus; SARS-CoV, Severe Acute Respiratory Virus-Coronavirus; MHV, Mouse Hepatitis Virus; MERS-CoV (Middle East Respiratory Syndrome-Coronavirus); IBV, Infectious Bronchitis Virus; EAV, Equine Arterivirus; CVB3, Coxsackie B3 Virus; FMDV, Foot and Mouth Disease Virus; MNV-1, Murine Norovirus-1; BYV, Beet Yellows Virus.
A common feature associated with the spherule/vesicle/vacuole/-type of rearranged membranes is their size (50–150 nm diameter) and the presence of a pore connecting the interior of the vesicle with the cytoplasm [15,20,60,88]. Since RNA replication occurs in the vesicle interior, the pore allows exchange of nucleotides and RNA products with the cytoplasm. The size of the pore is variable, ranging from 4.4 nm in case of IBV-induced spherules to ~10 nm in case of membrane invaginations induced by flaviviruses. In contrast, in the majority of DMVs no such channel or pore has been detected. Nevertheless, as exemplified with nidoviruses, the inner compartments enclosed by interconnected DMVs contain the bulk of dsRNA [63], and in some cases they depict an electron-dense core assumed to correspond to viral RNA (IBV and EAV) [67,69]. Although this represents a functional enigma in terms of RNA synthesis and transport, the presence of dsRNA in the DMV interior does not necessarily indicate active RNA replication. Assuming a temporal regulation, it is possible that DMVs might be sites of RNA synthesis as long as they are linked to the cytoplasm, but replication would stop upon closure of the vesicles. Yet, another strategy appears to be used by enteroviruses, where active RNA replication has been detected on the cytosolic side of the membranous structures [71,120], consistent with the membrane topology of the nonstructural proteins catalyzing RNA replication [121,122,123]. As described above, studies conducted with picornaviruses revealed that the exponential phase of viral RNA synthesis coincides with the accumulation of single-membrane tubules [79,80]. Importantly, pulse-radiolabeling experiments localized sites of active RNA replication to the outer surface of single-membrane tubules [71] and isolation of the membranous replication factories and their subsequent visualization by EM revealed that they form rosette-like structures composed of virus-induced cytoplasmic vesicles [124]. RNA replication is thought to occur at sites where the vesicles cluster, whereas RNA translation probably takes place on the exposed periphery of the vesicles. This raises the question, what the role of DMVs in the replication cycle of picornaviruses might be. It is possible that DMVs either support RNA synthesis or serve as RNA storage sites (especially in case of closed DMVs). In this manner, DMVs might be involved in regulating viral RNA replication: by complete sealing of the viral replicase inside the vesicle, it would be inactive, thus regulating overall RNA copy number in the infected cell. Alternatively, DMVs might be an epiphenomenon, resulting from the over-expression of membrane-active proteins that accumulate especially during the late stages of infection.
The mechanism responsible for DMV formation is not clear. In case of picornaviruses, it is thought that single-membrane structures are the precursors of DMVs [79,80]. Nevertheless, DMV formation might also involve the autophagy machinery, or at least several components thereof, by a process analogous to the formation of autophagic vacuoles [76]. This hypothesis is supported by the morphological resemblance of DMVs and autophagosomes. It has been shown that the inhibition or stimulation of autophagy results in a modest inhibition or stimulation of poliovirus and coxsackie B3 virus yield, respectively, and there are also data supporting the involvement of autophagy in the replication of rhinovirus 2 and 14 [125,126]. However, Brabec-Zaruba et al. [127] reported that replication of rhinovirus 2 was insensitive to pharmacological manipulation of autophagy and did not induce detectable modification of LC3. This discrepancy might be due to the use of different cell types and experimental conditions.
The mechanism of DMV formation in case of HCV and coronaviruses is also unclear. Biochemical analysis of isolated host cell membranes associated with HCV RNA and proteins identified markers of the autophagy machinery, including LC3-II, the lipidated form of LC3 (LC3-II) that is generated upon activation of the autophagy machinery [46]. However, the role of autophagy in the HCV replication cycle is also a matter of controversy. For instance, immunolabeling did not identify LC3-II at those sites where nonstructural proteins accumulate [48]. Moreover, different roles of autophagy for the HCV replication cycle have been proposed. These include a role of autophagy in HCV RNA translation [128], initiation of RNA replication [129,130], production of infectious virus particles [131] or suppression of the innate antiviral defense [132,133]. To clarify these discrepancies, future studies should combine biochemical and cell biological approaches with ultrastructural analyses.
The autophagy machinery might also be involved in the formation of virus-induced membrane invaginations/spherules. For instance, Lee and coworkers provided evidence that DENV infection enhances autophagolysosome formation and that inhibition of the autophagy machinery by 3-methyladenine (3-MA) reduces DENV particle production [134]. However, the effects were moderate, arguing that this pathway may contribute to DENV replication to only a minor extent. Moreover, autophagy includes membrane wrapping, leading to double-membrane compartments involved in lysosomal degradation whereas DENV-induced vesicles are invaginations. Finally, immunolabeling experiments failed to detect lamp-1 at these vesicles [15], arguing against the involvement of lysosomes in the formation of the DENV replication vesicles. It remains to be determined whether autophagy is actively induced by these viruses to provide a compartment favoring replication or induced as a bystander defense against infection leading to degradation of the replicase proteins [135].
Another membrane compartment frequently induced by (+) RNA viruses are convoluted membranes (CMs) that were observed e.g., in SARS-CoV-, MHV-, WNV- or DENV-infected cells [13,15,18,63,65,66,136]. Morphologically, CMs resemble smooth ER membranes, lack ribosomes and in case of DENV are induced by the sole expression of NS4A [137,138]. CMs are often associated with late stages of infection, suggesting that DMV formation might precede the development of CMs. In SARS-CoV-infected cells, DMVs appear to be connected with CMs [63], while in MHV-infected cells no such connections have been observed [65]. The role of CMs for the viral replication cycle is not well understood. In case of WNVKUN, CMs are supposed to be the site of polyprotein processing [18,136]. This conclusion is based primarily on the strong immunolabeling for NS2B and NS3 and the absence of NS1 and NS4B. Since polyprotein cleavage occurs co-translationally and thus, should happen at the rER, this model would require the formation of rather stable processing intermediates that are transferred from the rER to the CMs where further cleavage would occur. Alternatively, at least in case of DENV, CMs might represent a storage site for proteins and lipids involved in viral replication that can be recruited to vesicles upon demand. The fact that CMs are physically linked with ER-containing invaginations and contain NS3 would be consistent with this assumption [15]. Along these lines, the fact that insects are cholesterol auxotrophs and lack several enzymes in the cholesterol biosynthesis pathway [139], suggests that cholesterol might be a key component of CM structures, which would explain their absence in infected insect cells [16].
5. Role of Viral Proteins in the Formation of the Replication Organelles
Viral replication complexes are targeted to the respective membranous organelle primarily by nonstructural (NS) proteins rather than viral RNA [140]. These NS proteins seem to have some specificity in recognizing organelle subpopulations and often contain multiple hydrophobic domains implicated in membrane targeting and rearrangement. The molecular mechanisms orchestrating the formation of these complex structures are still poorly understood, but it is clear that NS proteins, often working in a concerted action, are key players in replication factory biogenesis.
5.1. Single-Membrane Vesicle Inducers
One well-studied example among the single-membrane vesicle inducers is BMV where it was shown that the sole expression of the NS protein 1a is sufficient to induce the formation of single-membrane spherules resembling the ones observed in infected cells. These spherules had a diameter of 50–70 nm, resided in the ER lumen and were shown to be the site of viral RNA synthesis [108]. In case of the insect nodavirus FHV, protein A and replication competent RNA were required for induction of the spherules. Expression of protein A alone induced only “zippering” of the surfaces of adjacent mitochondria, but did not induce spherules. Thus, protein A is necessary, but not sufficient for spherule formation. Moreover, spherules were not formed when replication-competent FHV RNA templates were expressed with a protein A mutant lacking polymerase activity or when wild-type protein A was expressed with a replication-incompetent FHV RNA template. Thus, the membranous FHV replication compartment requires both a viral protein and active RNA synthesis [141].
Feline Calicivirus (FCV) infection results in rearrangement of intracellular membranes and production of numerous membrane-bound vesicular structures on which viral genome replication is thought to occur. Expression of individual FCV nonstructural proteins revealed that p30 induces significant reorganization of the ER into large, fenestrated membrane networks, resembling the structures found in infected cells [142]. Moreover, expression of p39 and p32, two additional FCV NS proteins, induced extensive reorganization of the ER and the nuclear envelope suggesting that the ER is the primary source of the membranous replication factory [142].
Flavivirus membrane rearrangements are mainly induced by NS4A, as suggested by recent studies with WNVKUN and DENV [137,138], but it is unknown whether the same applies to NS4A of TBEV. In case of DENV, NS4A is thought to contain a central peripheral membrane domain that intercalates into the luminal leaflet of the ER membrane [138]. It is tempting to speculate that NS4A oligomers [143] might dilate the luminal leaflet, resulting in membrane invaginations towards the ER lumen. However, the sole expression of NS4A is not sufficient to induce ER membrane invaginations that have been detected in infected cells. Instead, expression of NS4A lacking the C-terminal 2K fragment (corresponding to fully processed NS4A) induced ER membrane rearrangements reminiscent of CMs, whereas unprocessed NS4A/2K did not induce membrane alterations [138]. These results provide strong evidence that processing at the NS4A-2K site is required for the induction of membrane alterations. The critical role of polyprotein cleavage for induction of membrane rearrangements is supported by studies conducted with WNVKUN. There it was shown that a regulated cleavage of a NS4A/2K/4B precursor by the viral NS2B/3 protease is needed for induction of membrane rearrangements [137]. However, the same study reported that expression of full-length (uncleaved) WNVKUN NS4A/2K led to membrane alterations similar to those induced in infected cells whereas the 2K fragment impaired the ability of NS4A to induce membrane rearrangements. Whether this reflects a biological difference between WNVKUN and DENV NS4A or is due to the use of alternative experimental approaches remains to be determined.
One of the most fascinating mechanisms employed by (+) RNA viruses to induce their replication factories is used by SFV. It was shown that the spherules of SFV arise by blebbing at the surface of the plasma membrane [93]. These blebs are internalized and after fusion with lysosomes give rise to large cytoplasmic vacuoles. Formation of these membrane alterations requires the viral protein nsP1, which has several functions. It has guanine-7-methyltransferase and guanylyltransferase activities and thus is critically involved in capping of the viral RNAs [144,145,146], but at the same time has affinity to lipids [89]. In fact, of the four NS proteins of SFV, only nsP1 has affinity for membranes, and when expressed alone, it is specifically targeted to the inner surface of the plasma membrane [147]. NsP1 is a monotopic membrane protein and its affinity for membranes is dictated by an amphipathic α-helix, located in the central region of the protein [148,149]. NsP1 has a specific affinity for negatively charged phospholipids, which might account for its prevalent localization to the plasma membrane, where such lipids are enriched. Membrane binding of nsP1 via its amphipathic α-helix is essential for alphavirus replication [93]. However, nsP1 is not sufficient for cytoplasmic vacuole formation. For instance, it was found that nsP3 contributes to the transport of the replicase polyprotein from the plasma membrane to the surface of endosomes [150]. These results indicate that nsp1 has to cooperate with other viral and cellular factors to allow formation of the cytoplasmic vacuoles. Furthermore, in a recent study Kallio et al. [151] have shown that the size of the spherule is dependent on the length of the RNA template, in contrast to what has been observed for FHV, another spherule-inducer [141]. These results indicate that in addition to the NS proteins, the viral RNA template itself critically determines the morphology of the membranous vesicles.
5.2. Double-Membrane Vesicle Inducers
In order to induce the variety of membrane alterations observed in CVB3 -infected cells (Figure 3F), several membrane-remodeling mechanisms are required: induction of membrane curvature, membrane fusion and membrane-membrane interactions [80]. These rearrangements require the enteroviral proteins 2BC and 3A; their coexpression generates ER membrane-derived structures mimicking those observed during viral infection [78]. Importantly, 2B and 2C both contain an amphipathic α-helix [152,153,154], a well-known curvature-inducing motif [155]. Along the same lines, FMDV 2B and 2BC locate to the ER when expressed on their own and cause a swelling of ER cisternae [156].
In case of HCV, we recently found that a concerted action of NS3/4A, NS4B, NS5A and NS5B is required to generate the membranous web. Furthermore, all these replicase components were capable of inducing membrane vesiculation with NS5A having the highest potential to trigger membrane curvature. Importantly, some of these NS5A-induced structures corresponded to DMVs [48]. In addition, NS4B also plays an important role in triggering rearrangements of intracellular membranes [45]. NS4B is an integral membrane protein containing two N-terminal amphipathic α-helices, a highly hydrophobic central core domain composed of four putative transmembrane segments, and a highly conserved C-terminal domain that is thought to harbor two α-helices (reviewed in [157]). A recent study has demonstrated that NS4B oligomerizes through multiple conserved determinants and that oligomerization appears to be required for membranous web induction [158]. Indeed, mutations affecting the highly conserved C-terminal domain impairing NS4B self-interaction resulted in the formation of aberrant DMVs arguing for a central role of NS4B in formation of functional replication compartments [159].
Studies on arteriviruses revealed that the sole expression of EAV nsp2 and nsp3 is sufficient to induce membrane structures similar to those generated during EAV infection [160]. Mutations within nsp3, which is a tetra-spanning integral membrane protein, alter membrane rearrangements, highlighting the importance of this protein for the biogenesis of EAV-induced DMVs [161]. In case of SARS-CoV, nsp3, nsp4 and nsp6 were found to be sufficient to induce the formation of DMVs that are similar to those observed in SARS-CoV-infected cells [162]. These DMVs were, however, smaller in diameter, suggesting a role for other viral proteins or the presence of viral RNA in determining the DMV morphology. Importantly, EM analysis of nsp4 mutants that are impaired in RNA replication and virus growth, revealed an aberrant morphology of DMVs as well as an increased prevalence of CMs [163]. Another important viral protein involved in inducing the SARS-CoV membranous replication factory is nsp6, which is predicted to contain seven transmembrane segments and a hydrophilic cytoplasmic domain [164]. Nsp6 was shown to induce vesicles containing Atg5 and LC3-II as well as phosphatidylinositol-3-phosphate, thus sharing many features with omegasomes, which are omega-shaped membrane compartments that are formed during activation of autophagy [164]. This result suggests that autophagy might contribute to the formation of the membranous replication site of SARS-CoV.
6. Conclusions and Future Perspectives
In the last couple of years, our knowledge of the architecture of the replication factories induced by (+) RNA viruses has increased substantially. This is primarily due to the more widespread use of ET and other high-resolution imaging methods. Nevertheless, our knowledge is still rather restricted to descriptions of the morphologies of these complex structures, whereas our understanding of their biogenesis in most cases is very rudimentary. More efforts are required to elucidate the role of the viral proteins in the formation of the replication vesicles, to identify the involved cellular components and the mechanisms used by these proteins to subvert and exploit cellular pathways to establish membranous replication factories. This includes determination of the 3D structure of involved (viral) proteins as well as evaluation of host cell factors and lipids contributing to biogenesis and activity of the replication compartment. In addition, further studies are needed to understand how viruses utilize these compartments to coordinate the different steps of their life cycle (replication, assembly and release) in space and time to achieve efficient replication.
Acknowledgments
The authors are very grateful to Erik J. Snijder (Leiden University Medical Center, Leiden, The Netherlands) and Kèvin Knoops (European Molecular Biology Laboratory, Grenoble, France) for providing the unpublished pictures depicted in Figure 3B and 3D and to Montserrat Bárcena (Leiden University Medical Center, Leiden, The Netherlands) and Tero Ahola (University of Helsinki, Helsinki, Finland) for providing the unpublished pictures depicted in Figure 3F and 3J, respectively. Figure 3A, 3C, 3E, 3G, 3H and 3I are reproduced with permission from [60], [67], [109], [79], [95] and [86], respectively.
We also would like to thank Jason M. Mackenzie (University of Queensland, Brisbane, Australia), Paul Ahlquist (Howard Hughes Medical Institute, Madison, WI, USA), Cristina Risco (Centro Nacional de Biotecnología, Madrid, Spain), Georg A. Belov (University of Maryland, College Park, MD, USA), Christiane Wobus (Washington University School of Medicine, St Louis, MO, USA), Paul Britton (The Pirbright Institute, Compton, UK) and Alexey A. Agranovsky (Moscow State University, Moscow, Russia) for providing other electron micrographs and 3D reconstructions.
Work in the authors’ laboratory was supported by the Deutsche Forschungsgemeinschaft (Sonder-forschungsbereich 638, TP5 and Transregional Collaborative Research Project TR83, TP13).
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The GB viruses: A review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J. Gen. Virol. 2011, 92, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. The Origin of Hepatitis C Virus. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy, 82nd ed.; Bartenschlager, R., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 2013; Volume 369. [Google Scholar]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Thiel, H.-J.; Rice, C.M. Flaviviridae: The Viruses and Their Replication. In Fields Virology, 5th ed.; Fields, B.N., Knipe, D.M., Howley, P., Eds.; Raven Press: New York, NY, USA, 2001; pp. 1001–1152. [Google Scholar]
- Mackenzie, J.S.; Gubler, D.J.; Petersen, L.R. Emerging flaviviruses: The spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 2004, 10, S98–S109. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Collett, M.S.; Gould, E.A.; Heinz, F.X.; Meyers, G.; Monath, A.; Pletnev, A.; Rice, C.M.; Stiasny, K.; et al. Family Flaviviridae. In Virus taxonomy: Classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Academic Press: San Diego, CA, USA, 2012; pp. 1003–1020. [Google Scholar]
- Miller, R.H.; Purcell, R.H. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. USA 1990, 87, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Stollar, V.; Schlesinger, R.W. Studies on the nature of dengue viruses. V. Structure and development of dengue virus in Vero cells. Virology 1971, 46, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Stohlman, S.A.; Wisseman, C.L., Jr.; Eylar, O.R.; Silverman, D.J. Dengue virus-induced modifications of host cell membranes. J. Virol. 1975, 16, 1017–1026. [Google Scholar] [PubMed]
- Hase, T.; Summers, P.L.; Eckels, K.H.; Baze, W.B. An electron and immunoelectron microscopic study of dengue-2 virus infection of cultured mosquito cells: Maturation events. Arch. Virol. 1987, 92, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.C.; Chen, C.C.; Hou, R.F. Ultrastructural studies on the reproductive system of male Aedes aegypti (Diptera: Culicidae) infected with dengue 2 virus. J. Med. Entomol. 1998, 35, 71–76. [Google Scholar] [PubMed]
- Grief, C.; Galler, R.; Cortes, L.M.; Barth, O.M. Intracellular localisation of dengue-2 RNA in mosquito cell culture using electron microscopic in situ hybridisation. Arch. Virol. 1997, 142, 2347–2357. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Improved membrane preservation of flavivirus-infected cells with cryosectioning. J. Virol. Methods 1996, 56, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell. Host. Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Shahar, A.; Lustig, S.; Akov, Y.; David, Y.; Schneider, P.; Friedmann, A.; Levin, R. West Nile virions aligned along myelin lamellae in organotypic spinal cord cultures. J. Neurosci. Res. 1990, 26, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: Colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Westaway, E.G. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Westaway, E.G. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus-infected cells. J. Virol. 1999, 73, 9555–9567. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Kenney, M.T.; Westaway, E.G. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J. Gen. Virol. 2007, 88, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.K.; Westaway, E.G. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Popov, V.L.; Niedrig, M.; Weber, F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 2010, 84, 8470–8483. [Google Scholar] [CrossRef]
- Pichlmair, A.; Reis e Sousa, C. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Matsumura, T.; Masuda, K.; Kanemura, K.; Fukunaga, T. Maturation site of dengue type 2 virus in cultured mosquito C6/36 cells and Vero cells. Kobe J. Med. Sci. 1998, 44, 65–79. [Google Scholar] [PubMed]
- Senigl, F.; Grubhoffer, L.; Kopecky, J. Differences in maturation of tick-borne encephalitis virus in mammalian and tick cell line. Intervirology 2006, 49, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS One 2012, 7, e47912. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, C.; Blair, C.D. Persistent infection of cultured mammalian cells by Japanese encephalitis virus. J. Virol. 1977, 24, 580–589. [Google Scholar] [PubMed]
- Brinton, M.A.; Fernandez, A.V. A replication-efficient mutant of West Nile virus is insensitive to DI particle interference. Virology 1983, 129, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Poidinger, M.; Coelen, R.J.; Mackenzie, J.S. Persistent infection of Vero cells by the flavivirus Murray Valley encephalitis virus. J. Gen. Virol. 1991, 72, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Hoenninger, V.M.; Rouha, H.; Orlinger, K.K.; Miorin, L.; Marcello, A.; Kofler, R.M.; Mandl, C.W. Analysis of the effects of alterations in the tick-borne encephalitis virus 3'-noncoding region on translation and RNA replication using reporter replicons. Virology 2008, 377, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Khromykh, A.A.; Westaway, E.G. Subgenomic replicons of the flavivirus Kunjin: Construction and applications. J. Virol. 1997, 71, 1497–1505. [Google Scholar] [PubMed]
- Jones, C.T.; Patkar, C.G.; Kuhn, R.J. Construction and applications of yellow fever virus replicons. Virology 2005, 331, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.; Robertson, T.; Kendrick, T.; Abdo, M.; Papadimitriou, J.; McMinn, P. Morphological features of Murray Valley encephalitis virus infection in the central nervous system of Swiss mice. Int. J. Exp. Pathol. 2000, 81, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Hase, T.; Summers, P.L.; Ray, P. Entry and replication of Japanese encephalitis virus in cultured neurogenic cells. J. Virol. Methods 1990, 30, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hase, T.; Summers, P.L.; Dubois, D.R. Ultrastructural changes of mouse brain neurons infected with Japanese encephalitis virus. Int. J. Exp. Pathol. 1990, 71, 493–505. [Google Scholar] [PubMed]
- Murphy, F.A.; Harrison, A.K.; Gary, G.W., Jr.; Whitfield, S.G.; Forrester, F.T. St. Louis encephalitis virus infection in mice. Electron microscopic studies of central nervous system. Lab. Investig. 1968, 19, 652–662. [Google Scholar] [PubMed]
- Whitfield, S.G.; Murphy, F.A.; Sudia, W.D. St. Louis encephalitis virus: An ultrastructural study of infection in a mosquito vector. Virology 1973, 56, 70–87. [Google Scholar] [CrossRef] [PubMed]
- McGavran, M.H.; White, J.D. Electron microscopic and immunofluorescent observations on monkey liver and tissue culture cells infected with the Asibi strain of yellow fever virus liver and tissue culture cells infected with the asibi strain of yellow fever virus. Am. J. Pathol. 1964, 45, 501–517. [Google Scholar] [PubMed]
- Egger, D.; Wolk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 2013, 70, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef] [PubMed]
- Chasey, D.; Roeder, P.L. Virus-like particles in bovine turbinate cells infected with bovine virus diarrhoea/mucosal disease virus. Arch. Virol. 1981, 67, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.W.; Nettleton, P.F. The ultrastructure of cell cultures infected with border disease and bovine virus diarrhoea viruses. J. Gen. Virol. 1987, 68, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Kubovicova, E.; Makarevich, A.V.; Pivko, J.; Chrenek, P.; Grafenau, P.; Riha, L.; Sirotkin, A.V.; Louda, F. Alteration in ultrastructural morphology of bovine embryos following subzonal microinjection of bovine viral diarrhea virus (BVDV). Zygote 2008, 16, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Dubovi, E.J.; Cohen-Gould, L.; Donis, R.; Szeto, H.H. Cytoplasmic vacuolization responses to cytopathic bovine viral diarrhoea virus. Virus Res. 2008, 132, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Weiskircher, E.; Aligo, J.; Ning, G.; Konan, K.V. Bovine viral diarrhea virus NS4B protein is an integral membrane protein associated with Golgi markers and rearranged host membranes. Virol. J. 2009, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, S.; Mast, J.; Thiel, H.J.; Konig, M. Morphogenesis of pestiviruses: New insights from ultrastructural studies of strain Giraffe-1. J. Virol. 2014, 88, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Keppler, O.T.; Habermann, A.; Allespach, I.; Krijnse-Locker, J.; Krausslich, H.G. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007, 3, e36. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Berghofer, B.; Bamberg, S.; Becker, S. Multivesicular bodies as a platform for formation of the Marburg virus envelope. J. Virol. 2004, 78, 12277–12287. [Google Scholar] [CrossRef] [PubMed]
- Mittler, E.; Kolesnikova, L.; Strecker, T.; Garten, W.; Becker, S. Role of the transmembrane domain of marburg virus surface protein GP in assembly of the viral envelope. J. Virol. 2007, 81, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Kopek, B.G.; Perkins, G.; Miller, D.J.; Ellisman, M.H.; Ahlquist, P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 2007, 5, e220. [Google Scholar] [CrossRef] [PubMed]
- Lanman, J.; Crum, J.; Deerinck, T.J.; Gaietta, G.M.; Schneemann, A.; Sosinsky, G.E.; Ellisman, M.H.; Johnson, J.E. Visualizing flock house virus infection in Drosophila cells with correlated fluorescence and electron microscopy. J. Struct. Biol. 2008, 161, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; van der Meer, Y.; Zevenhoven-Dobbe, J.; Onderwater, J.J.; van der Meulen, J.; Koerten, H.K.; Mommaas, A.M. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 2006, 80, 5927–5940. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; van, N.S.; Limpens, R.W.; Posthuma, C.C.; van der Meer, Y.; Barcena, M.; Haagmans, B.L.; et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. MBio 2013, 4, e00801–e00813. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [PubMed]
- Knoops, K.; Barcena, M.; Limpens, R.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural characterization of arterivirus replication structures: Reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Kallman, F.; Williams, R.C.; Dulbecco, R.; Vogt, M. Fine structure of changes produced in cultured cells sampled at specified intervals during a single growth cycle of polio virus. J. Biophys. Biochem. Cytol. 1958, 4, 301–308. [Google Scholar] [CrossRef]
- Bienz, K.; Egger, D.; Pasamontes, L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 1987, 160, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Bienz, K. Intracellular location and translocation of silent and active poliovirus replication complexes. J. Gen. Virol. 2005, 86, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.P.; Egger, D.; Bienz, K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Bienz, K.; Egger, D.; Rasser, Y.; Bossart, W. Intracellular distribution of poliovirus proteins and the induction of virus-specific cytoplasmic structures. Virology 1983, 131, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar] [PubMed]
- Bienz, K.; Egger, D.; Rasser, Y.; Bossart, W. Kinetics and location of poliovirus macromolecular synthesis in correlation to virus-induced cytopathology. Virology 1980, 100, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Barcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, P.; Cook, H.; Jackson, T.; Ryan, M.; Wileman, T. The ultrastructure of the developing replication site in foot-and-mouth disease virus-infected BHK-38 cells. J. Gen. Virol. 2004, 85, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.K. Molecular biology of rubella virus. Adv. Virus Res. 1994, 44, 69–160. [Google Scholar] [PubMed]
- Fontana, J.; Tzeng, W.P.; Calderita, G.; Fraile-Ramos, A.; Frey, T.K.; Risco, C. Novel replication complex architecture in rubella replicon-transfected cells. Cell. Microbiol. 2007, 9, 875–890. [Google Scholar] [CrossRef]
- Lee, J.Y.; Marshall, J.A.; Bowden, D.S. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 1994, 200, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Magliano, D.; Marshall, J.A.; Bowden, D.S.; Vardaxis, N.; Meanger, J.; Lee, J.Y. Rubella virus replication complexes are virus-modified lysosomes. Virology 1998, 240, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Lopez-Iglesias, C.; Tzeng, W.P.; Frey, T.K.; Fernandez, J.J.; Risco, C. Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579–591. [Google Scholar] [CrossRef]
- Risco, C.; Carrascosa, J.L.; Frey, T.K. Structural maturation of rubella virus in the Golgi complex. Virology 2003, 312, 261–269. [Google Scholar] [CrossRef]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell. Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.M.; Levin, J.G.; Grimley, P.M.; Berezesky, I.K. Membrane-associated replication complex in arbovirus infection. J. Virol. 1972, 10, 504–515. [Google Scholar]
- Grimley, P.M.; Berezesky, I.K.; Friedman, R.M. Cytoplasmic structures associated with an arbovirus infection: Loci of viral ribonucleic acid synthesis. J. Virol. 1968, 2, 1326–1338. [Google Scholar] [PubMed]
- Grimley, P.M.; Levin, J.G.; Berezesky, I.K.; Friedman, R.M. Specific membranous structures associated with the replication of group A arboviruses. J. Virol. 1972, 10, 492–503. [Google Scholar]
- Spuul, P.; Balistreri, G.; Kaariainen, L.; Ahola, T. Phosphatidylinositol 3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest Virus replication complexes from the plasma membrane to modified lysosomes. J. Virol. 2010, 84, 7543–7557. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y.; Chanock, R.M.; Kapikian, A.Z. Human caliciviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 841–874. [Google Scholar]
- Wobus, C.E.; Karst, S.M.; Thackray, L.B.; Chang, K.O.; Sosnovtsev, S.V.; Belliot, G.; Krug, A.; Mackenzie, J.M.; Green, K.Y.; Virgin, H.W. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004, 2, e432. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Sosnovtsev, S.V.; Green, K.Y.; Wobus, C.; Virgin, H.W.; Mackenzie, J.M. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. J. Virol. 2009, 83, 9709–9719. [Google Scholar] [CrossRef] [PubMed]
- Duizer, E.; Schwab, K.J.; Neill, F.H.; Atmar, R.L.; Koopmans, M.P.; Estes, M.K. Laboratory efforts to cultivate noroviruses. J. Gen. Virol. 2004, 85, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y.; Mory, A.; Fogg, M.H.; Weisberg, A.; Belliot, G.; Wagner, M.; Mitra, T.; Ehrenfeld, E.; Cameron, C.E.; Sosnovtsev, S.V. Isolation of enzymatically active replication complexes from feline calicivirus-infected cells. J. Virol. 2002, 76, 8582–8595. [Google Scholar] [CrossRef] [PubMed]
- Mas, P.; Beachy, R.N. Replication of tobacco mosaic virus on endoplasmic reticulum and role of the cytoskeleton and virus movement protein in intracellular distribution of viral RNA. J. Cell. Biol. 1999, 147, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Schaad, M.C.; Jensen, P.E.; Carrington, J.C. Formation of plant RNA virus replication complexes on membranes: Role of an endoplasmic reticulum-targeted viral protein. EMBO J. 1997, 16, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.A.; Sit, T.L.; Callaway, A.S.; Allen, N.S.; Lommel, S.A. Red clover necrotic mosaic virus replication proteins accumulate at the endoplasmic reticulum. Virology 2004, 320, 276–290. [Google Scholar] [CrossRef] [PubMed]
- De, G.M.; Coscoy, L.; Jaspars, E.M. Localization and biochemical characterization of alfalfa mosaic virus replication complexes. Virology 1993, 194, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.J.; Buck, K.W. Complete replication of a eukaryotic virus RNA in vitro by a purified RNA-dependent RNA polymerase. Cell 1990, 63, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Panavas, T.; Hawkins, C.M.; Panaviene, Z.; Nagy, P.D. The role of the p33:p33/p92 interaction domain in RNA replication and intracellular localization of p33 and p92 proteins of Cucumber necrosis tombusvirus. Virology 2005, 338, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lotfi, F.; Dietrich, A.; Russo, M.; Rubino, L. Mitochondrial targeting and membrane anchoring of a viral replicase in plant and yeast cells. J. Virol. 2002, 76, 10485–10496. [Google Scholar] [CrossRef]
- Restrepo-Hartwig, M.A.; Ahlquist, P. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol. 1996, 70, 8908–8916. [Google Scholar] [PubMed]
- Den Boon, J.A.; Chen, J.; Ahlquist, P. Identification of sequences in Brome mosaic virus replicase protein 1a that mediate association with endoplasmic reticulum membranes. J. Virol. 2001, 75, 12370–12381. [Google Scholar] [CrossRef]
- Schwartz, M.; Chen, J.; Janda, M.; Sullivan, M.; den, B.J.; Ahlquist, P. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell. 2002, 9, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Gushchin, V.A.; Solovyev, A.G.; Erokhina, T.N.; Morozov, S.Y.; Agranovsky, A.A. Beet yellows virus replicase and replicative compartments: Parallels with other RNA viruses. Front. Microbiol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Cronshaw, J.; Hoefert, L.; Esau, K. Ultrastructural features of Beta leaves infected with beet yellows virus. J. Cell. Biol. 1966, 31, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Esau, K.; Cronshaw, J.; Hoefert, L.L. Relation of beet yellows virus to the phloem and to movement in the sieve tube. J. Cell. Biol. 1967, 32, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Erokhina, T.N.; Vitushkina, M.V.; Zinovkin, R.A.; Lesemann, D.E.; Jelkmann, W.; Koonin, E.V.; Agranovsky, A.A. Ultrastructural localization and epitope mapping of the methyltransferase-like and helicase-like proteins of Beet yellows virus. J. Gen. Virol. 2001, 82, 1983–1994. [Google Scholar] [PubMed]
- Zinovkin, R.A.; Erokhina, T.N.; Lesemann, D.E.; Jelkmann, W.; Agranovsky, A.A. Processing and subcellular localization of the leader papain-like proteinase of Beet yellows closterovirus. J. Gen. Virol. 2003, 84, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Stewart, L.R.; Kiss, Z.; Falk, B.W. Lettuce infectious yellows virus (LIYV) RNA 1-encoded P34 is an RNA-binding protein and exhibits perinuclear localization. Virology 2010, 403, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Gonsalves, D.; Teliz, D.; Lee, K.W. Ultrastructure and mitochondrial vesiculation associated with closterovirus-like particles in leafroll-diseased grapevines. Phytopathology 1989, 79, 357–360. [Google Scholar] [CrossRef]
- Faoro, F.; Carzaniga, R. Cyotchemistry and immuno-cytochemistry of the inclusion bodies induced by grapevine leafroll-associated closteroviruses GLRaV-1 and GLRaV-3. Riv. Patol. Veg. 1995, 5, 85–94. [Google Scholar]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Kuhn, R.J.; Yang, C.F.; Takegami, T.; Wimmer, E. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J. Virol. 1986, 60, 43–53. [Google Scholar] [PubMed]
- Xiang, W.; Cuconati, A.; Hope, D.; Kirkegaard, K.; Wimmer, E. Complete protein linkage map of poliovirus P3 proteins: Interaction of polymerase 3Dpol with VPg and with genetic variants of 3AB. J. Virol. 1998, 72, 6732–6741. [Google Scholar] [PubMed]
- Wessels, E.; Notebaart, R.A.; Duijsings, D.; Lanke, K.; Vergeer, B.; Melchers, W.J.; van Kuppeveld, F.J. Structure-function analysis of the coxsackievirus protein 3A: Identification of residues important for dimerization, viral rna replication, and transport inhibition. J. Biol. Chem. 2006, 281, 28232–28243. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Krishnakumar, S.S.; Franco, D.; Paul, A.V.; London, E.; Wimmer, E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry 2007, 46, 5185–5199. [Google Scholar] [CrossRef] [PubMed]
- Bienz, K.; Egger, D.; Pfister, T.; Troxler, M. Structural and functional characterization of the poliovirus replication complex. J. Virol. 1992, 66, 2740–2747. [Google Scholar] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed]
- Brabec-Zaruba, M.; Berka, U.; Blaas, D.; Fuchs, R. Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 2007, 81, 10815–10817. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Autophagy: A novel guardian of HCV against innate immune response. Autophagy 2011, 7, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Lei, H.Y.; Liu, M.T.; Wang, J.R.; Chen, S.H.; Jiang-Shieh, Y.F.; Lin, Y.S.; Yeh, T.M.; Liu, C.C.; Liu, H.S. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008, 374, 240–248. [Google Scholar] [CrossRef]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Westaway, E.G.; Khromykh, A.A.; Kenney, M.T.; Mackenzie, J.M.; Jones, M.K. Proteins C and NS4B of the flavivirus Kunjin translocate independently into the nucleus. Virology 1997, 234, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Buhler, S.; Bartenschlager, R. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef]
- Rawson, R.B. The SREBP pathway--insights from Insigs and insects. Nat. Rev. Mol. Cell. Biol. 2003, 4, 631–640. [Google Scholar] [CrossRef]
- Salonen, A.; Ahola, T.; Kaariainen, L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar] [PubMed]
- Kopek, B.G.; Settles, E.W.; Friesen, P.D.; Ahlquist, P. Nodavirus-induced membrane rearrangement in replication complex assembly requires replicase protein a, RNA templates, and polymerase activity. J. Virol. 2010, 84, 12492–12503. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; Kaiser, W.J.; Hollinshead, M.; Moffat, K.; Chaudhry, Y.; Wileman, T.; Sosnovtsev, S.V.; Goodfellow, I.G. Feline calicivirus p32, p39 and p30 proteins localize to the endoplasmic reticulum to initiate replication complex formation. J. Gen. Virol. 2010, 91, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Stern, O.; Hung, Y.F.; Valdau, O.; Yaffe, Y.; Harris, E.; Hoffmann, S.; Willbold, D.; Sklan, E.H. An N-terminal amphipathic helix in dengue virus nonstructural protein 4A mediates oligomerization and is essential for replication. J. Virol. 2013, 87, 4080–4085. [Google Scholar] [CrossRef]
- Ahola, T.; Kaariainen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Laakkonen, P.; Vihinen, H.; Kaariainen, L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997, 71, 392–397. [Google Scholar] [PubMed]
- Laakkonen, P.; Ahola, T.; Kaariainen, L. The effects of palmitoylation on membrane association of Semliki forest virus RNA capping enzyme. J. Biol. Chem. 1996, 271, 28567–28571. [Google Scholar] [CrossRef] [PubMed]
- Peranen, J.; Laakkonen, P.; Hyvonen, M.; Kaariainen, L. The alphavirus replicase protein nsP1 is membrane-associated and has affinity to endocytic organelles. Virology 1995, 208, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Lampio, A.; Auvinen, P.; Kaariainen, L. Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J. 1999, 18, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Lampio, A.; Kilpelainen, I.; Pesonen, S.; Karhi, K.; Auvinen, P.; Somerharju, P.; Kaariainen, L. Membrane binding mechanism of an RNA virus-capping enzyme. J. Biol. Chem. 2000, 275, 37853–37859. [Google Scholar] [CrossRef]
- Salonen, A.; Vasiljeva, L.; Merits, A.; Magden, J.; Jokitalo, E.; Kaariainen, L. Properly folded nonstructural polyprotein directs the semliki forest virus replication complex to the endosomal compartment. J. Virol. 2003, 77, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.; Hellstrom, K.; Balistreri, G.; Spuul, P.; Jokitalo, E.; Ahola, T. Template RNA length determines the size of replication complex spherules for Semliki Forest virus. J. Virol. 2013, 87, 9125–9134. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; Molla, A.; Wimmer, E. Studies of a putative amphipathic helix in the N-terminus of poliovirus protein 2C. Virology 1994, 199, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Echeverri, A.C.; Dasgupta, A. Amino terminal regions of poliovirus 2C protein mediate membrane binding. Virology 1995, 208, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Van Kuppeveld, F.J.; Melchers, W.J.; Kirkegaard, K.; Doedens, J.R. Structure-function analysis of coxsackie B3 virus protein 2B. Virology 1997, 227, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.; Wileman, T. Inhibition of the secretory pathway by foot-and-mouth disease virus 2BC protein is reproduced by coexpression of 2B with 2C, and the site of inhibition is determined by the subcellular location of 2C. J. Virol. 2007, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Penin, F.; Moradpour, D. Hepatitis C virus nonstructural protein 4B: A journey into unexplored territory. Rev. Med. Virol. 2010, 20, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Roingeard, P.; Penin, F.; Moradpour, D. Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J. Virol. 2010, 84, 12529–12537. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Romero-Brey, I.; Gouttenoire, J.; Stoitsova, S.; Krijnse-Locker, J.; Moradpour, D.; Bartenschlager, R. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 2011, 85, 6963–6976. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; van, T.H.; Roos, N.; Pedersen, K.W. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J. Gen. Virol. 2001, 82, 985–994. [Google Scholar] [PubMed]
- Posthuma, C.C.; Pedersen, K.W.; Lu, Z.; Joosten, R.G.; Roos, N.; Zevenhoven-Dobbe, J.C.; Snijder, E.J. Formation of the arterivirus replication/transcription complex: A key role for nonstructural protein 3 in the remodeling of intracellular membranes. J. Virol. 2008, 82, 4480–4491. [Google Scholar] [CrossRef] [PubMed]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio 2013, 4. [Google Scholar] [CrossRef]
- Gadlage, M.J.; Sparks, J.S.; Beachboard, D.C.; Cox, R.G.; Doyle, J.D.; Stobart, C.C.; Denison, M.R. Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function. J. Virol. 2010, 84, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Cottam, E.M.; Maier, H.J.; Manifava, M.; Vaux, L.C.; Chandra-Schoenfelder, P.; Gerner, W.; Britton, P.; Ktistakis, N.T.; Wileman, T. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 2011, 7, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).