Immunogenetic Factors Affecting Susceptibility of Humans and Rodents to Hantaviruses and the Clinical Course of Hantaviral Disease in Humans


Abstract
:
1. Introduction
1.1. Immunogenetics and Diseases
1.2. Hantavirus Infection and Disease
1.3. Potential Applications
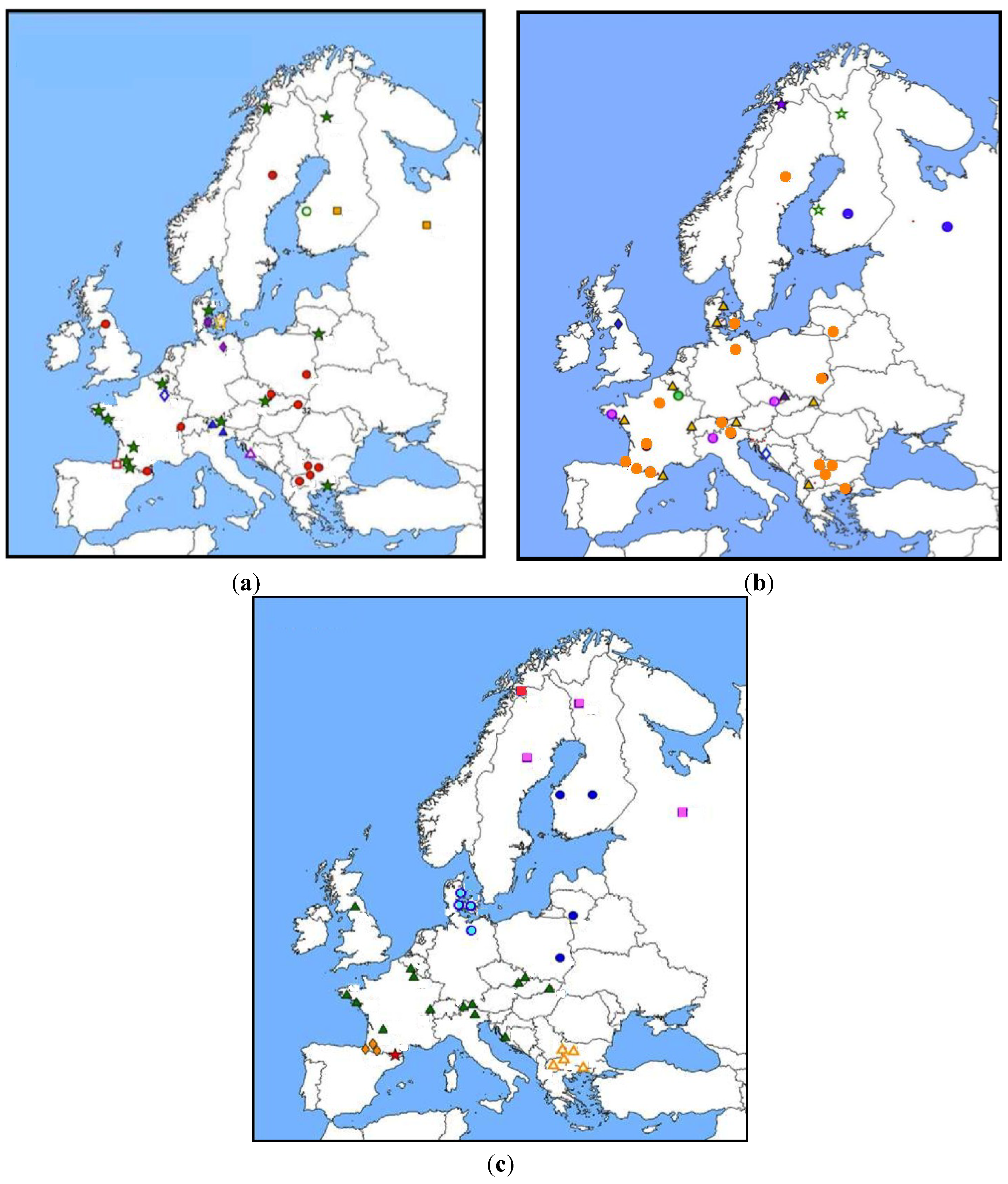
2. Impact of Genetic Factors in Human Hantavirus Infections
2.1. Sequence Polymorphism of Immunity-Related Genes and the Severity of Human Hantavirus Infections

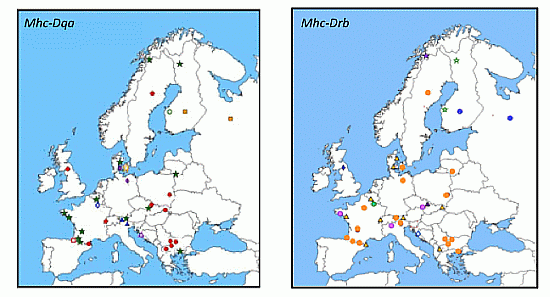
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Country | Haplotype | Expression | Hantavirus | Relevance with Disease Severity (−: Mild form; +: More Severe) | Relevance with Infection (P: Protective; R: Risk) | |
|---|---|---|---|---|---|---|---|
| HFRS | HLA | Finland | HLA-B*08 | PUUV | + | ||
| HLA-DRB1*0301 | PUUV | + | |||||
| HLA-B*27 | PUUV | − | |||||
| Slovenia | HLA-DRB1*15 | PUUV | + | ||||
| HLA-DRB1*13 | PUUV & DOBV | + (PUUV) | PUUV > DOBV | ||||
| HLA-B*35 | PUUV & DOBV | + (DOBV) | DOBV > PUUV | ||||
| HLA-B*07 | PUUV | P | |||||
| China | HLA-B*46 | HTNV | + | ||||
| HLA-B*46/DRB1*09 | HTNV | + | R | ||||
| HLA-B*51/DRB1*09 | HTNV | + | |||||
| HLA-DRB1*12 | HTNV | − | P | ||||
| TNF | Finland | −308 | PUUV | + | |||
| Belgium | −238 | PUUV | + | ||||
| C4A | Finland | Deletion | PUUV | + | |||
| HPA-3 | China | 3b | HTNV | + | R | ||
| PAI-1 | Finland | GG | PUUV | + | |||
| Gp1A | Finland | C | PUUV | + | |||
| VE-CDH5 | Russia | *T/*T | PUUV | + | |||
| GATA-3 | Finland | Higher | PUUV | + | |||
| IL1-RA | China | */* | HTNV | R | |||
| IL1-1b | China | −511 | HTNV | R | |||
| IL1 | Finland | x | PUUV | None | |||
| HPA-1 | China | x | HTNV | None | |||
| CD3e | Finland | x | PUUV | None | |||
| T-BET | Finland | x | PUUV | None | |||
| HCPS | HLA | US | HLA-B*3501 | SNV | + | ||
| HLA-DRB1*1402 | SNV | + | |||||
| HLA-B*35 | ANDV | − | |||||
| Chile | HLA-B*08 | ANDV | + | ||||
| HLA-DRB1*15 | ANDV | − | |||||
| TNF | Brazil | −308G/A | ARAV | + |
2.2. Variability in Immunity-Related Gene Expression and Severity of Human Hantavirus Infections
2.3. Polymorphism of Immunity-Related Genes and Human Susceptibility to Hantavirus Infections
3. Impact of Immunity-Related Genes on the Risk of Hantavirus Infection in Rodents
3.1. Kinetics of Immunity-Related Gene Expression During Hantavirus Infection in Rodents
3.1.1. Immunity-Related Gene Expression and Sex Differences in Hantavirus Infections
3.1.2. Immunity-Related Gene Expression and Persistence/Clearance of Hantavirus Infections
3.2. Immunogenetics and Rodent Susceptibility to Hantavirus Infections
3.2.1. Sequence Polymorphism of Immunity-Related Genes between Reservoir and Non-Reservoir Species and Their Association with Susceptibility to Hantavirus Infection
3.2.2. Sequence/Expression Variability of Immunity-Related Genes between Rodent Populations Sampled in Endemic and Non-Endemic Areas and Their Associations with Susceptibility to Hantavirus Infection
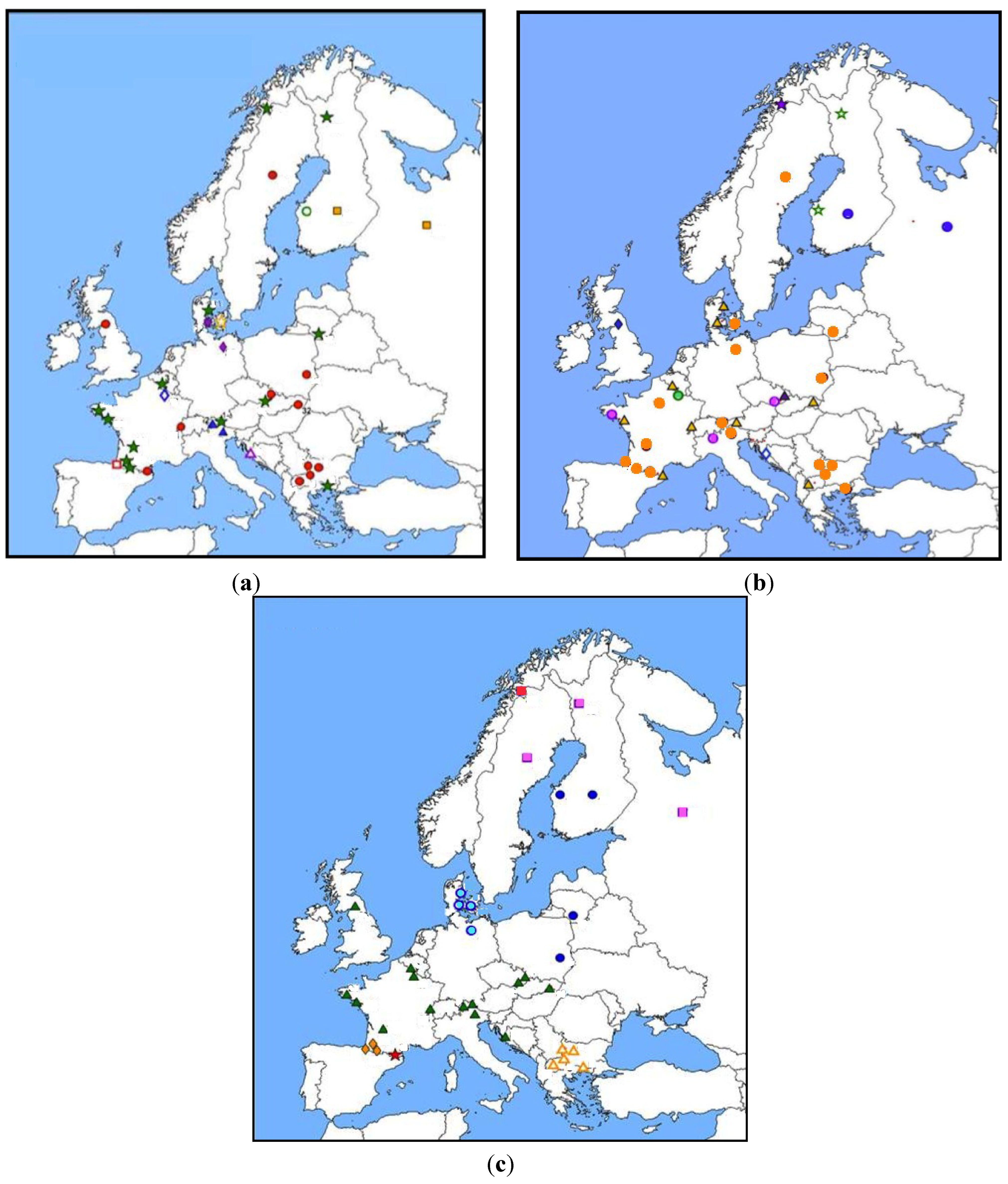
3.2.2.1. Mhc Class II Genes: Drb, Dqa
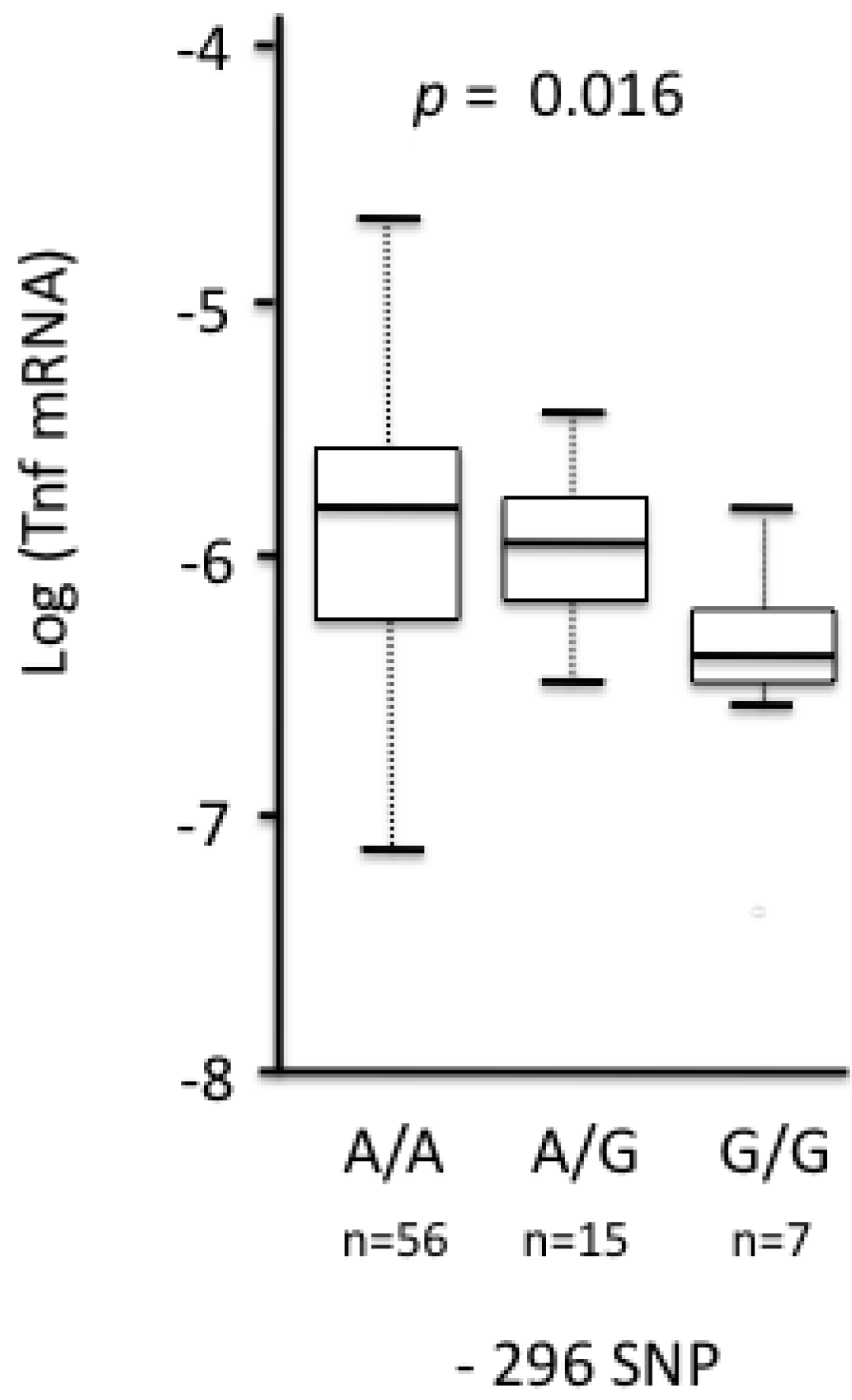
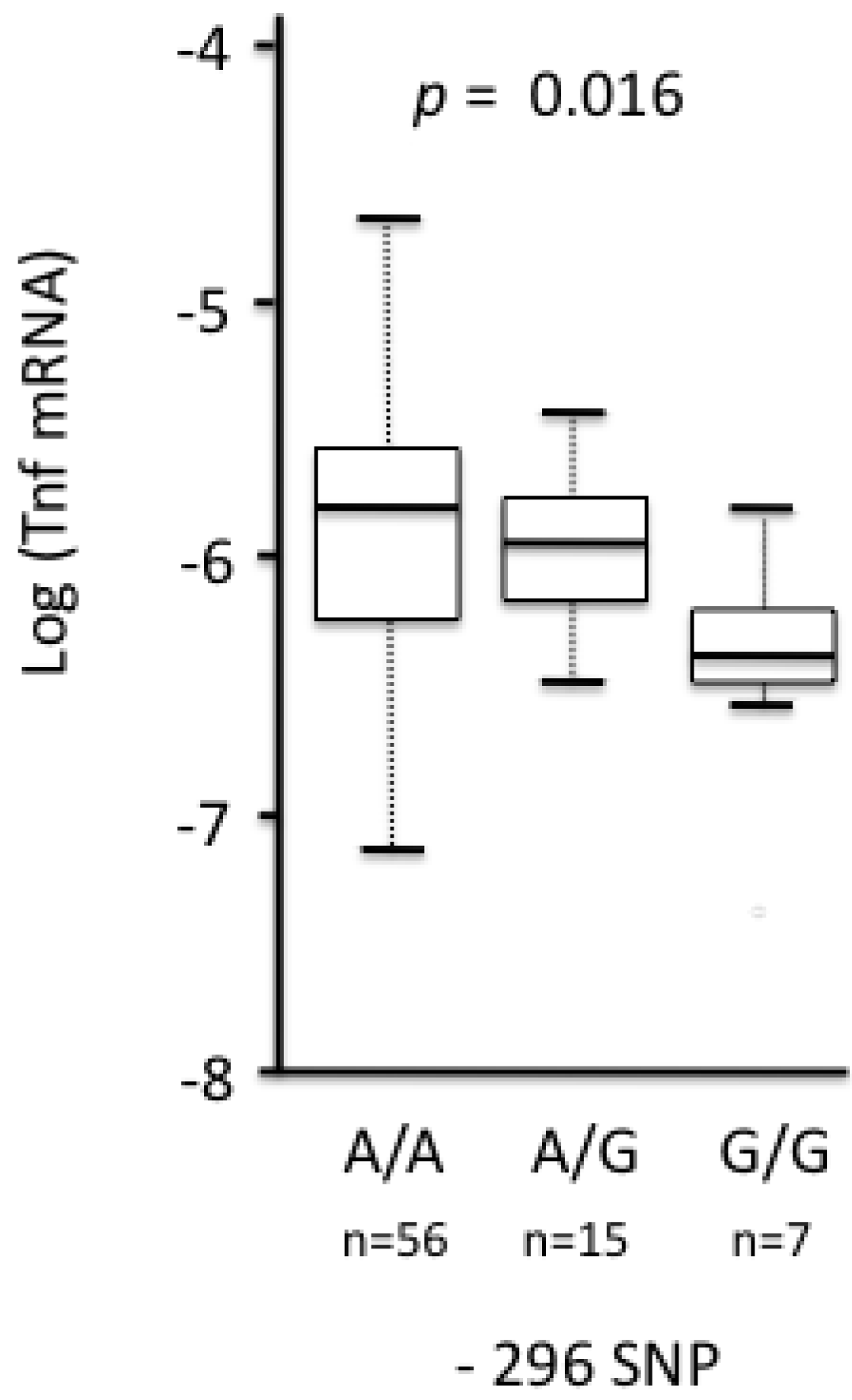
3.2.2.2. Tnf

3.2.2.3. Other Genes (Tlr4, Tlr7, Mx2, β3 Integrin)
4. Discussion: The Evolutionary Perspectives
4.1. Geographic Distribution of Susceptible Haplotypes and the Risk of Hantavirus Emergence
4.2. Evolution of Tolerance in Rodents and Its Epidemiological Consequences
4.3. Difficulties to Define What Is a Non-Reservoir Species for Hantaviruses
4.4. Differences in Hantavirus Virulence
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References and Notes
- Chapman, S.J.; Hill, A.V.S. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 2012, 13, 175–188. [Google Scholar]
- Geraghty, D.E.; Daza, R.; Williams, L.M.; Vu, Q.; Ishitani, A. Genetics of the immune response: Identifying immune variation within the mhc and throughout the genome. Immunol. Rev. 2002, 190, 69–85. [Google Scholar] [CrossRef]
- Cooke, G.S.; Hill, A.V.S. Genetics of susceptibility to human infectious disease. Nat. Rev. Genet. 2001, 2, 967–977. [Google Scholar] [CrossRef]
- Trowsdale, J.; Knight, J.C. Major histocompatibility complex genomics and human disease. Genom. Hum. Genet. 2013, 14, 301–323. [Google Scholar] [CrossRef]
- Do Valle, T.Z.; Billecocq, A.; Guillemot, L.; Alberts, R.; Gommet, C.; Geffers, R.; Calabrese, K.; Schughart, K.; Bouloy, M.; Montagutelli, X.; et al. A new mouse model reveals a critical role for host innate immunity in resistance to rift valley fever. J. Immunol. 2010, 185, 6146–6156. [Google Scholar] [CrossRef]
- Finlay, E.K.; Berry, D.P.; Wickham, B.; Gormley, E.P.; Bradley, D.G. A genome wide association scan of bovine tuberculosis susceptibility in holstein-friesian dairy cattle. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Acevedo-Whitehouse, K.; Cunningham, A.A. Is mhc enough for understanding wildlife immunogenetics? Trends Ecol. Evol. 2006, 21, 433–438. [Google Scholar] [CrossRef]
- Tschirren, B.; Andersson, M.; Scherman, K.; Westerdahl, H.; Raberg, L. Contrasting patterns of diversity and population differentiation at the innate immunity gene toll-like receptor 2 (TLR2) in two sympatric rodent species. Evolution 2012, 66, 720–731. [Google Scholar] [CrossRef]
- Turner, A.K.; Begon, M.; Jackson, J.A.; Paterson, S. Evidence for selection at cytokine loci in a natural population of field voles (Microtus agrestis). Mol. Ecol. 2012, 21, 1632–1646. [Google Scholar] [CrossRef]
- Tollenaere, C.; Duplantier, J.M.; Rahalison, L.; Ranjalahy, M.; Brouat, C. Aflp genome scan in the black rat (Rattus rattus) from Madagascar: Detecting genetic markers undergoing plague-mediated selection. Mol. Ecol. 2011, 20, 1026–1038. [Google Scholar] [CrossRef]
- Bonneaud, C.; Balenger, S.L.; Zhang, J.; Edwards, S.V.; Hill, G.E. Innate immunity and the evolution of resistance to an emerging infectious disease in a wild bird. Mol. Ecol. 2012, 21, 2628–2639. [Google Scholar] [CrossRef]
- Jensen, L.F.; Hansen, M.M.; Mensberg, K.L.; Loeschcke, V. Spatially and temporally fluctuating selection at non-mhc immune genes: Evidence from tap polymorphism in populations of brown trout (Salmo trutta, l.). Heredity 2008, 100, 79–91. [Google Scholar] [CrossRef]
- Tschirren, B.; Andersson, M.; Scherman, K.; Westerdahl, H.; Mittl, P.R.E.; Raberg, L. Polymorphisms at the innate immune receptor TLR2 are associated with borrelia infection in a wild rodent population. Proc. Roy. Soc. Lond. B 2013, 280. [Google Scholar] [CrossRef]
- Bradbury, J. Ancient footsteps in our genes: Evolution and human disease. Gene variants selected during evolution may underlie many common diseases. Lancet 2004, 363, 952–953. [Google Scholar] [CrossRef]
- Guivier, E.; Galan, M.; Henttonen, H.; Cosson, J.F.; Charbonnel, N. Landscape features and helminth co-infection shape bank vole immunoheterogeneity, with consequences for Puumala virus epidemiology. Heredity 2014, in press. [Google Scholar]
- Tollenaere, C.; Bryja, J.; Galan, M.; Cadet, P.; Deter, J.; Chaval, Y.; Berthier, K.; Ribas Salvador, A.; Voutilainen, L.; Laakkonen, J.; et al. Multiple parasites mediate balancing selection at mhc class ii genes: Insights from multivariate analyses and population genetics in the fossorial water vole. J. Evol. Biol. 2008, 21, 1307–1320. [Google Scholar] [CrossRef]
- Vasseur, E.; Quintana-Murci, L. The impact of natural selection on health and disease: Uses of the population genetics approach in humans. Evol. Appl. 2013, 6, 596–607. [Google Scholar] [CrossRef]
- Jonsson, C.B.; Figueiredo, L.T.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef]
- Schmaljohn, C.; Hjelle, B. Hantaviruses: A global disease problem. Emerg. Infect. Dis. 1997, 3, 95–104. [Google Scholar] [CrossRef]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef]
- Mäkelä, S.; Mustonen, J.; Ala-Houhala, I.; Hurme, M.; Partanen, J.; Vapalahti, O.; Vaheri, A.; Pasternack, A. Human leukocyte antigen-B8-DR3 is a more important risk factor for severe Puumala hantavirus infection than the tumor necrosis factor-alpha(-308) g/a polymorphism. J. Infect. Dis. 2002, 186, 843–846. [Google Scholar] [CrossRef]
- Lundkvist, A.; Plyusnin, A. Molecular epidemiology of hantavirus infections. In The Molecular Epidemiology of Human Viruses; Leitner, T., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 2002; pp. 351–384. [Google Scholar]
- Makary, P.; Kanerva, M.; Ollgren, J.; Virtanen, M.J.; Vapalahti, O.; Lyytikäinen, O. Disease burden of Puumala virus infections, 1995–2008. Epidemiol. Infect. 2010, 138, 1484–1492. [Google Scholar] [CrossRef]
- Childs, J.E.; Glass, G.E.; Korch, G.W.; LeDuc, J.W. Effects of hantaviral infection on survival, growth and fertility in wild rat (Rattus norvegicus) populations of Baltimore, Maryland. J. Wildl. Dis. 1989, 25, 469–476. [Google Scholar] [CrossRef]
- Meyer, B.J.; Schmaljohn, C.S. Persistent hantavirus infections: Characteristics and mechanisms. Trends Microbiol. 2000, 8, 61–67. [Google Scholar] [CrossRef]
- Kallio, E.R.; Voutilainen, L.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Koskela, E.; Mappes, T. Endemic hantavirus infection impairs the winter survival of its rodent host. Ecology 2007, 88, 1911–1916. [Google Scholar] [CrossRef]
- Tersago, K.; Crespin, L.; Verhagen, R.; Leirs, H. Impact of Puumala virus infection on maturation and survival in bank voles: A capture-mark-recapture analysis. J. Wildl. Dis. 2012, 48, 148–156. [Google Scholar] [CrossRef]
- Luis, A.D.; Douglass, R.J.; Hudson, P.J.; Mills, J.N.; Björnstad, O.N. Sin Nombre hantavirus decreases survival of male deer mice. Oecologia 2012, 169, 431–439. [Google Scholar] [CrossRef]
- Kallio, E.R.; Klingström, J.; Gustafsson, E.; Manni, T.; Vaheri, A.; Henttonen, H.; Vapalahti, O.; Lundkvist, A. Prolonged survival of Puumala hantavirus outside the host: Evidence for indirect transmission via the environment. J. Gen. Virol. 2006, 87, 2127–2134. [Google Scholar] [CrossRef]
- Hardestam, J.; Karlsson, M.; Falk, K.I.; Olsson, G.; Klingström, J.; Lundkvist, A. Puumala hantavirus excretion kinetics in bank voles (Myodes glareolus). Emerg. Inf. Dis. 2008, 14, 1209–1215. [Google Scholar]
- Schountz, T.; Shaw, T.I.; Glenn, T.C.; Feldmann, H.; Prescott, J. Expression profiling of lymph node cells from deer mice infected with Andes virus. BMC Immunol. 2013, 14, 18. [Google Scholar] [CrossRef]
- The Mhc sequencing consortium. Complete sequence and genemap of a human major histocompatibility complex. Nature 1999, 401, 921–923. [Google Scholar] [CrossRef]
- Klein, J. The Natural History of the Major Histocompatibility Complex; John Wiley and Sons: New York, NY, USA, 1986. [Google Scholar]
- Robinson, J.; Halliwell, J.A.; McWilliam, H.; Lopez, R.; Parham, P.; Marsh, S.G.E. The IMGT/HLA Database. Nucl. Acids Res. 2013, 41, D1222–D1227. [Google Scholar] [CrossRef]
- Bernatchez, L.; Landry, C. Mhc studies in nonmodel vertebrates: What have we learned about natural selection in 15 years. J. Evol. Biol. 2003, 16, 363–377. [Google Scholar] [CrossRef]
- Mustonen, J.; Partanen, J.; Kanerva, M.; Pietilä, K.; Vapalahti, O.; Pasternack, A.; Vaheri, A. Genetic susceptibility to severe course of nephropathia epidemica caused by Puumala hantavirus. Kidney Int. 1996, 49, 217–221. [Google Scholar] [CrossRef]
- Plyusnin, A.; Hörling, J.; Kanerva, M.; Mustonen, J.; Cheng, Y.; Partanen, J.; Vapalahti, O.; Kukkonen, S.K.; Niemimaa, J.; Henttonen, H.; et al. Puumala hantavirus genome in patients with nephropathia epidemica: Correlation of PCR positivity with HLA haplotype and link to viral sequences in local rodents. J. Clin. Microbiol. 1997, 35, 1090–1096. [Google Scholar]
- Mustonen, J.; Partanen, J.; Kanerva, M.; Pietilä, K.; Vapalahti, O.; Pasternack, A.; Vaheri, A. Association of HLA-B27 with benign clinical course of nephropathia epidemica caused by Puumala hantavirus. Scand. J. Immunol. 1998, 47, 277–279. [Google Scholar] [CrossRef]
- Korva, M.; Saksida, A.; Kunilo, S.; Jeras, B.V.; Avsic-Zupanc, T. HLA-associated hemorrhagic fever with renal syndrome disease progression in Slovenian patients. Clin. Vacc. Immunol. 2011, 18, 1435–1440. [Google Scholar] [CrossRef]
- Ma, Y.; Yuan, B.; Yi, J.; Zhuang, R.; Wang, J.; Zhang, Y.; Xu, Z.; Zhang, Y.; Liu, B.; Wei, C.; et al. The genetic polymorphisms of HLA are strongly correlated with the disease severity after Hantaan virus infection in the Chinese Han population. Clin. Dev. Immunol. 2012. [Google Scholar] [CrossRef]
- Koster, F.; Foucar, K.; Hjelle, B.; Scott, A.; Chong, Y.Y.; Larson, R.; McCabe, M. Rapid presumptive diagnosis of hantavirus cardiopulmonary syndrome by peripheral blood smear review. Am. J. Clin. Pathol. 2001, 116, 665–672. [Google Scholar] [CrossRef]
- Kilpatrick, E.D.; Terajima, M.; Koster, F.T.; Catalina, M.D.; Cruz, J.; Ennis, F.A. Role of specific CD8(+) T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 2004, 172, 3297–3304. [Google Scholar] [CrossRef]
- Terajima, M.; Ennis, F.A. T cells and pathogenesis of hantavirus cardiopulmonary syndrome and hemorrhagic fever with renal syndrome. Viruses 2011, 3, 1059–1073. [Google Scholar] [CrossRef]
- Manigold, T.; Mori, A.; Graumann, R.; Llop, E.; Simon, V.; Ferres, M.; Valdivieso, F.; Castillo, C.; Hjelle, B.; Vial, P. Highly differentiated, resting Gn-specific memory CD8 + T cells persist years after infection by Andes hantavirus. PLoS Pathog. 2010. [Google Scholar] [CrossRef]
- Ferrer, C.P.; Vial, C.P.A.; Ferres, G.M.; Godoy, M.P.; Cuiza, V.A.; Marco, C.C.; Castillo, H.C.; Umana, C.M.E.; Rothhammer, E.F.; Llop, R.E. Genetic susceptibility to Andes hantavirus: Association between severity of disease and HLA alleles in Chilean patients. Revist. Chilena Infect. 2007, 24, 351–359. [Google Scholar]
- Wang, M.L.; Lai, J.H.; Zhu, Y.; Zhang, H.B.; Li, C.; Wang, J.P.; Li, Y.M.; Yang, A.G.; Jin, B.Q. Genetic susceptibility to haemorrhagic fever with renal syndrome caused by Hantaan virus in Chinese Han population. Int. J. Immunogenet. 2009, 36, 227–229. [Google Scholar] [CrossRef]
- Candore, G.; Cigna, D.; Gervasi, F.; Colucci, A.T.; Modica, M.A.; Caruso, C. In vitro cytokine production by hla-b8,dr3 positive subjects. Autoimmunity 1994, 18, 121–132. [Google Scholar]
- Rudwaleit, M.; Siegert, S.; Yin, Z.; Eick, J.; Thiel, A.; Radbruch, A.; Sieper, J.; Braun, J. Low T cell production of TNF-alpha and IFN-gamma in ankylosing spondylitis: Its relation to HLA-B27 and influence of the TNF-308 gene polymorphism. Ann. Rheum. Dis. 2001, 60, 36–42. [Google Scholar]
- Kanerva, M.; Vaheri, A.; Mustonen, J.; Partanen, J. High-producer allele of tumour necrosis factor-alpha is part of the susceptibility MHC haplotype in severe Puumala virus-induced nephropathia epidemica. Scand. J. Inf. Dis. 1998, 30, 532–534. [Google Scholar] [CrossRef]
- Temonen, M.; Mustonen, J.; Helin, H.; Pasternack, A.; Vaheri, A.; Holthofer, H. Cytokines, adhesion molecules, and cellular infiltration in nephropathia epidemica kidneys: An immunohistochemical study. Clin. Immunol. Immunopath. 1996, 78, 47–55. [Google Scholar] [CrossRef]
- Maes, P.; Clement, J.; Groeneveld, P.H.P.; Colson, P.; Huizinga, T.W.J.; Van Ranst, M. Tumor necrosis factor-alpha genetic predisposing factors can influence clinical severity in nephropathia epidemica. Viral Immunol. 2006, 19, 558–564. [Google Scholar]
- Maes, P.; Clement, J.; Gavrilovskaya, I.; Van Ranst, M. Hantaviruses: Immunology, treatment, and prevention. Viral Immunol. 2004, 17, 481–497. [Google Scholar] [CrossRef]
- Borges, A.A.; Donadi, E.A.; Campos, G.M.; Moreli, M.L.; de Sousa, R.L.M.; Saggioro, F.P.; de Figueiredo, G.G.; Badra, S.J.; Deghaide, N.H.S.; Figueiredo, L.T.M. Association of-308G/A polymorphism in the tumor necrosis factor-alpha gene promoter with susceptibility to development of hantavirus cardiopulmonary syndrome in the Ribeiro Preto region, Brazil. Arch. Virol. 2010, 155, 971–975. [Google Scholar] [CrossRef]
- Sane, J.; Laine, O.; Mäkelä, S.; Paakkala, A.; Jarva, H.; Mustonen, J.; Vapalahti, O.; Meri, S.; Vaheri, A. Complement activation in Puumala hantavirus infection correlates with disease severity. Ann. Med. 2012, 44, 468–475. [Google Scholar] [CrossRef]
- Plyusnina, A.; Razzauti, M.; Sironen, T.; Niemimaa, J.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Plyusnin, A. Analysis of complete Puumala virus genome, Finland. Emerg. Inf. Dis. 2012, 18, 2070–2072. [Google Scholar] [CrossRef]
- Mäkelä, S.; Hurme, M.; Ala-Houhala, I.; Mustonen, J.; Koivisto, A.M.; Partanen, J.; Vapalahti, O.; Vaheri, A.; Pasternack, A. Polymorphism of the cytokine genes in hospitalized patients with Puumala hantavirus infection. Nephrol. Dial. Transpl. 2001, 16, 1368–1373. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, M.; Han, Q.; Lou, S.; Fang, J. Platelet glycoprotein iib/iiia (HPA-1 and HPA-3) polymorphisms in patients with hemorrhagic fever with renal syndrome. Hum. Immunol. 2009, 70, 452–456. [Google Scholar] [CrossRef]
- Laine, O.; Joutsi-Korhonen, L.; Mäkelä, S.; Mikkelsson, J.; Pessi, T.; Tuomisto, S.; Huhtala, H.; Libraty, D.; Vaheri, A.; Karhunen, P.; et al. Polymorphisms of PAI-1 and platelet GP-ia may associate with impairment of renal function and thrombocytopenia in Puumala hantavirus infection. Thromb. Res. 2012, 129, 611–615. [Google Scholar] [CrossRef]
- Baigil’dina, A.A.; Islamgulov, D.V. Genetic determining of the change in VE-cadherin expression and intensified vessel deendothelisation during hemorrhagic fever with renal syndrome. Mol. Genet. Microbiol. Virol. 2012, 27, 160–166. [Google Scholar] [CrossRef]
- Mäkelä, S.; Mustonen, J.; Ala-Houhala, I.; Hurme, M.; Koivisto, A.M.; Vaheri, A.; Pasternack, A. Urinary excretion of interleukin-6 correlates with proteinuria in acute Puumala hantavirus-induced nephritis. Am. J. Kidney Dis. 2004, 43, 809–816. [Google Scholar] [CrossRef]
- Libraty, D.H.; Mäkelä, S.; Vlk, J.; Hurme, M.; Vaheri, A.; Ennis, F.A.; Mustonen, J. The degree of leukocytosis and urine gata-3 mRNA levels are risk factors for severe acute kidney injury in Puumala virus nephropathia epidemica. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Outinen, T.K.; Mäkelä, S.M.; Ala-Houhala, I.O.; Huhtala, H.S.A.; Hurme, M.; Paakkala, A.S.; Porsti, I.H.; Syrjänen, J.T.; Mustonen, J.T. The severity of Puumala hantavirus induced nephropathia epidemica can be better evaluated using plasma interleukin-6 than C-reactive protein determinations. BMC Inf. Dis. 2010, 10. [Google Scholar] [CrossRef]
- Sadeghi, M.; Eckerle, I.; Daniel, V.; Burkhardt, U.; Opelz, G.; Schnitzler, P. Cytokine expression during early and late phase of acute Puumala hantavirus infection. BMC Immunol. 2011, 12. [Google Scholar] [CrossRef]
- Kyriakidis, I.; Papa, A. Serum TNF-alpha, STNFR1, IL-6, IL-8 and IL-10 levels in hemorrhagic fever with renal syndrome. Virus Res. 2013, 175, 91–94. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, M.; Han, Q.; Fang, J.; Zhao, Q.; Zhang, N. Intensity of platelet beta (3) integrin in patients with hemorrhagic fever with renal syndrome and its correlation with disease severity. Virus Immunol. 2008, 21, 255–261. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, B.; Yuan, B.; Wang, J.; Yu, H.; Zhang, Y.; Xu, Z.; Zhang, Y.; Yi, J.; Zhang, C.; et al. Sustained high level of serum VEGF at convalescent stage contributes to the renal recovery after HTNV infection in patients with hemorrhagic fever with renal syndrome. Clin. Dev. Immunol. 2012, 2012. [Google Scholar] [CrossRef]
- Kotlik, P.; Deffontaine, V.; Mascheretti, S.; Zima, J.; Michaux, J.R.; Searle, J.B. A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc. Nat. Acad. Sci. USA 2006, 103, 14860–14864. [Google Scholar] [CrossRef]
- Botten, J.; Mirowsky, K.; Kusewitt, D.; Bharadwaj, M.; Yee, J.; Ricci, R.; Feddersen, R.M.; Hjelle, B. Experimental infection model for Sin Nombre hantavirus in the deer mouse (Peromyscus maniculatus). Proc. Nat. Acad. Sci. USA 2000, 97, 10578–10583. [Google Scholar] [CrossRef]
- Voutilainen, L. Interactions between Puumala Hantavirus and Its Host, the Bank Vole, in the Boreal Zone. Ph.D. Thesis, Helsinki University, Helsinki, Filand, 2013. [Google Scholar]
- Bernshtein, A.D.; Apekina, N.S.; Mikhailova, T.V.; Myasnikov, Y.A.; Khlyap, L.A.; Korotkov, Y.S.; Gavrilovskaya, I.N. Dynamics of Puumala hantavirus infection in naturally infected bank voles (Clethrionomys glareolus). Arch. Virol. 1999, 144, 2415–2428. [Google Scholar] [CrossRef]
- Deter, J.; Chaval, Y.; Galan, M.; Gauffre, B.; Morand, S.; Henttonen, H.; Laakkonen, J.; Voutilainen, L.; Charbonnel, N.; Cosson, J.F. Kinship, dispersal and hantavirus transmission in bank and common voles. Arch. Virol. 2008, 153, 435–444. [Google Scholar] [CrossRef]
- Mills, J.N.; Ksiazek, T.G.; Ellis, B.A.; Rollin, P.E.; Nichol, S.T.; Yates, T.L.; Gannon, W.L.; Levy, C.E.; Engelthaler, D.M.; Davis, T.; et al. Patterns of association with host and habitat: Antibody reactive with Sin Nombre virus in small mammals in the major biotic communities of the southwestern united states. Am. J. Trop. Med. Hyg. 1997, 56, 273–284. [Google Scholar]
- Klein, S.L.; Cernetich, A.; Hilmer, S.; Hoffman, E.P.; Scott, A.L.; Glass, G.E. Differential expression of immunoregulatory genes in male and female norway rats following infection with Seoul virus. J. Med. Virol. 2004, 74, 180–190. [Google Scholar] [CrossRef]
- Hannah, M.F.; Bajic, V.B.; Klein, S.L. Sex differences in the recognition of and innate antiviral responses to seoul virus in norway rats. Brain Behav. Immun. 2008, 22, 503–516. [Google Scholar] [CrossRef]
- Botten, J.; Mirowsky, K.; Kusewitt, D.; Ye, C.Y.; Gottlieb, K.; Prescott, J.; Hjelle, B. Persistent sin nombre virus infection in the deer mouse (Peromyscus maniculatus) model: Sites of replication and strand-specific expression. J. Virol. 2003, 77, 1540–1550. [Google Scholar]
- Schountz, T.; Prescott, J.; Cogswell, A.C.; Oko, L.; Mirowsky-Garcia, K.; Galvez, A.P.; Hjelle, B. Regulatory T cell-like responses in deer mice persistently infected with Sin Nombre virus. Proc. Nat. Acad. Sci. USA 2007, 104, 15496–15501. [Google Scholar]
- Easterbrook, J.D.; Zink, M.C.; Klein, S.L. Regulatory T cells enhance persistence of the zoonotic pathogen Seoul virus in its reservoir host. Proc. Nat. Acad. Sci. USA 2007, 104, 15502–15507. [Google Scholar] [CrossRef]
- Easterbrook, J.D.; Klein, S.L. Seoul virus enhances regulatory and reduces proinflammatory responses in male norway rats. J. Med. Virol. 2008, 80, 1308–1318. [Google Scholar] [CrossRef]
- Schonrich, G.; Rang, A.; Lutteke, N.; Raftery, M.J.; Charbonnel, N.; Ulrich, R.G. Hantavirus-induced immunity in rodent reservoirs and humans. Immunol. Rev. 2008, 225, 163–189. [Google Scholar] [CrossRef]
- Klingström, J.; Heyman, P.; Escutenaire, S.; Sjölander, K.B.; De Jaegere, F.; Henttonen, H.; Lundkvist, A. Rodent host specificity of European hantaviruses: Evidence of Puumala virus interspecific spillover. J. Med. Virol. 2002, 68, 581–588. [Google Scholar] [CrossRef]
- Gavrilovskaya, I.N.; Brown, E.J.; Ginsberg, M.H.; Mackow, E.R. Cellular entry of hantaviruses which cause hemorrhagic fever with renal syndrome is mediated by beta3 integrins. J. Virol. 1999, 73, 3951–3959. [Google Scholar]
- Gavrilovskaya, I.N.; Peresleni, T.; Geimonen, E.; Mackow, E.R. Pathogenic hantaviruses selectively inhibit beta3 integrin directed endothelial cell migration. Arch. Virol. S 2002, 147, 1913–1931. [Google Scholar] [CrossRef]
- Gavrilovskaya, I.N.; Shepley, M.; Shaw, R.; Ginsberg, M.H.; Mackow, E.R. Beta3 integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc. Nat. Acad. Sci. USA 1998, 95, 7074–7079. [Google Scholar] [CrossRef]
- Raymond, T.; Gorbunova, E.; Gavrilovskaya, I.N.; Mackow, E.R. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc. Nat. Acad. Sci. USA 2005, 102, 1163–1168. [Google Scholar] [CrossRef]
- Mou, D.L.; Wang, Y.P.; Huang, C.X.; Li, G.Y.; Pan, L.; Yang, W.S.; Bai, X.F. Cellular entry of Hantaan virus A9 strain: Specific interactions with beta 3 integrins and a novel 70 kda protein. Biochem. Bioph. Res. Co. 2006, 339, 611–617. [Google Scholar] [CrossRef]
- Matthys, V.S.; Gorbunova, E.E.; Gavrilovskaya, I.N.; Mackow, E.R. Andes virus recognition of human and syrian hamster beta (3) integrins is determined by an l33p substitution in the PSI domain. J. Virol. 2010, 84, 352–360. [Google Scholar] [CrossRef]
- Krautkramer, E.; Zeier, M. Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55). J. Virol. 2008, 82, 4257–4264. [Google Scholar] [CrossRef]
- Choi, Y.; Kwon, Y.-C.; Kim, S.-I.; Park, J.-M.; Lee, K.-H.; Ahn, B.-Y. A hantavirus causing hemorrhagic fever with renal syndrome requires gc1qr/p32 for efficient cell binding and infection. Virology 2008, 381, 178–183. [Google Scholar] [CrossRef]
- Bagamian, K.H.; Towner, J.S.; Kuenzi, A.J.; Douglass, R.J.; Rollin, P.E.; Waller, L.A.; Mills, J.N. Transmission ecology of Sin Nombre hantavirus in naturally infected North American deer mouse populations in outdoor enclosures. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Lyubsky, S.; Gavrilovskaya, I.; Luft, B.; Mackow, E. Histopathology of Peromyscus leucopus naturally infected with pathogenic NY-1 hantaviruses: pathologic markers of HPS viral infection in mice. Lab. Invest. 1996, 74, 627–633. [Google Scholar]
- Netski, D.; Thran, B.H.; St Jeor, S.C. Sin Nombre virus pathogenesis in Peromyscus maniculatus. J. Virol. 1999, 73, 585–591. [Google Scholar]
- Guivier, E.; Galan, M.; Male, P.J.G.; Kallio, E.R.; Voutilainen, L.; Henttonen, H.; Olsson, G.E.; Lundkvist, A.; Tersago, K.; Augot, D.; et al. Associations between MHC genes and Puumala virus infection in Myodes glareolus are detected in wild populations, but not from experimental infection data. J. Gen. Virol. 2010, 91, 2507–2512. [Google Scholar] [CrossRef]
- Spurgin, L.G.; Richardson, D.S. How pathogens drive genetic diversity: Mhc, mechanisms and misunderstandings. Proc. Roy. Soc. Lond. B 2010, 277, 979–988. [Google Scholar] [CrossRef]
- Male, P.-J.G.; Martin, J.-F.; Galan, M.; Deffontaine, V.; Bryja, J.; Cosson, J.-F.; Michaux, J.; Charbonnel, N. Discongruence of MHC and cytochrome b phylogeographical patterns in Myodes glareolus (rodentia: Cricetidae). Biol. J. Linn. Soc. 2012, 105, 881–899. [Google Scholar] [CrossRef]
- Rohfritsch, A.; Guivier, E.; Galan, M.; Chaval, Y.; Charbonnel, N. Apport de l’immunogénétique à la compréhension des interactions entre le campagnol roussâtre Myodes glareolus et l’hantavirus Puumala. Bulletin de l'académie vétérinaire de France 2013, 166, 165–176. [Google Scholar]
- Dupanloup, I.; Schneider, S.; Excoffier, L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol. 2002, 11, 2571–2581. [Google Scholar] [CrossRef]
- Guivier, E. Variabilité de la résistance/tolérance des campagnols roussatres à l’hantavirus Puumala et conséquences épidémiologiques. Ph.D. Thesis, Université Montpellier 2, Montpellier, France, 2010. [Google Scholar]
- Deffontaine, V.; Libois, R.; Kotlik, P.; Sommer, R.; Nieberding, C.; Paradis, E.; Searle, J.B.; Michaux, J.R. Beyond the mediterranean peninsulas: Evidence of central european glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol. Ecol. 2005, 14, 1727–1739. [Google Scholar] [CrossRef]
- Nemirov, K.; Leirs, H.; Lundkvist, A.; Olsson, G.E. Puumala hantavirus and Myodes glareolus in northern Europe: No evidence of co-divergence between genetic lineages of virus and host. J. Gen. Virol. 2010, 91, 1262–1274. [Google Scholar] [CrossRef]
- Deter, J.; Chaval, Y.; Galan, M.; Henttonen, H.; Laakkonen, J.; Voutilainen, L.; Ribas Salvador, A.; Bryja, J.; Morand, S.; Cosson, J.F.; et al. Association between the DQA MHC class II gene and Puumala virus infection in the specific reservoir Myodes glareolus, the bank vole. Inf. Genet. Evol. 2008, 8, 450–458. [Google Scholar] [CrossRef]
- Ribas Salvador, A.; Guivier, E.; Xuereb, A.; Chaval, Y.; Cadet, P.; Poulle, M.L.; Sironen, T.; Voutilainen, L.; Henttonen, H.; Cosson, J.F.; et al. Concomitant influence of helminth infection and landscape on the distribution of Puumala hantavirus in its reservoir, Myodes glareolus. BMC Microbiol. 2011, 11. [Google Scholar] [CrossRef]
- Guivier, E.; Galan, M.; Ribas Salvador, A.; Xuéreb, A.; Chaval, Y.; Olsson, G.; Essbauer, S.; Henttonen, H.; Voutilainen, L.; Cosson, J.F.; et al. Tnf-α expression and promoter sequences reflect the balance of tolerance/resistance to Puumala virus infection in European bank vole populations. Inf. Genet. Evol. 2010, 10, 1208–1217. [Google Scholar] [CrossRef]
- Haldane, J.B.S. The estimation and significance of the logarithm of a ratio of frequencies. Ann. Hum. Genet. 1956, 20, 309–311. [Google Scholar] [CrossRef]
- Råberg, L.; Graham, A.L.; Read, A.F. Decomposing health: Tolerance and resistance to parasites in animals. Philos. Trans. Roy. Soc. Lond. B 2009, 364, 37–49. [Google Scholar] [CrossRef]
- Jin, H.K.; Yoshimatsu, K.; Takada, A.; Ogino, M.; Asano, A.; Arikawa, J.; Watanabe, T. Mouse Mx2 protein inhibits hantavirus but not influenza virus replication. Arch. Virol. 2001, 146, 41–49. [Google Scholar] [CrossRef]
- Li, Y.; Youssoufian, H. Mxa overexpression reveals a common genetic link among four fanconi anemia complementation groups. J. Clin. Investig. 1997, 100, 2873–2880. [Google Scholar]
- Porter, B.F.; Ambrus, A.; Storts, R.W. Immunohistochemical evaluation of Mx protein expression in canine encephalitides. Vet. Pathol. 2006, 43, 981–987. [Google Scholar] [CrossRef]
- Guivier, E.; Galan, M.; Chaval, Y.; Xuereb, A.; Ribas Salvador, A.; Poulle, M.L.; Charbonnel, N.; Cosson, J.F. Landscape genetics highlights the role of bank vole metapopulation dynamics in the epidemiology of Puumala hantavirus. Mol. Ecol. 2011, 20, 3569–3583. [Google Scholar]
- The International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature. 2010, 467, 52–58. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1092 human genomes. Nature. 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Patin, E.; Neyrolles, O. The heritage of pathogen pressures and ancient demography in the human innate-immunity CD209/CD209l region. Am. J. Hum. Genet. 2005, 77, 869–886. [Google Scholar] [CrossRef]
- Hedrick, P.W. Resistance to malaria in humans: The impact of strong, recent selection. Malaria J. 2012, 11. [Google Scholar] [CrossRef]
- Schountz, T.; Acuna-Retamar, M.; Feinstein, S.; Prescott, J.; Torres-Perez, F.; Podell, B.; Peters, S.; Ye, C.; Black, W.C.; Hjelle, B. Kinetics of immune responses in deer mice experimentally infected with Sin Nombre virus. J. Virol. 2012, 86, 10015–10027. [Google Scholar] [CrossRef]
- Schneider, D.S.; Ayres, J.S. Two ways to survive infection: What resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 2008, 8, 889–895. [Google Scholar] [CrossRef]
- Lloyd-Smith, J.O.; Schreiber, S.J.; Kopp, P.E.; Getz, W.M. Superspreading and the effect of individual variation on disease emergence. Nature 2005, 438, 355–359. [Google Scholar] [CrossRef]
- Stein, R.A. Super-spreaders in infectious diseases. Int. J. Inf. Dis. 2011, 15, E510–E513. [Google Scholar] [CrossRef]
- Klempa, B.; Witkowski, P.T.; Popugaeva, E.; Auste, B.; Koivogui, L.; Fichet-Calvet, E.; Strecker, T.; ter Meulen, J.; Krueger, D.H. Sangassou virus, the first hantavirus isolate from Africa, displays genetic and functional properties distinct from those of other murinae-associated hantaviruse. J. Virol. 2012, 86, 3819–3827. [Google Scholar] [CrossRef]
- Milazzo, M.L.; Eyzaguirre, E.J.; Molina, C.P.; Fulhorst, C.F. Maporal viral infection in the syrian golden hamster: A model of hantavirus pulmonary syndrome. J. Infect. Dis. 2002, 186, 1390–1395. [Google Scholar] [CrossRef]
- Safronetz, D.; Ebihara, H.; Feldmann, H.; Hooper, J.W. The syrian hamster model of hantavirus pulmonary syndrome. Antivir. Res. 2012, 95, 282–292. [Google Scholar]
- Klempa, B.; Avsic-Zupanc, T.; Clement, J.; Dzagurova, T.K.; Henttonen, H.; Heyman, P.; Jakab, F.; Kruger, D.H.; Maes, P.; Papa, A.; et al. Complex evolution and epidemiology of Dobrava-Belgrade hantavirus: Definition of genotypes and their characteristics. Arch. Virol. 2013, 158, 521–529. [Google Scholar] [CrossRef]
- Mackow, E.R.; Gavrilovskaya, I.N. Cellular receptors and hantavirus pathogenesis. Hantaviruses 2001, 256, 91–115. [Google Scholar]
- Schultze, D.; Lundkvist, Å.; Blauenstein, U.; Heyman, P. Tula virus infection associated with fever and exanthema after a wild rodent bite. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 304–306. [Google Scholar] [CrossRef]
- Klempa, B.; Koivogui, L.; Sylla, O.; Koulemou, K.; Auste, B.; Ulrich, K.; Kruger, D.H.; Meulen, J. Serological evidence of human hantavirus infections in Guinea, West Africa. J. Inf. Dis. 2010, 201, 1031–1034. [Google Scholar] [CrossRef]
- Zelená, H.; Mrázek, J.; Kuhn, T. Tula hantavirus infection in immunocompromised host, Czech Republic. Emerg. Infect. Dis. 2013, 19, 1873–1876. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Charbonnel, N.; Pagès, M.; Sironen, T.; Henttonen, H.; Vapalahti, O.; Mustonen, J.; Vaheri, A. Immunogenetic Factors Affecting Susceptibility of Humans and Rodents to Hantaviruses and the Clinical Course of Hantaviral Disease in Humans. Viruses 2014, 6, 2214-2241. https://doi.org/10.3390/v6052214
Charbonnel N, Pagès M, Sironen T, Henttonen H, Vapalahti O, Mustonen J, Vaheri A. Immunogenetic Factors Affecting Susceptibility of Humans and Rodents to Hantaviruses and the Clinical Course of Hantaviral Disease in Humans. Viruses. 2014; 6(5):2214-2241. https://doi.org/10.3390/v6052214
Chicago/Turabian StyleCharbonnel, Nathalie, Marie Pagès, Tarja Sironen, Heikki Henttonen, Olli Vapalahti, Jukka Mustonen, and Antti Vaheri. 2014. "Immunogenetic Factors Affecting Susceptibility of Humans and Rodents to Hantaviruses and the Clinical Course of Hantaviral Disease in Humans" Viruses 6, no. 5: 2214-2241. https://doi.org/10.3390/v6052214
APA StyleCharbonnel, N., Pagès, M., Sironen, T., Henttonen, H., Vapalahti, O., Mustonen, J., & Vaheri, A. (2014). Immunogenetic Factors Affecting Susceptibility of Humans and Rodents to Hantaviruses and the Clinical Course of Hantaviral Disease in Humans. Viruses, 6(5), 2214-2241. https://doi.org/10.3390/v6052214