Human Papillomavirus Species-Specific Interaction with the Basement Membrane-Resident Non-Heparan Sulfate Receptor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids and Pseudovirions (PsVs)
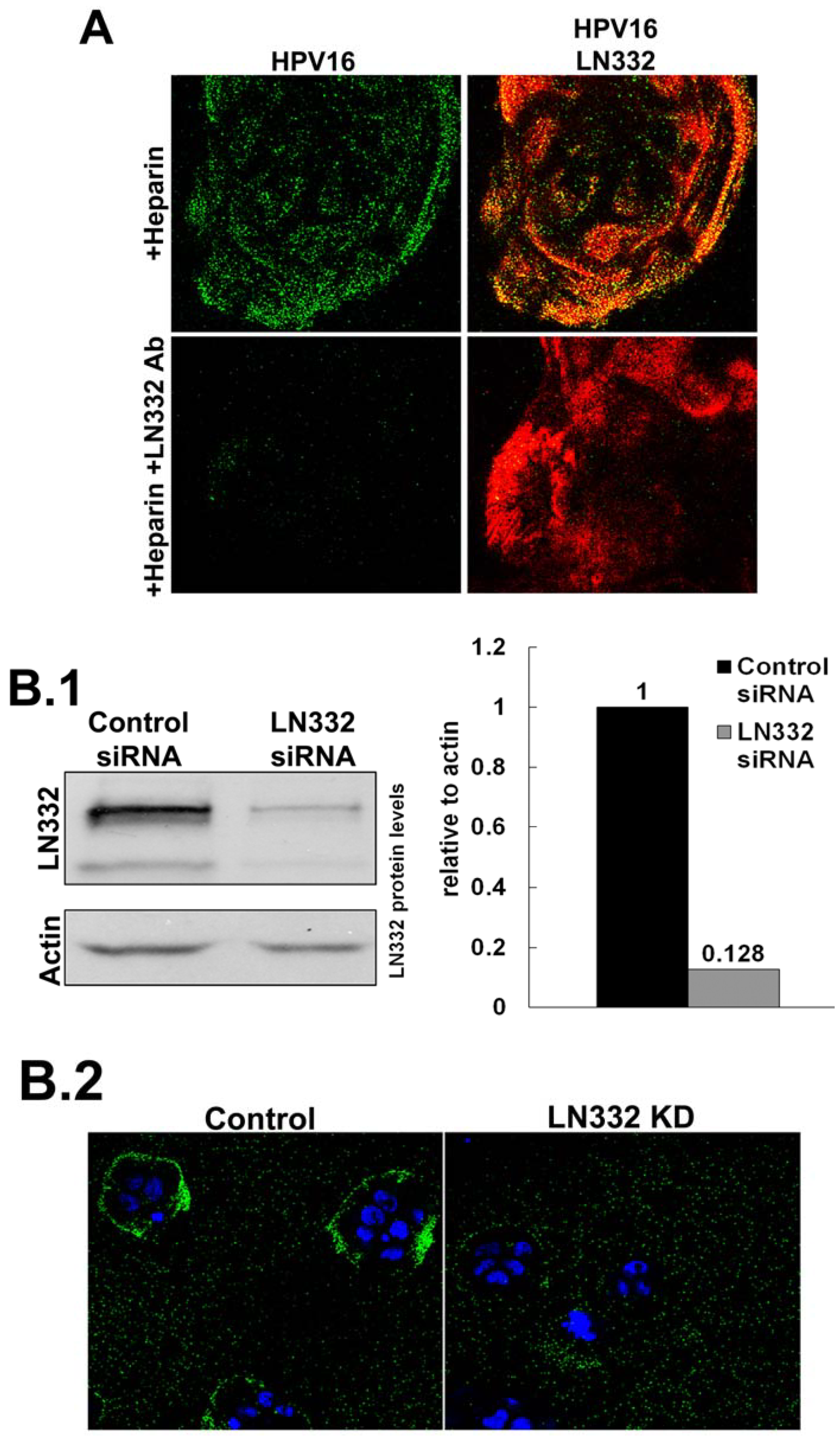
2.3. siRNA Knockdown of LN332
2.4. Antibodies, Inhibitors and Reagents
2.5. RNA Isolation and Quantification
2.6. ECM Cell Transfer Infection Assay
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Immunofluorescence (IF)
3. Results
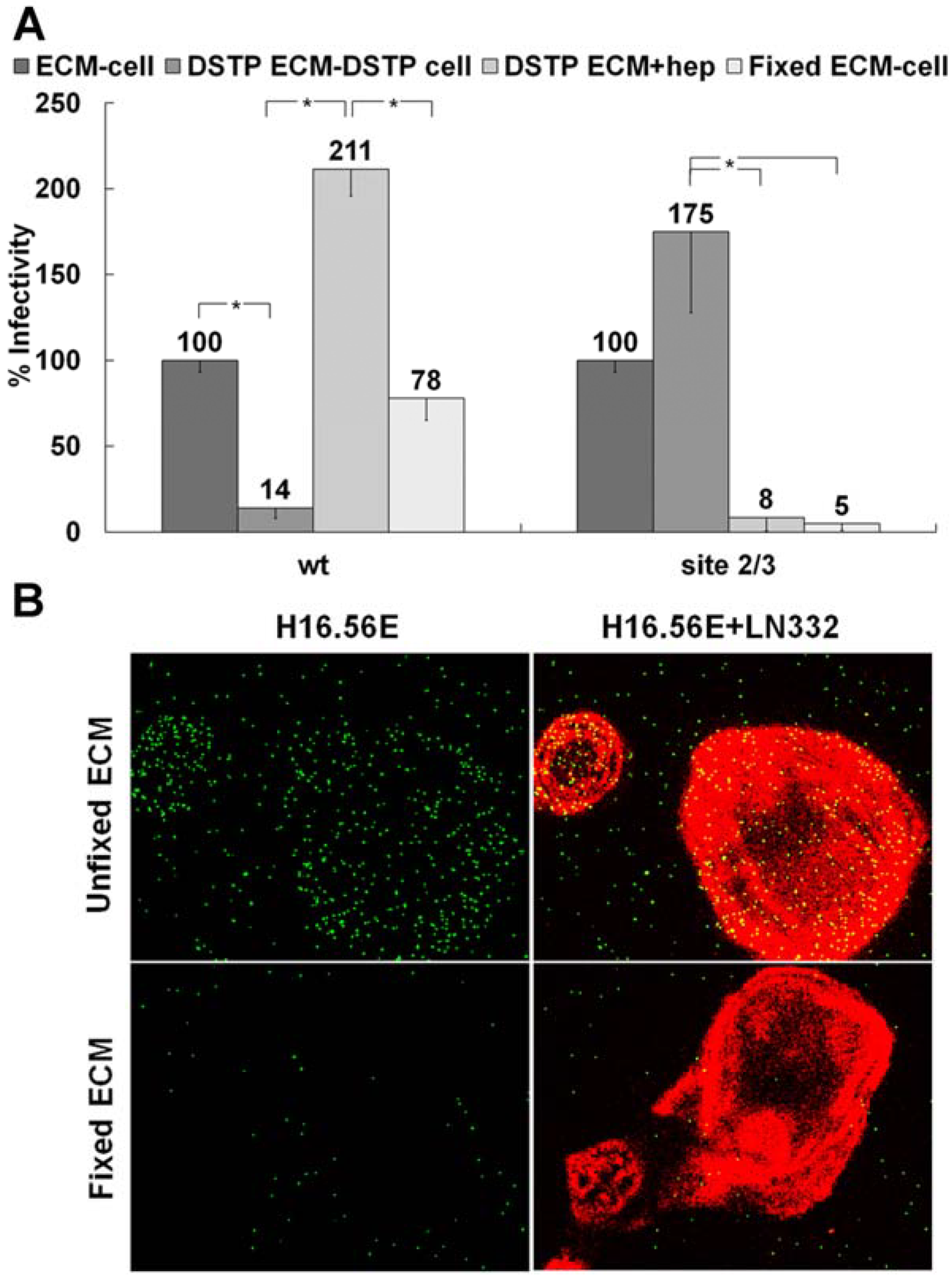
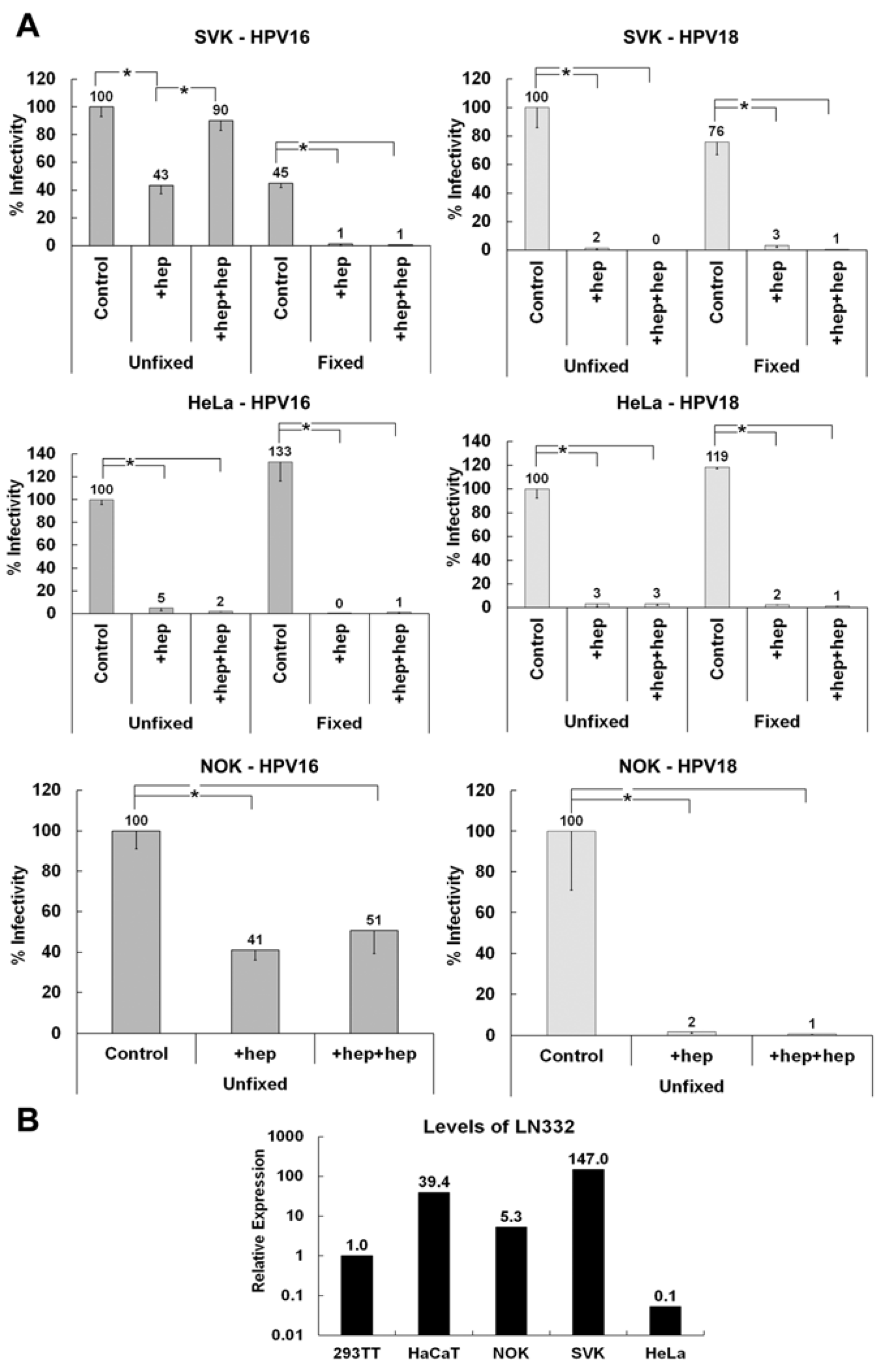
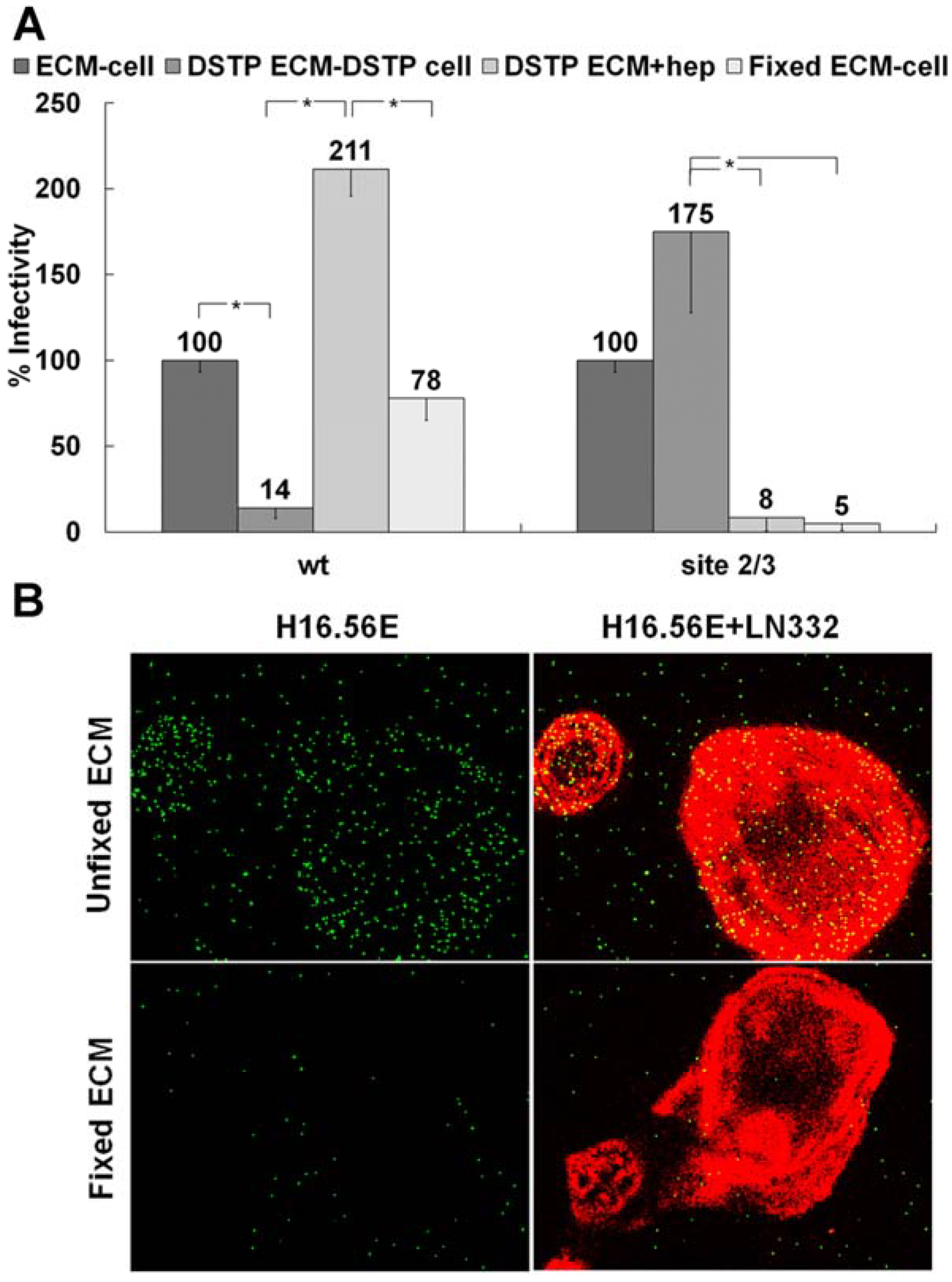
3.1. Presence of an ECM Receptor that Supports Infectious Transfer of HPV from the ECM to the Cell Surface
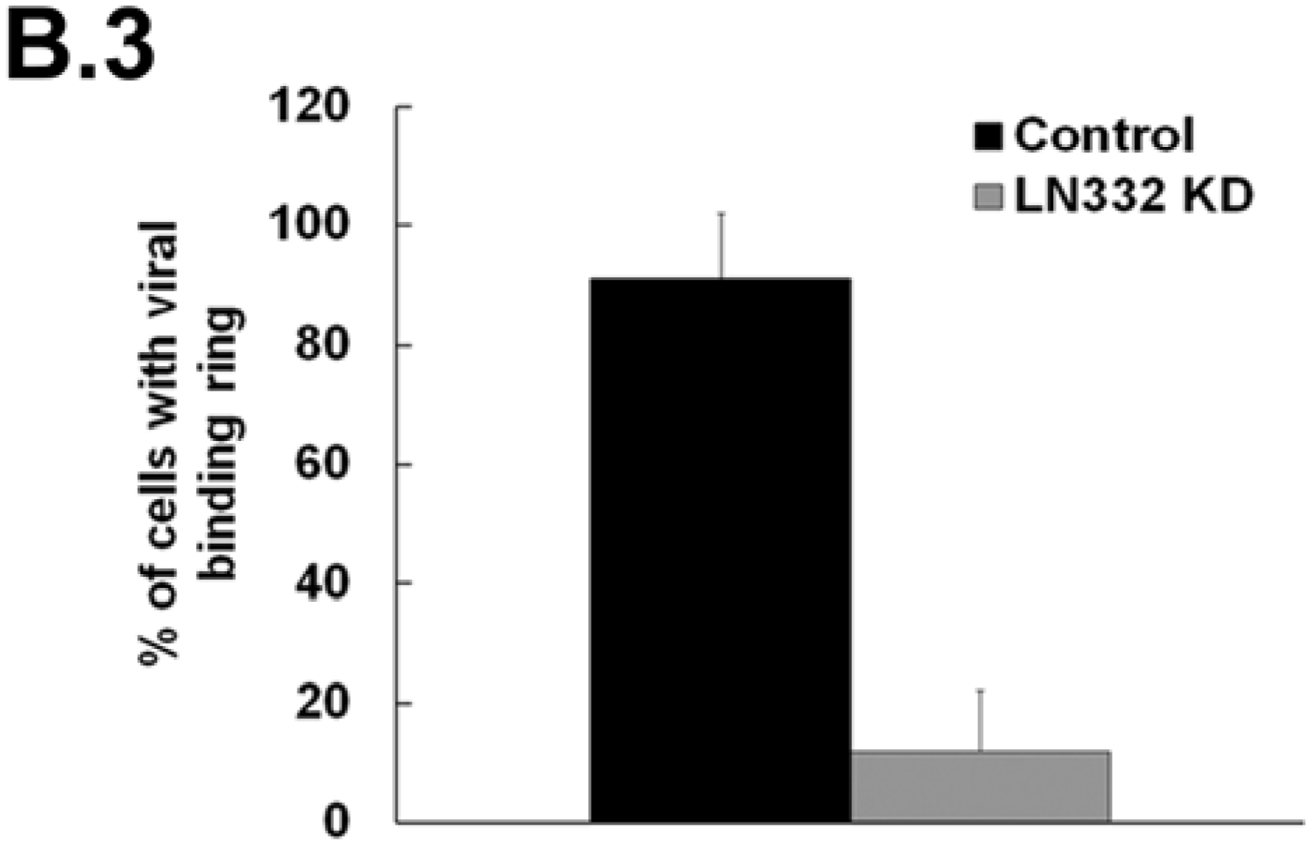
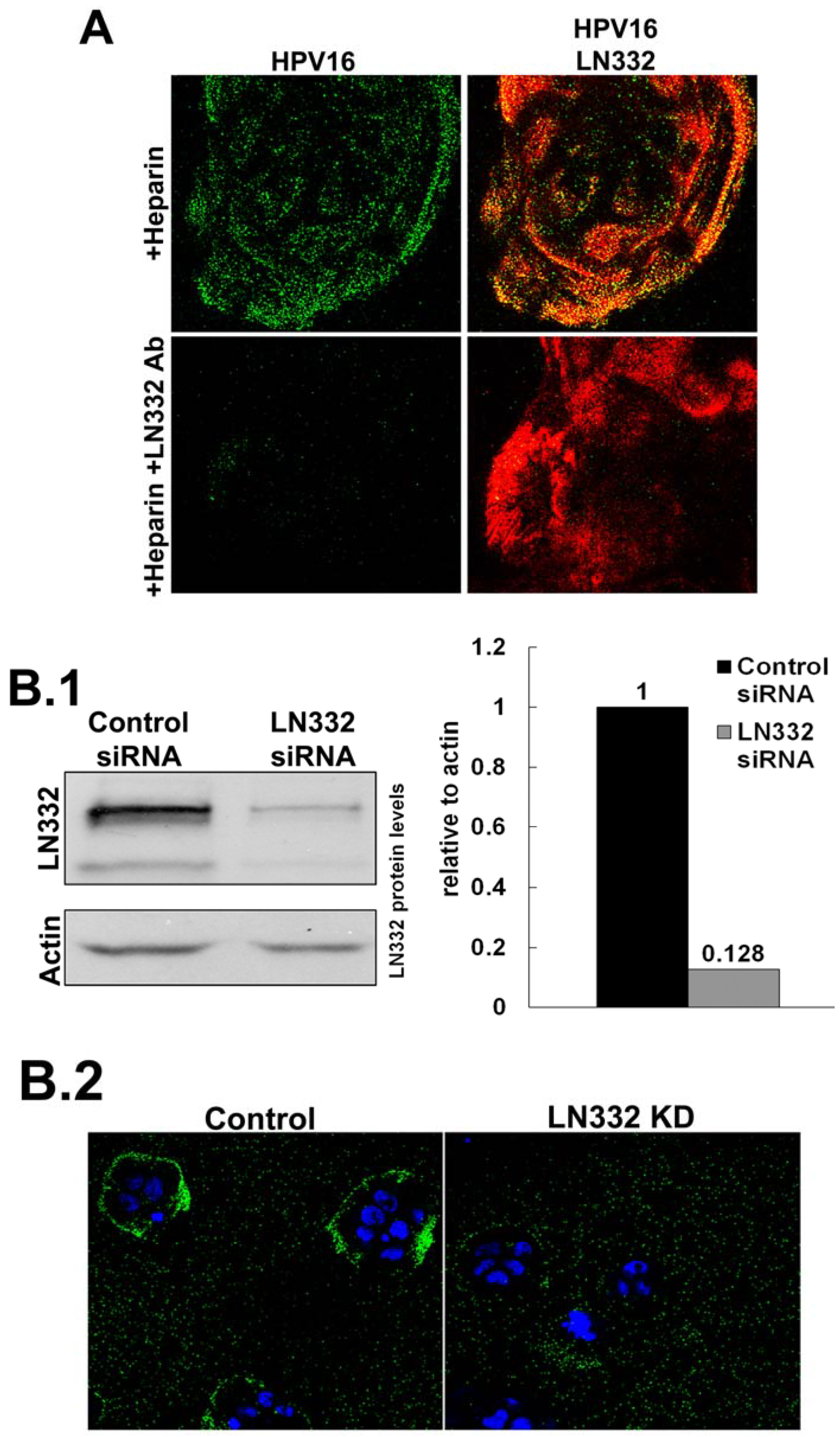
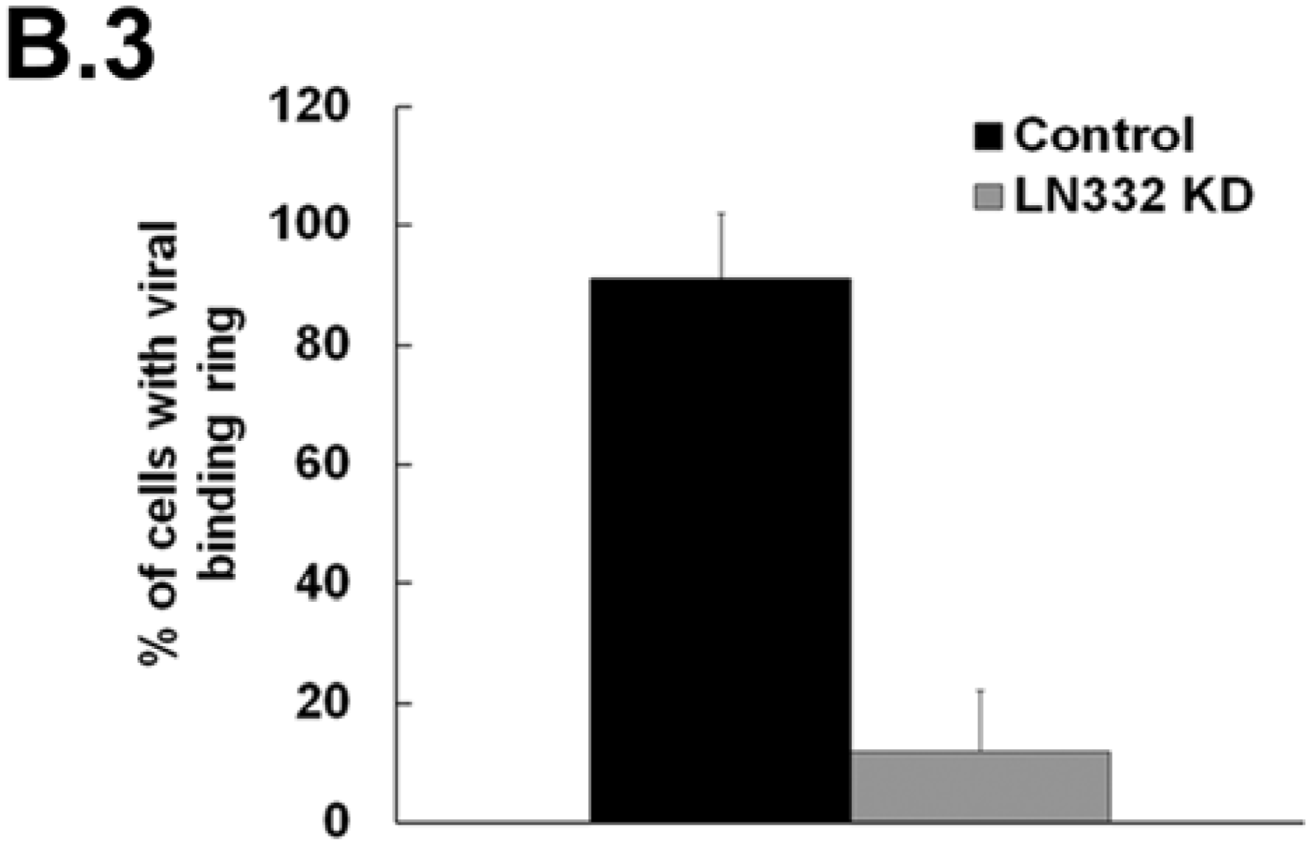
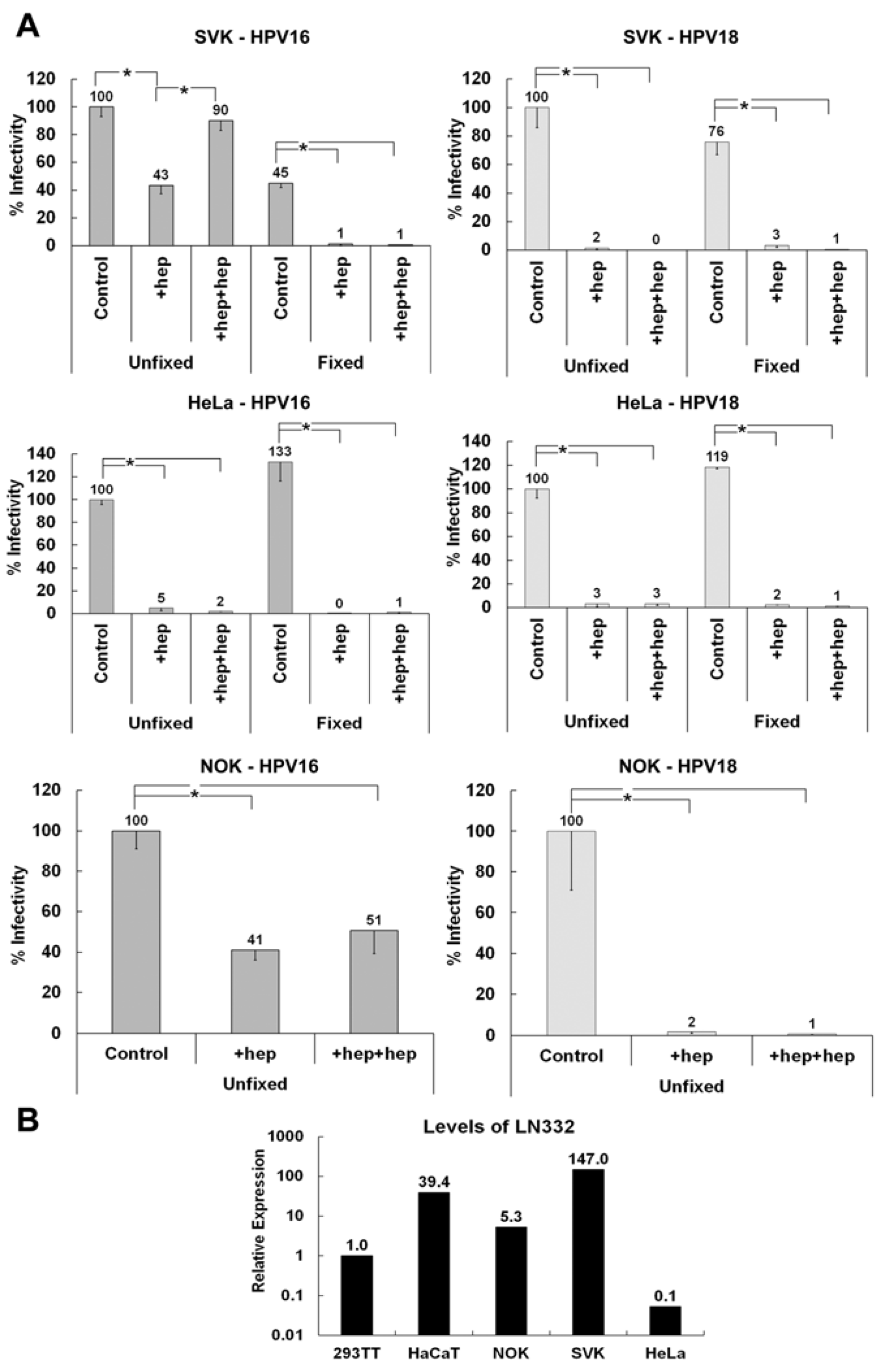
3.2. LN332 is Required for Infectious Transfer of HPV16 in the Presence of Heparin
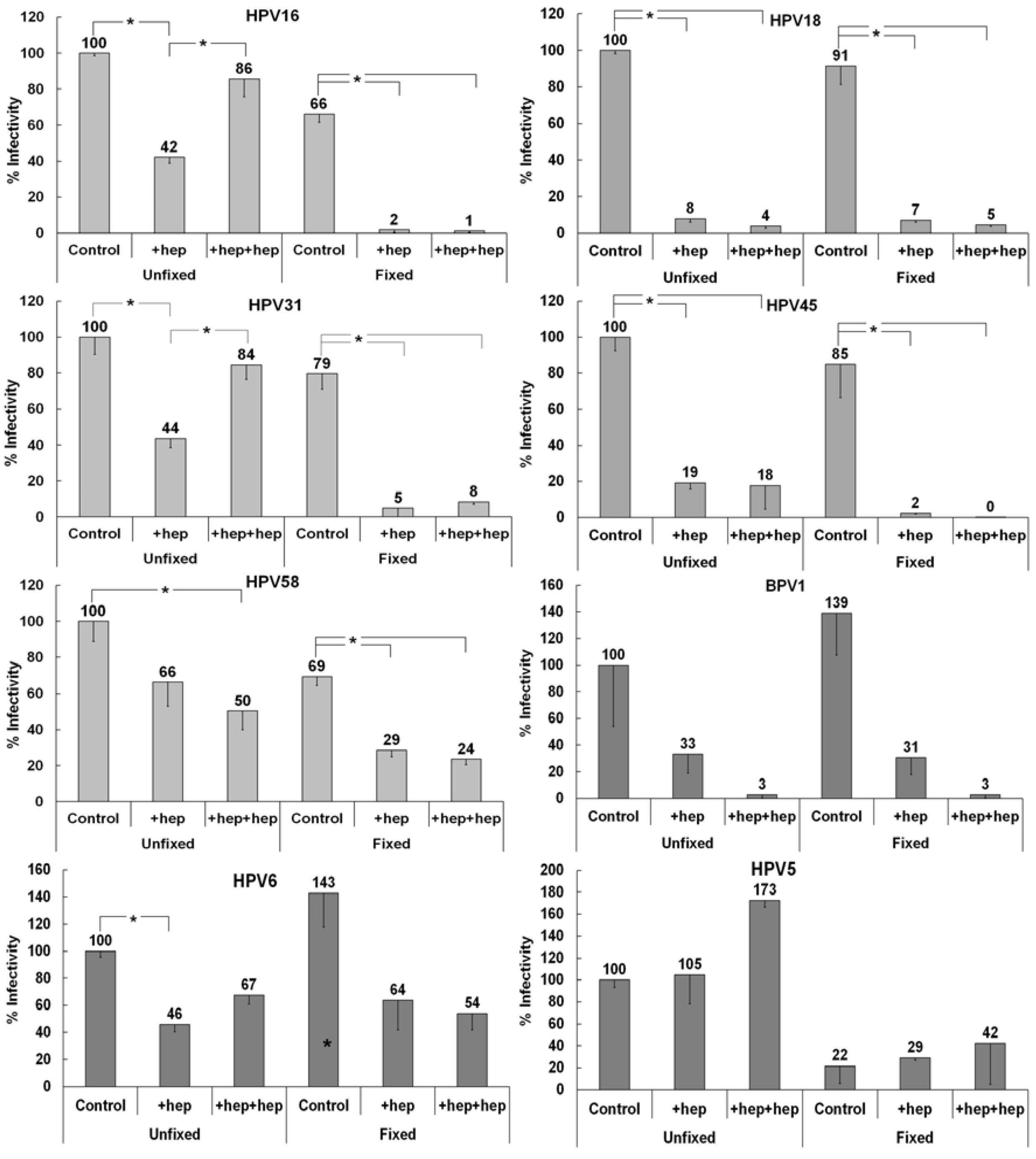
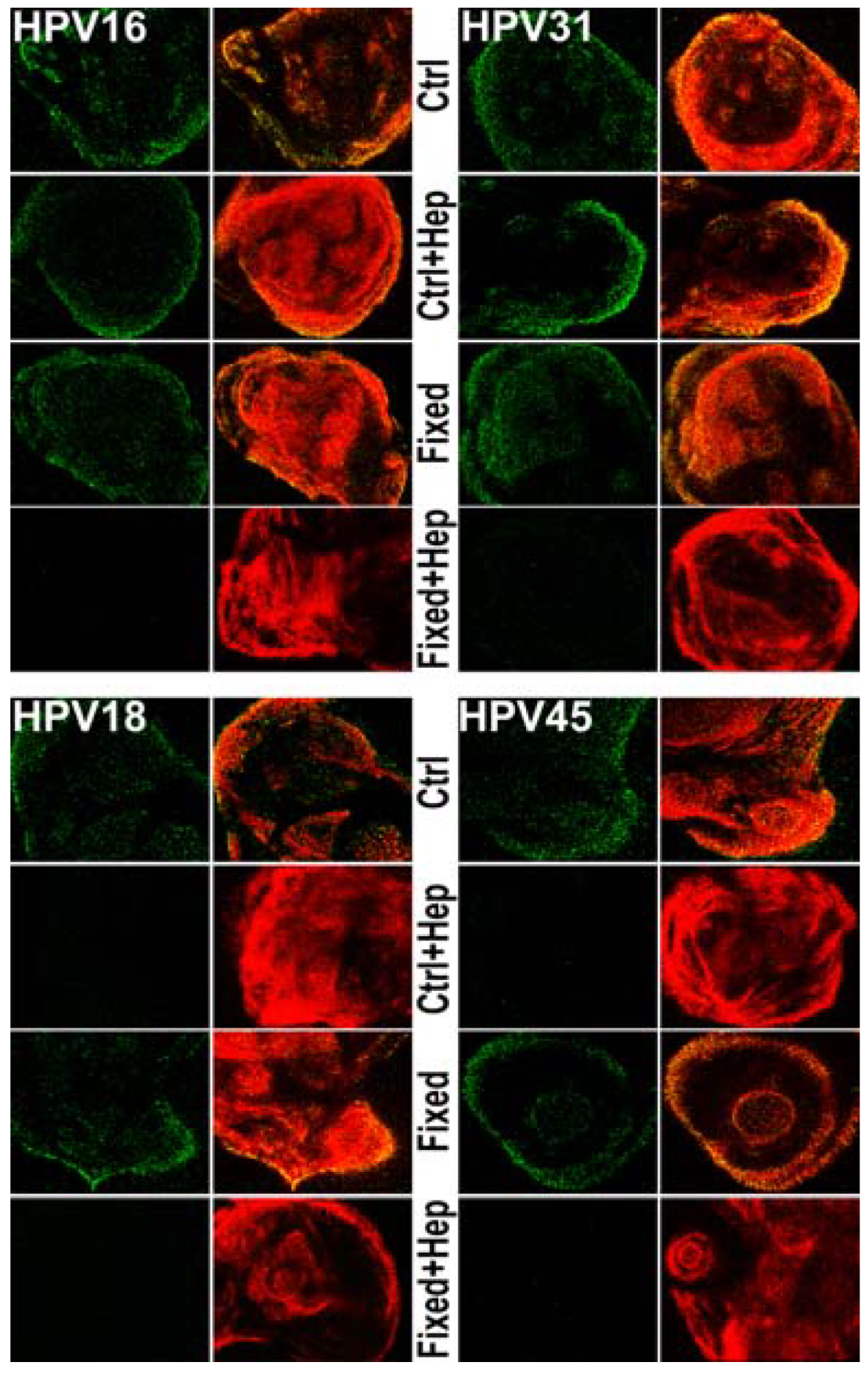
3.3. Investigating the Ability of Multiple HPV Types to Use a Non-HS Receptor in the ECM
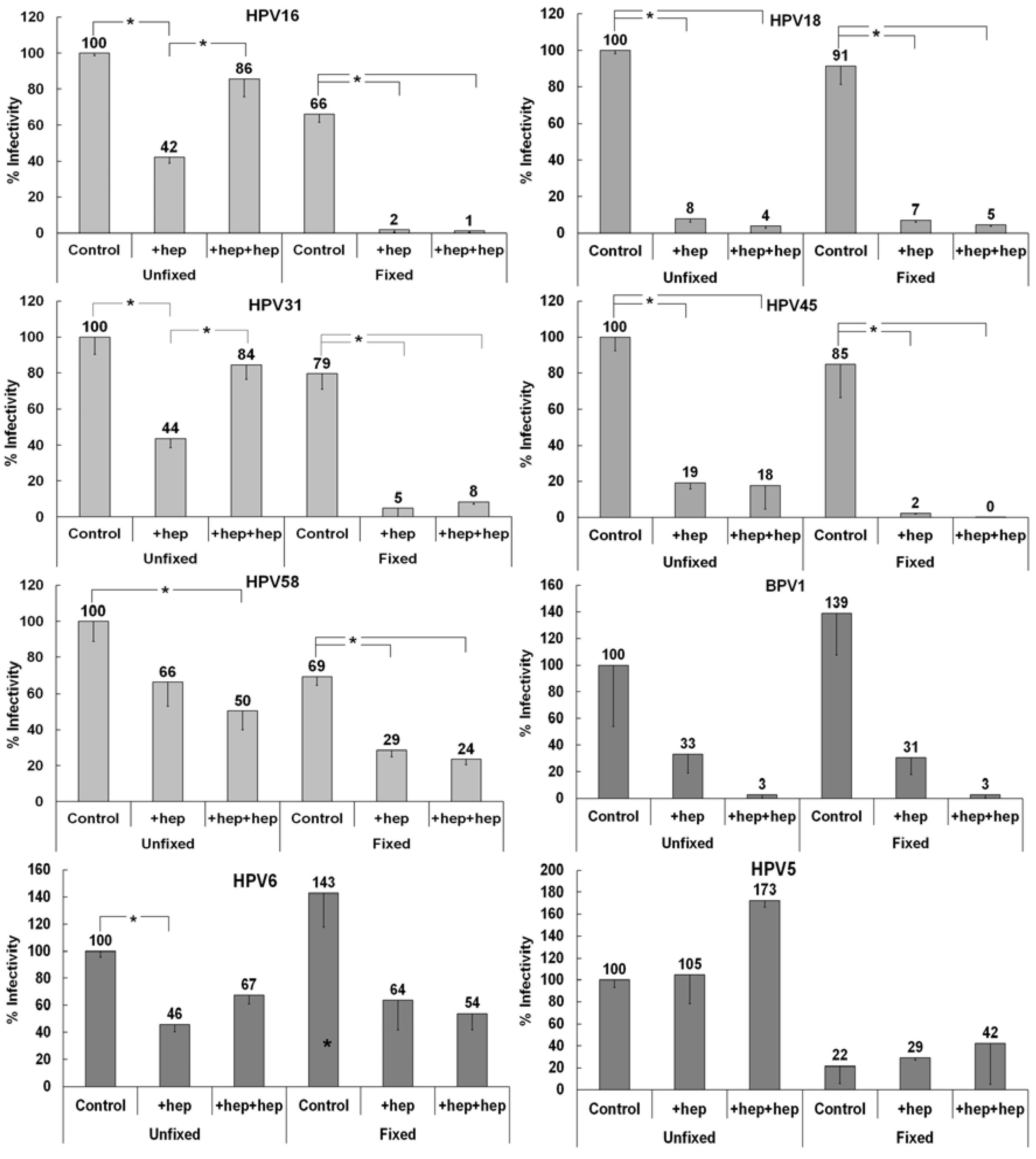
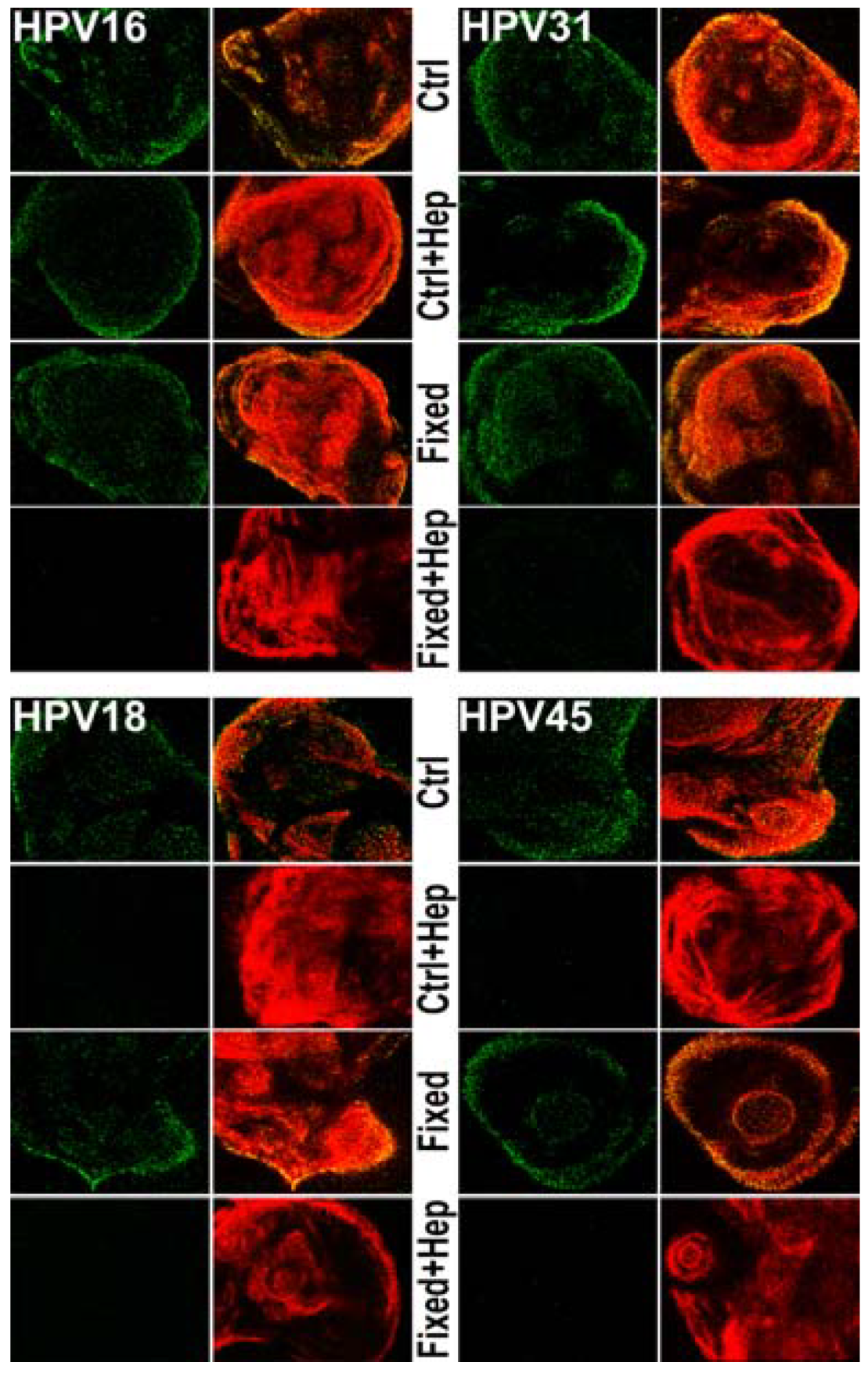
3.4. Monitoring ECM Binding Patterns for Different HPV Types
3.5. Determining if the Non-HS ECM Receptor is Found in the Secreted ECM of Various Keratinocytes
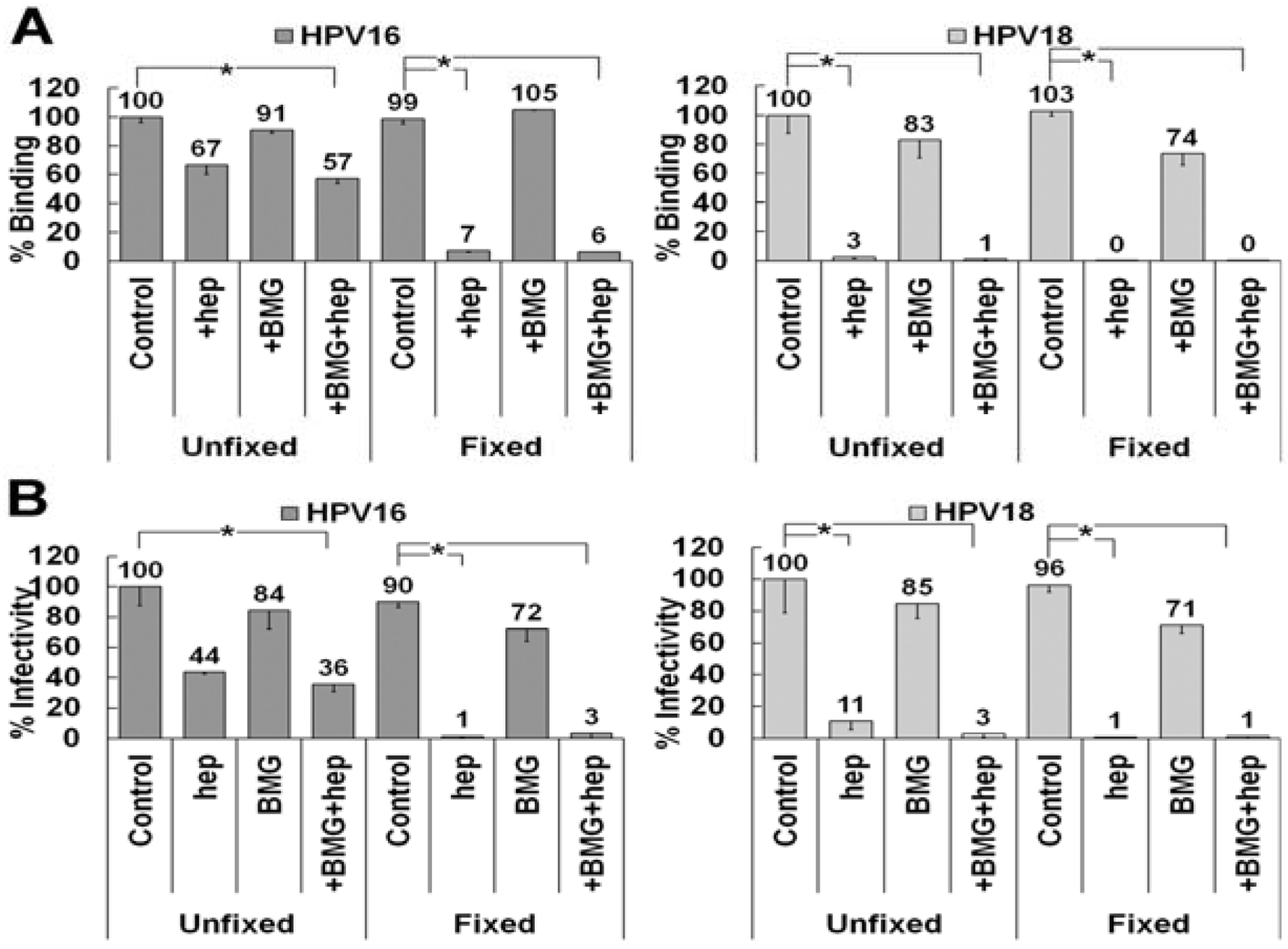
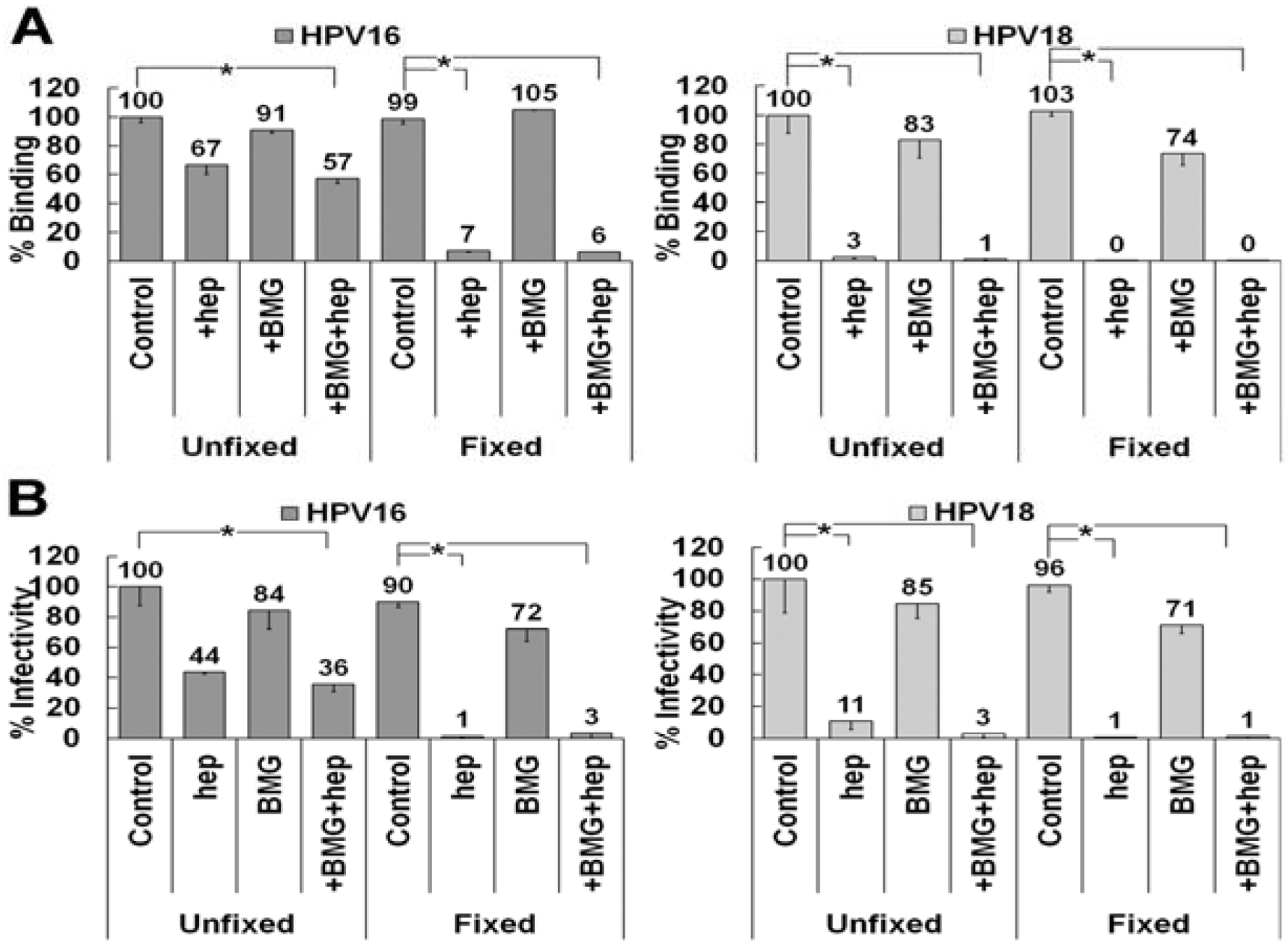
3.6. Matrix Metalloproteinase Processing of LN332 Does Not Affect HPV18 Dependence on HS
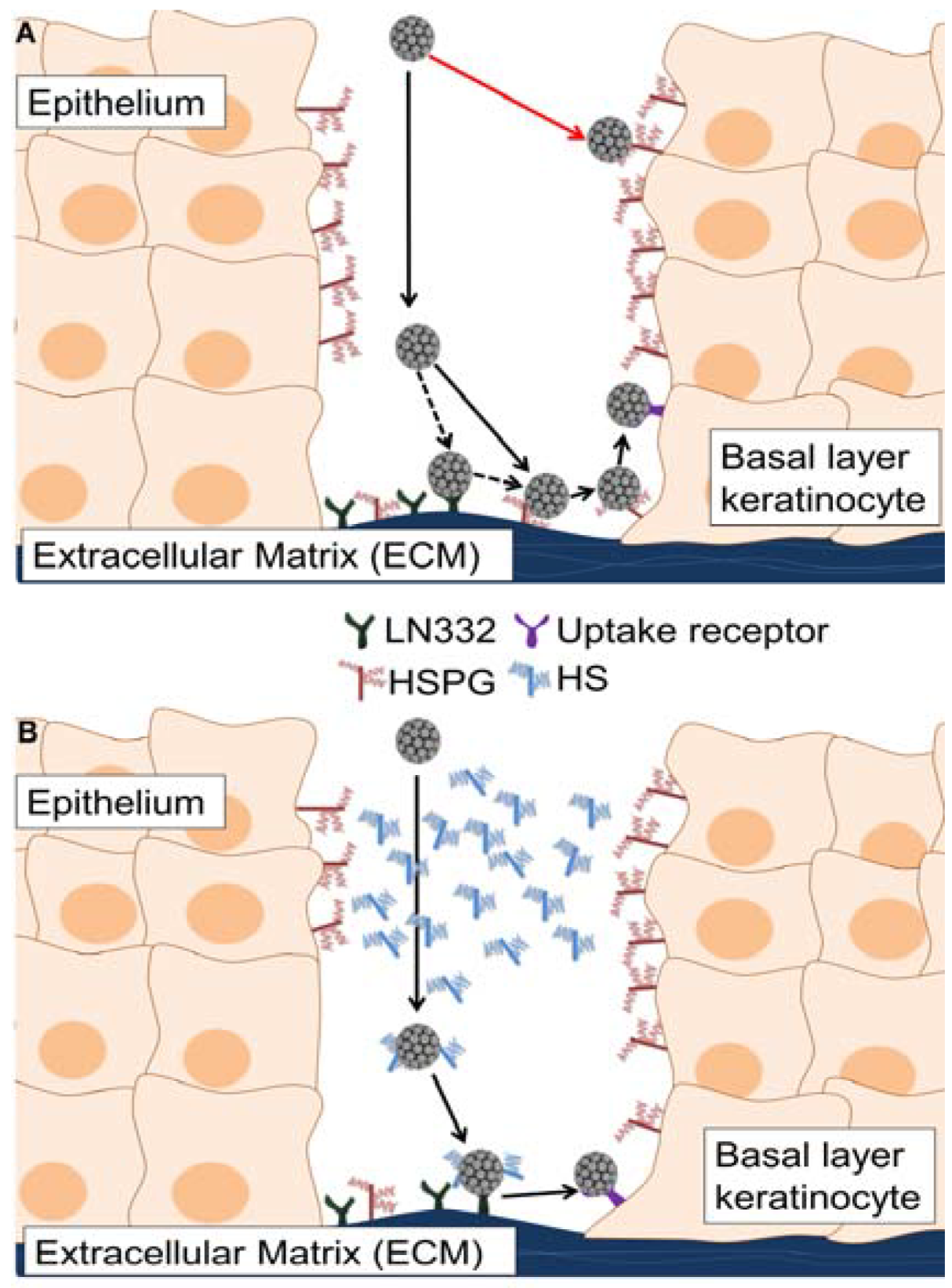
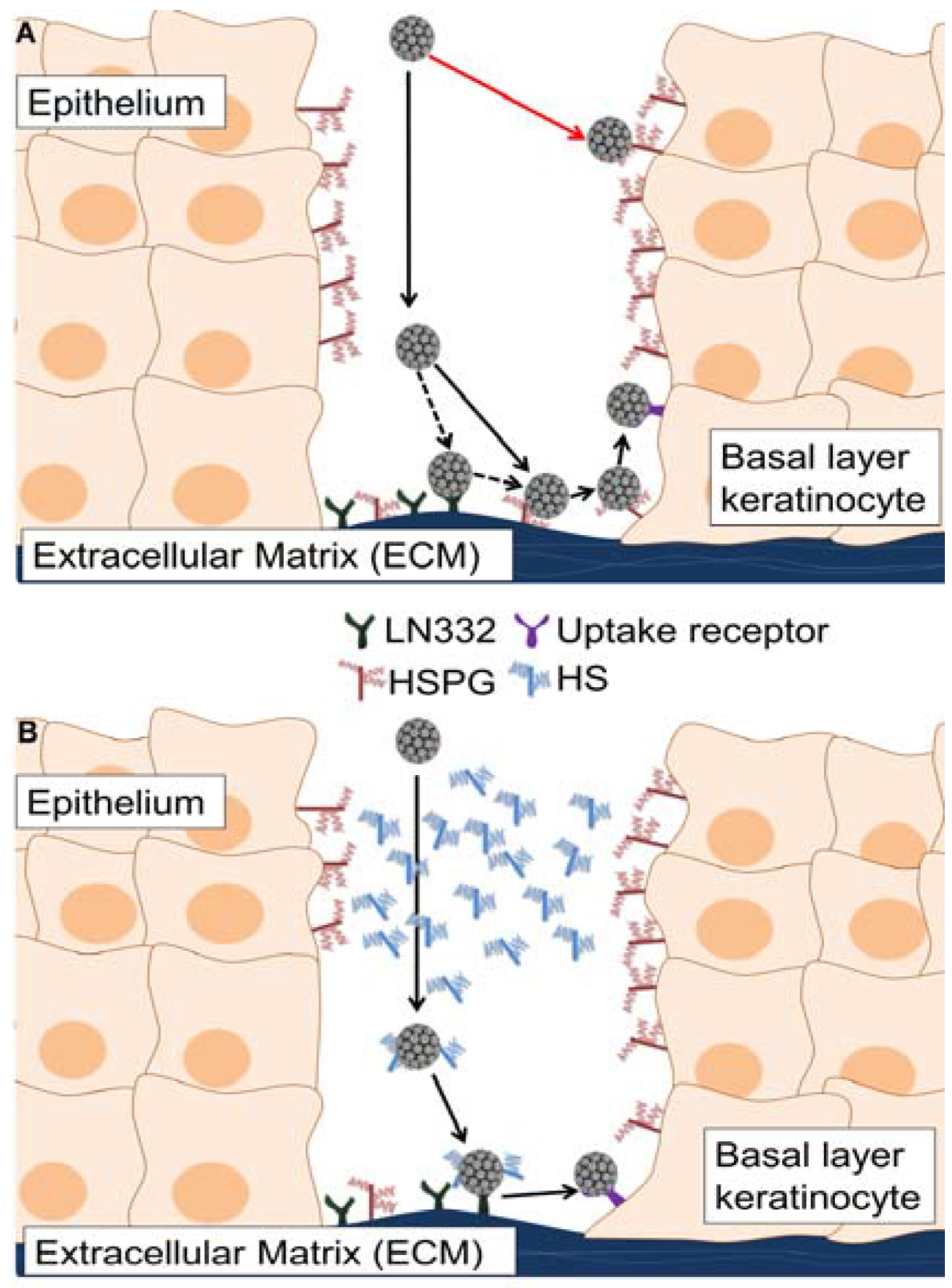
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Burchell, A.N.; Schiffman, M.; Giuliano, A.R.; de Sanjose, S.; Bruni, L.; Tortolero-Luna, G.; Kjaer, S.K.; Munoz, N. Epidemiology and natural history of human papillomavirus infections and type-specific implications in cervical neoplasia. Vaccine 2008, 26 (Suppl. 10), K1–K16. [Google Scholar] [PubMed]
- Zur Hausen, H. Human papillomavirus & cervical cancer. Indian J. Med. Res. 2009, 130, 209. [Google Scholar] [PubMed]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 2007, 121, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Manos, M.M.; Munoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.; Shah, K.V. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Natl. Cancer Inst. 1995, 87, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S.; Lodi, G.; von Bultzingslowen, I.; Aliko, A.; Arduino, P.; Campisi, G.; Challacombe, S.; Ficarra, G.; Flaitz, C.; Zhou, H.M.; et al. Human papillomaviruses in oral carcinoma and oral potentially malignant disorders: A systematic review. Oral. Dis. 2011, 17 (Suppl. 1), 58–72. [Google Scholar] [CrossRef] [PubMed]
- Cubie, H.A. Diseases associated with human papillomavirus infection. Virology 2013, 445, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Baker, T.S.; Newcomb, W.W.; Olson, N.H.; Cowsert, L.M.; Olson, C.; Brown, J.C. Structures of bovine and human papillomaviruses. Analysis by cryoelectron microscopy and three-dimensional image reconstruction. Biophys. J. 1991, 60, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Mol. Cell. 2000, 5, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Modis, Y.; Trus, B.L.; Harrison, S.C. Atomic model of the papillomavirus capsid. EMBO J. 2002, 21, 4754–4762. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Beard, P.; Estes, P.A.; Lyon, M.K.; Garcea, R.L. Intercapsomeric disulfide bonds in papillomavirus assembly and disassembly. J. Virol. 1998, 72, 2160–2167. [Google Scholar] [PubMed]
- Sapp, M.; Fligge, C.; Petzak, I.; Harris, J.R.; Streeck, R.E. Papillomavirus assembly requires trimerization of the major capsid protein by disulfides between two highly conserved cysteines. J. Virol. 1998, 72, 6186–6189. [Google Scholar]
- Buck, C.B.; Cheng, N.; Thompson, C.D.; Lowy, D.R.; Steven, A.C.; Schiller, J.T.; Trus, B.L. Arrangement of L2 within the papillomavirus capsid. J. Virol. 2008, 82, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.N.; Sun, X.Y.; Frazer, I.H.; Zhou, J. DNA packaging by L1 and L2 capsid proteins of bovine papillomavirus type 1. Virology 1998, 243, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Roden, R.B.; Day, P.M.; Bronzo, B.K.; Yutzy, W.H.T.; Yang, Y.; Lowy, D.R.; Schiller, J.T. Positively charged termini of the L2 minor capsid protein are necessary for papillomavirus infection. J. Virol. 2001, 75, 10493–10497. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Thompson, C.D.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Maturation of papillomavirus capsids. J. Virol. 2005, 79, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Leder, C.; Kleinschmidt, J.A.; Wiethe, C.; Muller, M. Enhancement of capsid gene expression: Preparing the human papillomavirus type 16 major structural gene L1 for DNA vaccination purposes. J. Virol. 2001, 75, 9201–9209. [Google Scholar] [CrossRef] [PubMed]
- Raff, A.B.; Woodham, A.W.; Raff, L.M.; Skeate, J.G.; Yan, L.; da Silva, D.M.; Schelhaas, M.; Kast, W.M. The evolving field of human papillomavirus receptor research: A review of binding and entry. J. Virol. 2013, 87, 6062–6072. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Giroglou, T.; Florin, L.; Schafer, F.; Streeck, R.E.; Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.G.; Tung, J.S.; Przysiecki, C.T.; Cook, J.C.; Lehman, E.D.; Sands, J.A.; Jansen, K.U.; Keller, P.M. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 1999, 274, 5810–5822. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, J.; Bienkowska-Haba, M.; Ortega, M.E.; Patel, H.D.; Bodevin, S.; Spillmann, D.; Bishop, B.; Sapp, M.; Chen, X.S. Structural basis of oligosaccharide receptor recognition by human papillomavirus. J. Biol. Chem. 2011, 286, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Knappe, M.; Bodevin, S.; Selinka, H.C.; Spillmann, D.; Streeck, R.E.; Chen, X.S.; Lindahl, U.; Sapp, M. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J. Biol. Chem. 2007, 282, 27913–27922. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.F.; Bienkowska-Haba, M.; Dasgupta, J.; Chen, X.S.; Sapp, M. Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. J. Virol. 2013, 87, 11426–11437. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef]
- Spoden, G.; Kuhling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol 2013, 87, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Abban, C.Y.; Meneses, P.I. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.S.; Kim, K.D.; Park, S.N.; Cheong, S.W. α6 integrin is the main receptor of human papillomavirus type 16 VLP. Biochem. Biophys. Res. Commun. 2001, 283, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Evander, M.; Frazer, I.H.; Payne, E.; Qi, Y.M.; Hengst, K.; McMillan, N.A. Identification of the alpha6 integrin as a candidate receptor for papillomaviruses. J. Virol. 1997, 71, 2449–2456. [Google Scholar] [PubMed]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and caveolin-independent entry of human papillomavirus type 16—Involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 2008, 3, e3313. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS One 2012, 7, e43519. [Google Scholar] [CrossRef] [PubMed]
- Dziduszko, A.; Ozbun, M.A. Annexin A2 and S100A10 Regulate Human Papillomavirus Type 16 Entry and Intracellular Trafficking in Human Keratinocytes. J. Virol. 2013, 87, 7502–7515. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins Facilitate Dissociation of the Human Papillomavirus Type 16 Capsid Protein L1 from the L2/DNA Complex following Virus Entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.A.; Poffenberger, A.C.; Nelson, C.D.; Atwood, W.J.; et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large Scale RNAi Reveals the Requirement of Nuclear Envelope Breakdown for Nuclear Import of Human Papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Hilbig, L.; Sapp, M. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 2014, 458–459, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Bulk, S.; Berkhof, J.; Bulkmans, N.W.; Zielinski, G.D.; Rozendaal, L.; van Kemenade, F.J.; Snijders, P.J.; Meijer, C.J. Preferential risk of HPV16 for squamous cell carcinoma and of HPV18 for adenocarcinoma of the cervix compared to women with normal cytology in The Netherlands. Br. J. Cancer 2006, 94, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Kusche-Gullberg, M.; Kjellen, L. Regulated diversity of heparan sulfate. J. Biol. Chem. 1998, 273, 24979–24982. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef] [PubMed]
- Selinka, H.C.; Florin, L.; Patel, H.D.; Freitag, K.; Schmidtke, M.; Makarov, V.A.; Sapp, M. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J. Virol. 2007, 81, 10970–10980. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, C.; Liu, Y.; Kuhling, L.; Chai, W.; Hafezi, W.; van Kuppevelt, T.H.; Kuhn, J.E.; Feizi, T.; Schelhaas, M. Heparin increases the infectivity of Human Papillomavirus type 16 independent of cell surface proteoglycans and induces L1 epitope exposure. Cell. Microbiol. 2013, 15, 1818–1836. [Google Scholar] [PubMed]
- Rommel, O.; Dillner, J.; Fligge, C.; Bergsdorf, C.; Wang, X.; Selinka, H.C.; Sapp, M. Heparan sulfate proteoglycans interact exclusively with conformationally intact HPV L1 assemblies: Basis for a virus-like particle ELISA. J. Med. Virol. 2005, 75, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Volpers, C.; Sapp, M.; Snijders, P.J.; Walboomers, J.M.; Streeck, R.E. Conformational and linear epitopes on virus-like particles of human papillomavirus type 33 identified by monoclonal antibodies to the minor capsid protein L2. J. Gen. Virol. 1995, 76 (Pt. 11), 2661–2667. [Google Scholar] [CrossRef] [PubMed]
- Bergsdorf, C.; Beyer, C.; Umansky, V.; Werr, M.; Sapp, M. Highly efficient transport of carboxyfluorescein diacetate succinimidyl ester into COS7 cells using human papillomavirus-like particles. FEBS Lett. 2003, 536, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, M.; Karger, A.; Meerbach, A.; Egerer, R.; Stelzner, A.; Makarov, V. Binding of a N,N'-bisheteryl derivative of dispirotripiperazine to heparan sulfate residues on the cell surface specifically prevents infection of viruses from different families. Virology 2003, 311, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, M.; Riabova, O.; Dahse, H.M.; Stelzner, A.; Makarov, V. Synthesis, cytotoxicity and antiviral activity of N,N'-bis-5-nitropyrimidyl derivatives of dispirotripiperazine. Antiviral. Res. 2002, 55, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Buck, C.B.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J. Virol. 2007, 81, 8784–8792. [Google Scholar] [CrossRef] [PubMed]
- Culp, T.D.; Budgeon, L.R.; Christensen, N.D. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 2006, 347, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, A.N.; Multhaupt, H.A.; Couchman, J.R. Syndecans in wound healing, inflammation and vascular biology. Int. J. Biochem. Cell. Biol. 2007, 39, 505–528. [Google Scholar] [CrossRef] [PubMed]
- Elenius, K.; Vainio, S.; Laato, M.; Salmivirta, M.; Thesleff, I.; Jalkanen, M. Induced expression of syndecan in healing wounds. J. Cell. Biol. 1991, 114, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Falk-Marzillier, J.; Schiraldi, O.; Stetler-Stevenson, W.G.; Quaranta, V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 1997, 277, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Giannelli, G.; Cirulli, V.; Miyazaki, K.; Quaranta, V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J. Cell. Biol. 2000, 148, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Amano, S.; Scott, I.C.; Takahara, K.; Koch, M.; Champliaud, M.F.; Gerecke, D.R.; Keene, D.R.; Hudson, D.L.; Nishiyama, T.; Lee, S.; et al. Bone morphogenetic protein 1 is an extracellular processing enzyme of the laminin 5 gamma 2 chain. J. Biol. Chem 2000, 275, 22728–22735. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.L.; Nagano, T.; Nakamura, M.; Kumagai, N.; Mishima, H.; Nishida, T. Effect of galardin on collagen degradation by Pseudomonas aeruginosa. Exp. Eye Res. 1999, 69, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Foley, E.M.; Gonzales, J.C.; Gordts, P.L.; Li, Y.; Esko, J.D. Shedding of syndecan-1 from human hepatocytes alters very low density lipoprotein clearance. Hepatology 2012, 55, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef] [PubMed]
- Broutian, T.R.; Brendle, S.A.; Christensen, N.D. Differential binding patterns to host cells associated with particles of several human alphapapillomavirus types. J. Gen. Virol. 2010, 91, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.J.; Buckley, C.H.; Wells, M. Aetiology, pathogenesis, and pathology of cervical neoplasia. J. Clin. Pathol. 1998, 51, 96–103. [Google Scholar] [CrossRef] [PubMed]
- McCluggage, W.G. New developments in endocervical glandular lesions. Histopathology 2013, 62, 138–160. [Google Scholar] [CrossRef] [PubMed]
- Castellsague, X.; Diaz, M.; de Sanjose, S.; Munoz, N.; Herrero, R.; Franceschi, S.; Peeling, R.W.; Ashley, R.; Smith, J.S.; Snijders, P.J.; et al. Worldwide human papillomavirus etiology of cervical adenocarcinoma and its cofactors: Implications for screening and prevention. J. Natl. Cancer Inst. 2006, 98, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Handisurya, A.; Day, P.M.; Thompson, C.D.; Buck, C.B.; Kwak, K.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Murine skin and vaginal mucosa are similarly susceptible to infection by pseudovirions of different papillomavirus classifications and species. Virology 2012, 433, 385–394. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richards, K.F.; Mukherjee, S.; Bienkowska-Haba, M.; Pang, J.; Sapp, M. Human Papillomavirus Species-Specific Interaction with the Basement Membrane-Resident Non-Heparan Sulfate Receptor. Viruses 2014, 6, 4856-4879. https://doi.org/10.3390/v6124856
Richards KF, Mukherjee S, Bienkowska-Haba M, Pang J, Sapp M. Human Papillomavirus Species-Specific Interaction with the Basement Membrane-Resident Non-Heparan Sulfate Receptor. Viruses. 2014; 6(12):4856-4879. https://doi.org/10.3390/v6124856
Chicago/Turabian StyleRichards, Kathleen F., Santanu Mukherjee, Malgorzata Bienkowska-Haba, Jia Pang, and Martin Sapp. 2014. "Human Papillomavirus Species-Specific Interaction with the Basement Membrane-Resident Non-Heparan Sulfate Receptor" Viruses 6, no. 12: 4856-4879. https://doi.org/10.3390/v6124856
APA StyleRichards, K. F., Mukherjee, S., Bienkowska-Haba, M., Pang, J., & Sapp, M. (2014). Human Papillomavirus Species-Specific Interaction with the Basement Membrane-Resident Non-Heparan Sulfate Receptor. Viruses, 6(12), 4856-4879. https://doi.org/10.3390/v6124856