Alpharetroviral Vectors: From a Cancer-Causing Agent to a Useful Tool for Human Gene Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
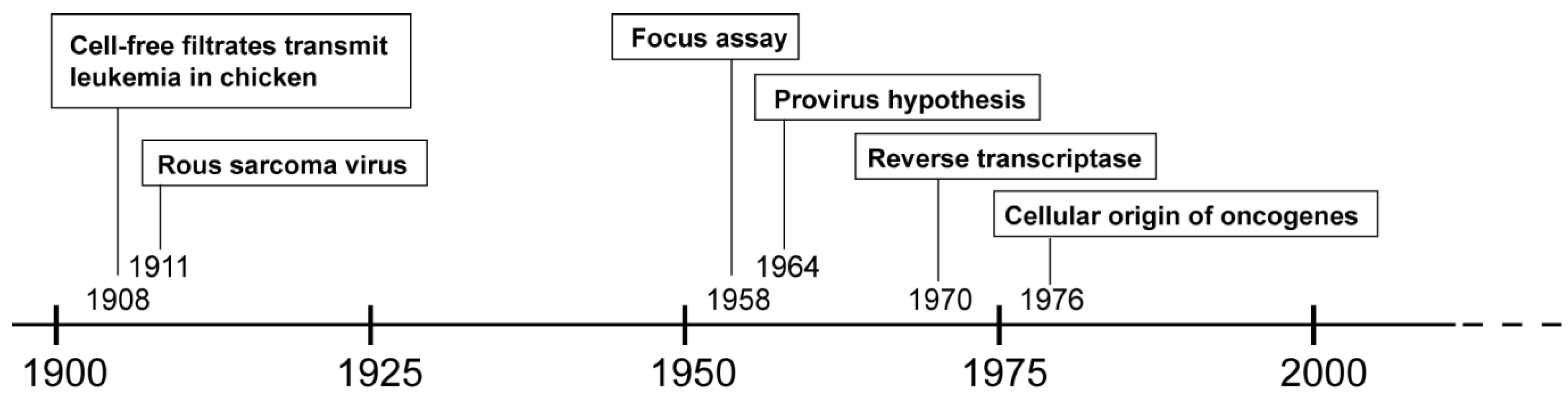
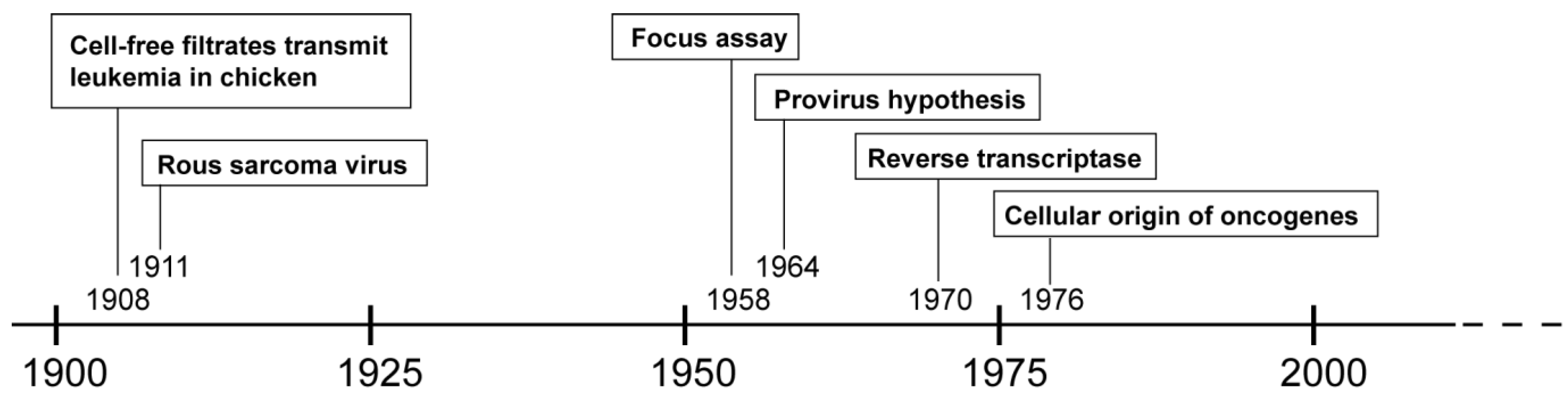
2. History of (Alpha-) Retroviruses
3. From the Virus to the Vector
3.1. Taking a Different Perspective: Retroviruses for Human Gene Therapy
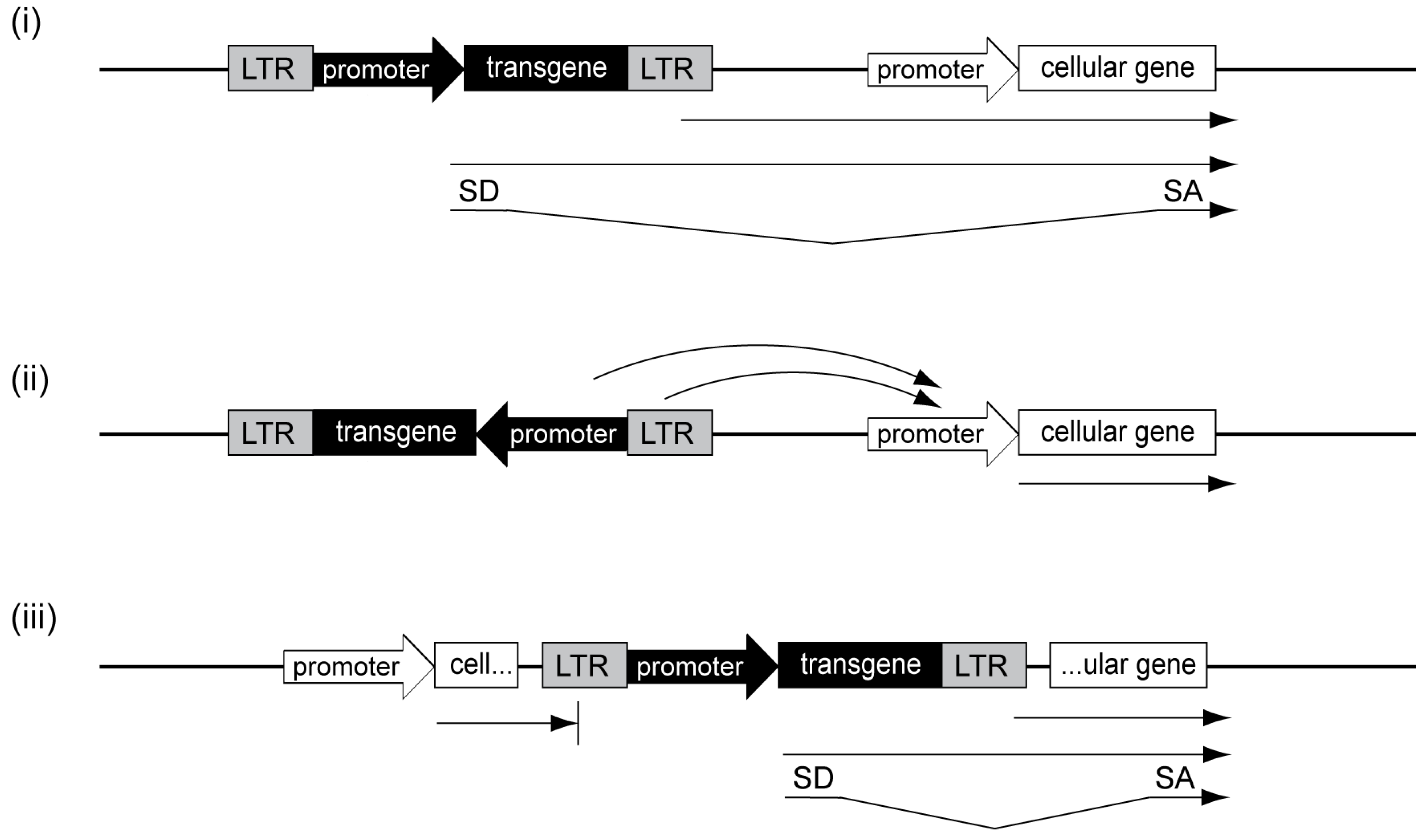
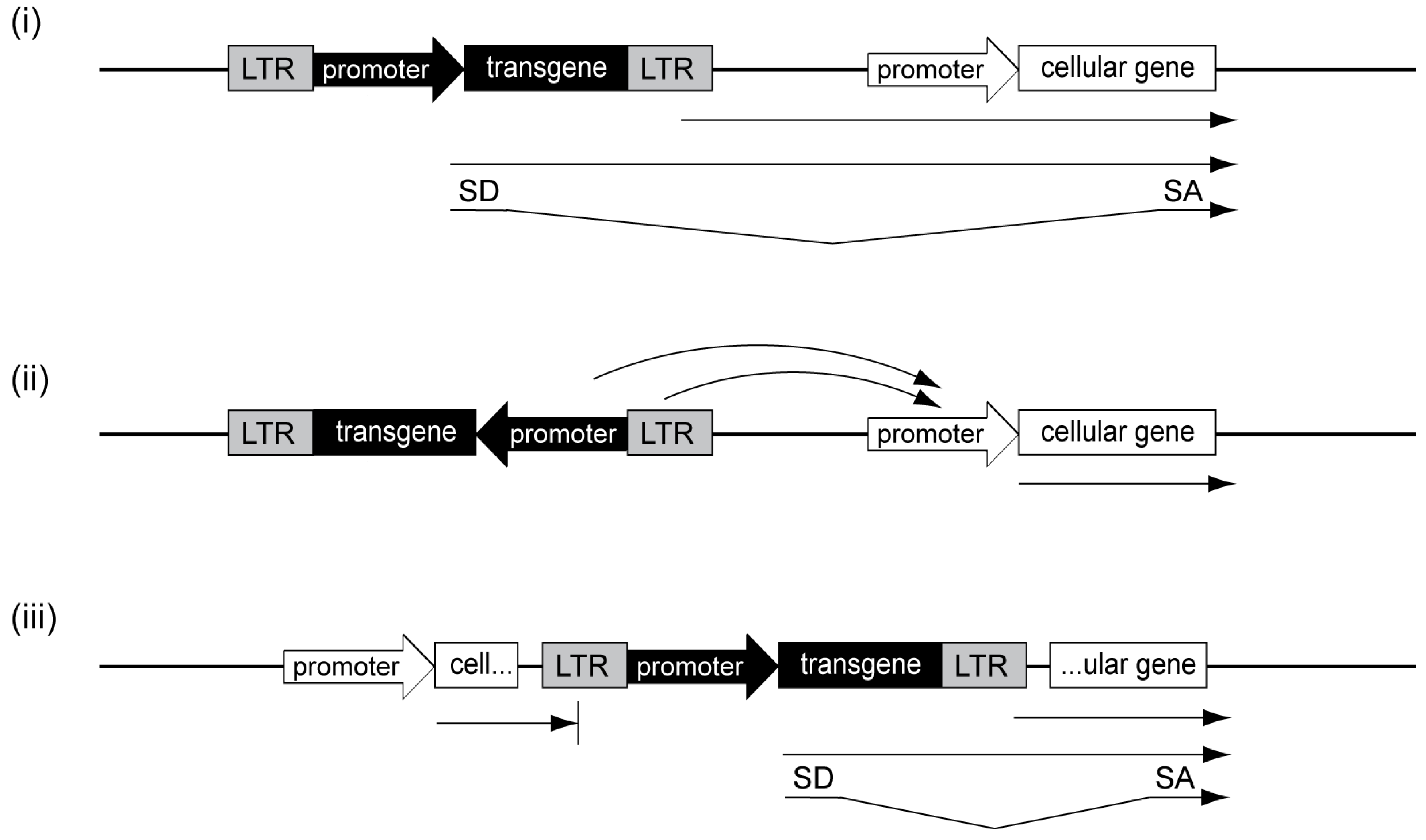
3.2. Genotoxicity of Retroviral Vectors
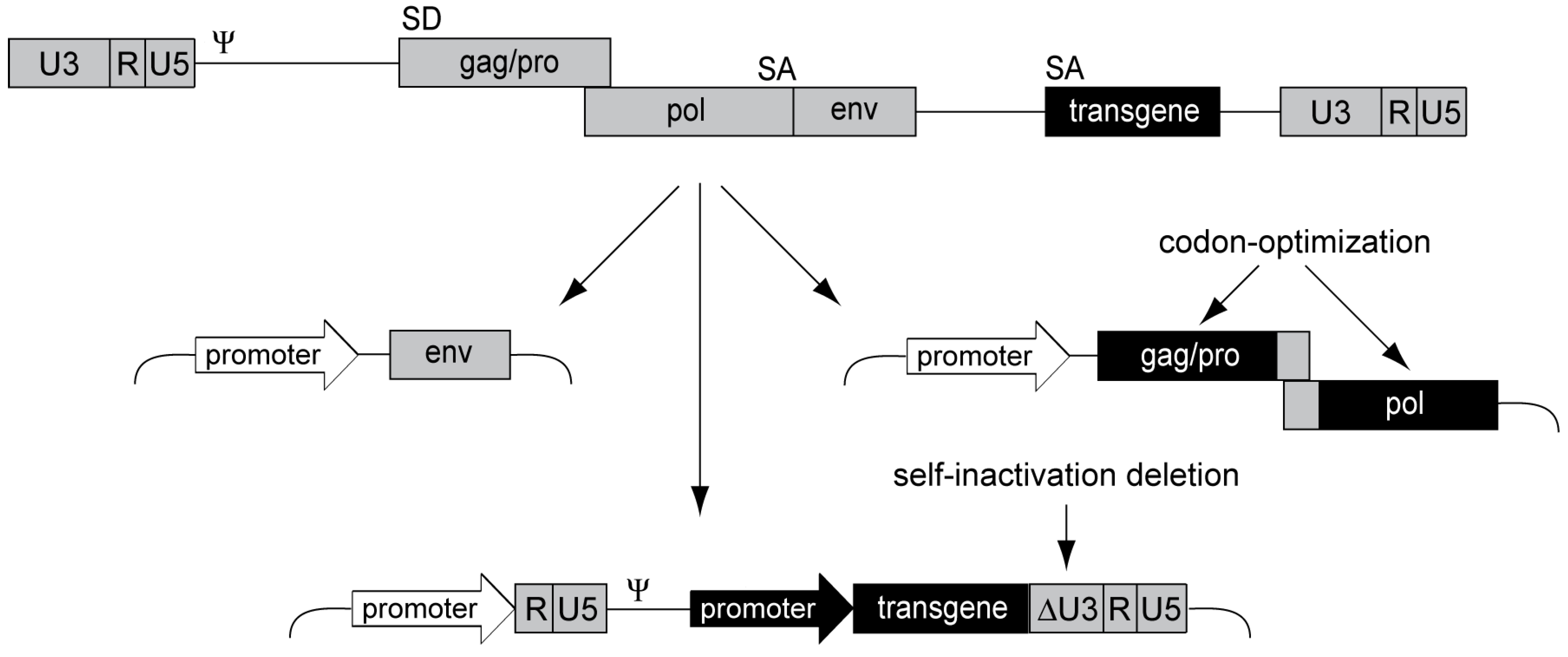
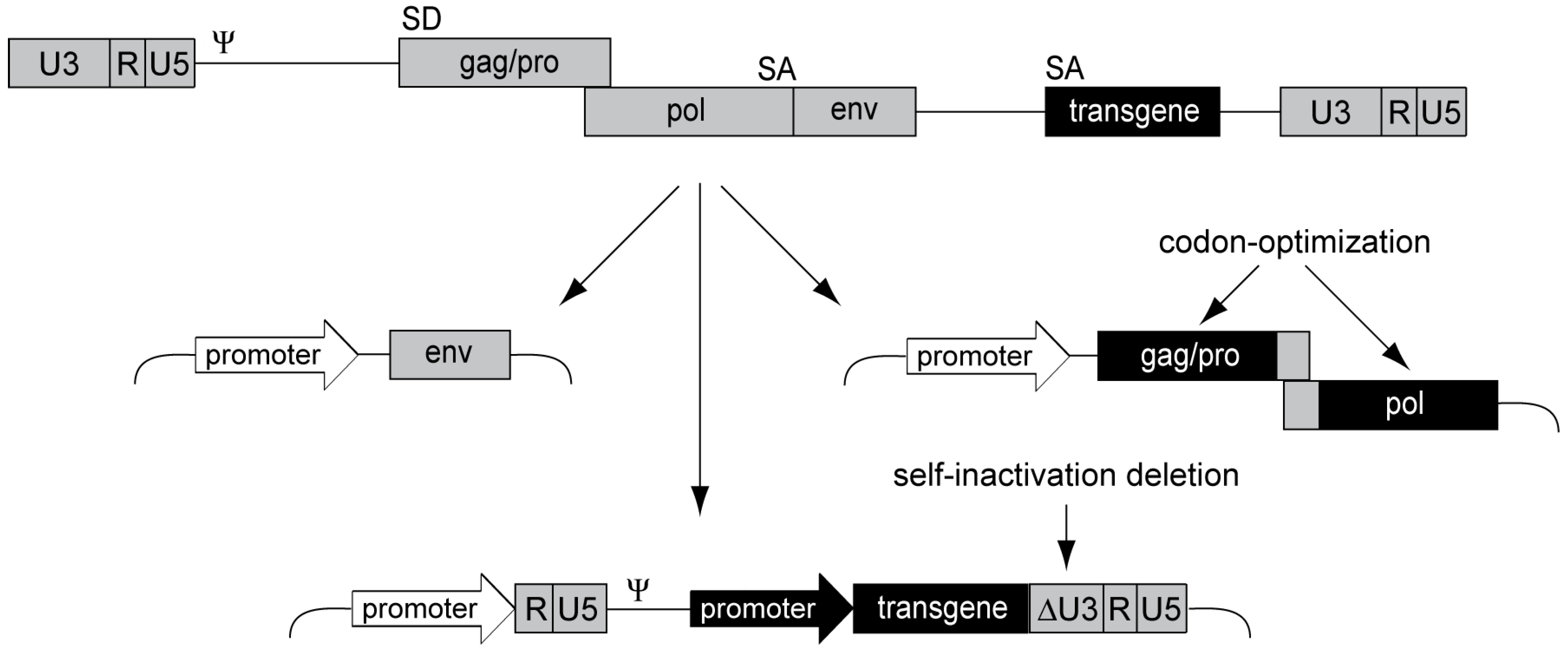
3.4. Alpharetroviral Vector Developments
5. From Bench to Bedside: Regulatory Requirements for Clinical Translation
7. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References and Notes
- Ellermann, V.; Bang, O. Experimentelle Leukämie bei Hühnern. Zentralbl. Bakteriol. Parasitenkd. Infektionskr. Hyg. Abt. I. 1908, 46, 595–609. [Google Scholar]
- Rous, P. A transmissible avian neoplasm. (sarcoma of the common fowl.). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Diamond, L.; Wolman, S.R. Charlotte friend, ph.D. 1921–1987. A scientist’s life. Ann. N. Y. Acad. Sci. 1989, 567, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Rubin, H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology 1958, 6, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Bather, R. The nucleic acid of partially purified Rous No. I sarcoma virus. Br. J. Cancer 1957, 11, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Rubin, H.; Temin, H.M. A radiological study of cell-virus interaction in the Rous sarcoma. Virology 1959, 7, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M. The Participation of DNA in Rous Sarcoma Virus Production. Virology 1964, 23, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, J.; Chyle, P.; Simkovic, D.; Hilgert, I. Demonstration of the absence of infectious Rous virus in rat tumour XC, whose structurally intact cells produce Rous sarcoma when transferred to chicks. Folia Biol. 1963, 9, 77–81. [Google Scholar]
- Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Temin, H.M.; Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef] [PubMed]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Hayward, W.S.; Neel, B.G.; Astrin, S.M. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 1981, 290, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Hayward, W.S.; Robinson, H.L.; Fang, J.; Astrin, S.M. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: Oncogenesis by promoter insertion. Cell 1981, 23, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.; Mulligan, R.C.; Baltimore, D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell 1983, 33, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A.; Lemischka, I.R.; Nathan, D.G.; Mulligan, R.C. Introduction of new genetic material into pluripotent haematopoietic stem cells of the mouse. Nature 1984, 310, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E.; Magli, M.C.; Huszar, D.; Phillips, R.A.; Bernstein, A. Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/Wv mice. Cell 1985, 42, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B. Update on gene therapy for immunodeficiencies. Clin. Immunol. 2010, 135, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kuhlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Kramer, A.; Schwable, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin Invest. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Gohring, G.; Steinemann, D.; et al. Gene therapy for Wiskott-Aldrich syndrome—Long-term efficacy and genotoxicity. Sci. Transl. Med. 2014, 6, 227ra233. [Google Scholar] [CrossRef]
- Lazo, P.A.; Lee, J.S.; Tsichlis, P.N. Long-distance activation of the Myc protooncogene by provirus insertion in Mlvi-1 or Mlvi-4 in rat T-cell lymphomas. Proc. Natl. Acad. Sci. USA 1990, 87, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, C.; Ihle, J.N. Retroviral insertions 90 kilobases proximal to the Evi-1 myeloid transforming gene activate transcription from the normal promoter. Mol. Cell Biol. 1991, 11, 1820–1828. [Google Scholar] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Heckl, D.; Schwarzer, A.; Haemmerle, R.; Steinemann, D.; Rudolph, C.; Skawran, B.; Knoess, S.; Krause, J.; Li, Z.; Schlegelberger, B.; et al. Lentiviral vector induced insertional haploinsufficiency of Ebf1 causes murine leukemia. Mol. Ther. 2012, 20, 1187–1195. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Sokol, M.; Wabl, M.; Ruiz, I.R.; Pedersen, F.S. Novel principles of gamma-retroviral insertional transcription activation in murine leukemia virus-induced end-stage tumors. Retrovirology 2014, 11, 36. [Google Scholar] [CrossRef]
- Mooslehner, K.; Karls, U.; Harbers, K. Retroviral integration sites in transgenic Mov mice frequently map in the vicinity of transcribed DNA regions. J. Virol. 1990, 64, 3056–3058. [Google Scholar] [PubMed]
- Scherdin, U.; Rhodes, K.; Breindl, M. Transcriptionally active genome regions are preferred targets for retrovirus integration. J. Virol. 1990, 64, 907–912. [Google Scholar] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Hong, B.K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef]
- Barr, S.D.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. Integration targeting by avian sarcoma-leukosis virus and human immunodeficiency virus in the chicken genome. J. Virol. 2005, 79, 12035–12044. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Laufs, S.; Blake, J.; Schwager, C.; Wu, X.; Zeller, J.W.; Ho, A.D.; Fruehauf, S. Retroviral integration sites correlate with expressed genes in hematopoietic stem cells. Stem Cells 2005, 23, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Beard, B.C.; Dickerson, D.; Beebe, K.; Gooch, C.; Fletcher, J.; Okbinoglu, T.; Miller, D.G.; Jacobs, M.A.; Kaul, R.; Kiem, H.P.; et al. Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells. Mol. Ther. 2007, 15, 1356–1365. [Google Scholar] [CrossRef]
- Beard, B.C.; Keyser, K.A.; Trobridge, G.D.; Peterson, L.J.; Miller, D.G.; Jacobs, M.; Kaul, R.; Kiem, H.P. Unique integration profiles in a canine model of long-term repopulating cells transduced with gammaretrovirus, lentivirus, or foamy virus. Hum. Gene Ther. 2007, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Brady, T.; Agosto, L.M.; Malani, N.; Berry, C.C.; O’Doherty, U.; Bushman, F. HIV integration site distributions in resting and activated CD4+ T cells infected in culture. Aids 2009, 23, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS One 2007, 2, e1340. [Google Scholar] [CrossRef] [PubMed]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; de Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 determines cellular trafficking of diverse lentiviral but not murine oncoretroviral integrase proteins and is a component of functional lentiviral preintegration complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [PubMed]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The bet family of proteins targets moloney murine leukemia virus integration near transcription start sites. Cell. Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Gijsbers, R.; Ronen, K.; Vets, S.; Malani, N.; de Rijck, J.; McNeely, M.; Bushman, F.D.; Debyser, Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010, 18, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Vets, S.; De Rijck, J.; Brendel, C.; Grez, M.; Bushman, F.; Debyser, Z.; Gijsbers, R. Transient Expression of an LEDGF/p75 Chimera Retargets Lentivector Integration and Functionally Rescues in a Model for X-CGD. Mol. Ther. Nucleic Acids 2013, 2, e77. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Shun, M.C.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog. 2009, 5, e1000259. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A.; Diamond, T.L.; Hwang, Y.; Marshall, H.M.; Bushman, F.D. Modulating target site selection during human immunodeficiency virus DNA integration in vitro with an engineered tethering factor. Hum. Gene Ther. 2006, 17, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Ferris, A.L.; Wu, X.; Hughes, C.M.; Stewart, C.; Smith, S.J.; Milne, T.A.; Wang, G.G.; Shun, M.C.; Allis, C.D.; Engelman, A.; et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. Natl. Acad. Sci. USA 2010, 107, 3135–3140. [Google Scholar] [CrossRef] [PubMed]
- Aiyer, S.; Swapna, G.V.; Malani, N.; Aramini, J.M.; Schneider, W.M.; Plumb, M.R.; Ghanem, M.; Larue, R.C.; Sharma, A.; Studamire, B.; et al. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 2014, 42, 5917–5928. [Google Scholar] [CrossRef] [PubMed]
- El Ashkar, S.; de Rijck, J.; Demeulemeester, J.; Vets, S.; Madlala, P.; Cermakova, K.; Debyser, Z.; Gijsbers, R. BET-independent MLV-based vectors target away from promoters and regulatory elements. Mol. Ther. Nucleic Acids 2014, 3, e179. [Google Scholar]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Narezkina, A.; Taganov, K.D.; Litwin, S.; Stoyanova, R.; Hayashi, J.; Seeger, C.; Skalka, A.M.; Katz, R.A. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 2004, 78, 11656–11663. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Renaud, G.; Gomes, T.J.; Ferris, A.; Hendrie, P.C.; Donahue, R.E.; Hughes, S.H.; Wolfsberg, T.G.; Russell, D.W.; Dunbar, C.E. Reduced genotoxicity of avian sarcoma leukosis virus vectors in rhesus long-term repopulating cells compared to standard murine retrovirus vectors. Mol. Ther. 2008, 16, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Maetzig, T.; Brugman, M.H.; Heinz, N.; Appelt, J.U.; Kaufmann, K.B.; Schmidt, M.; Grez, M.; Modlich, U.; Baum, C.; et al. Alpharetroviral self-inactivating vectors: Long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol. Ther. 2012, 20, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Moiani, A.; Suerth, J.D.; Gandolfi, F.; Rizzi, E.; Severgnini, M.; de Bellis, G.; Schambach, A.; Mavilio, F. Genome-wide analysis of alpharetroviral integration in human hematopoietic stem/progenitor cells. Genes 2014, 5, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Donahue, R.E.; Kessler, S.W.; Bodine, D.; McDonagh, K.; Dunbar, C.; Goodman, S.; Agricola, B.; Byrne, E.; Raffeld, M.; Moen, R. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J. Exp. Med. 1992, 176, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi Sergi, L.; Sanvito, F.; et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Temin, H.M. Construction of a helper cell line for avian reticuloendotheliosis virus cloning vectors. Mol. Cell Biol. 1983, 3, 2241–2249. [Google Scholar] [PubMed]
- Miller, A.D.; Rosman, G.J. Improved retroviral vectors for gene transfer and expression. Biotechniques 1989, 7, 980–990. [Google Scholar] [PubMed]
- Soneoka, Y.; Cannon, P.M.; Ramsdale, E.E.; Griffiths, J.C.; Romano, G.; Kingsman, S.M.; Kingsman, A.J. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995, 23, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; von Ruden, T.; Kantoff, P.W.; Garber, C.; Seiberg, M.; Ruther, U.; Anderson, W.F.; Wagner, E.F.; Gilboa, E. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3194–3198. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [PubMed]
- Modlich, U.; Bohne, J.; Schmidt, M.; von Kalle, C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.; Dullmann, J.; Li, Z.; Fehse, B.; Meyer, J.; Williams, D.A.; von Kalle, C. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood 2003, 101, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Willemsen, R.; van Elzakker, P.; van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Akatsuka, Y.; Nawa, A.; Kuzushima, K.; Tsujimura, K.; Tanimoto, M.; Kodera, Y.; Morishima, Y.; Kuzuya, K.; Takahashi, T. Retroviral vector backbone immunogenicity: Identification of cytotoxic t-cell epitopes in retroviral vector-packaging sequences. Gene Ther. 2005, 12, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, M.J.; Hughes, S.H. Retroviral gene delivery. Methods Cell Biol. 1997, 52, 179–214. [Google Scholar] [PubMed]
- Hughes, S.H. The RCAS vector system. Folia Biol. 2004, 50, 107–119. [Google Scholar]
- Svoboda, J. Molecular biology of cell nonpermissiveness to retroviruses. Has the time come? Gene 1998, 206, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ferris, A.; Larochelle, A.; Krouse, A.E.; Metzger, M.E.; Donahue, R.E.; Hughes, S.H.; Dunbar, C.E. Transduction of rhesus macaque hematopoietic stem and progenitor cells with avian sarcoma and leukosis virus vectors. Hum. Gene Ther. 2007, 18, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Roth, M.G.; Hunter, E. A chimeric avian retrovirus containing the influenza virus hemagglutinin gene has an expanded host range. J. Virol. 1992, 66, 7374–7382. [Google Scholar] [PubMed]
- Barsov, E.V.; Hughes, S.H. Gene transfer into mammalian cells by a Rous sarcoma virus-based retroviral vector with the host range of the amphotropic murine leukemia virus. J. Virol. 1996, 70, 3922–3929. [Google Scholar] [PubMed]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [PubMed]
- Kraunus, J.; Schaumann, D.H.S.; Meyer, J.; Modlich, U.; Fehse, B.; Brandenburg, G.; von Laer, D.; Klump, H.; Schambach, A.; Bohne, J.; et al. Self-inactivating retroviral vectors with improved RNA processing. Gene Therapy 2004, 11, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Mueller, D.; Galla, M.; Verstegen, M.M.; Wagemaker, G.; Loew, R.; Baum, C.; Bohne, J. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006, 13, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Self-inactivating alpharetroviral vectors with a split-packaging design. J. Virol. 2010, 84, 6626–6635. [Google Scholar] [CrossRef] [PubMed]
- Ruddell, A. Transcription regulatory elements of the avian retroviral long terminal repeat. Virology 1995, 206, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Russ, J.L.; Eiden, M.V. Evaluation of residual promoter activity in gamma-retroviral self-inactivating (SIN) vectors. Mol. Ther. 2012, 20, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.B.; Brendel, C.; Suerth, J.D.; Mueller-Kuller, U.; Chen-Wichmann, L.; Schwable, J.; Pahujani, S.; Kunkel, H.; Schambach, A.; Baum, C.; et al. Alpharetroviral vector-mediated gene therapy for X-CGD: Functional correction and lack of aberrant splicing. Mol. Ther. 2012, 21, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Barklis, E.; Mulligan, R.C.; Jaenisch, R. Chromosomal position or virus mutation permits retrovirus expression in embryonal carcinoma cells. Cell 1986, 47, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.R.; Krieg, A.M.; Max, E.E.; Khan, A.S. Negative control region at the 5' end of murine leukemia virus long terminal repeats. Mol. Cell Biol. 1989, 9, 739–746. [Google Scholar] [PubMed]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Katz, R.A.; Ishov, A.M.; Maul, G.G.; Skalka, A.M. The cellular protein daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J. Virol. 2005, 79, 4610–4618. [Google Scholar] [CrossRef] [PubMed]
- Poleshko, A.; Palagin, I.; Zhang, R.; Boimel, P.; Castagna, C.; Adams, P.D.; Skalka, A.M.; Katz, R.A. Identification of cellular proteins that maintain retroviral epigenetic silencing: Evidence for an antiviral response. J. Virol. 2008, 82, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.A.; Jack-Scott, E.; Narezkina, A.; Palagin, I.; Boimel, P.; Kulkosky, J.; Nicolas, E.; Greger, J.G.; Skalka, A.M. High-frequency epigenetic repression and silencing of retroviruses can be antagonized by histone deacetylase inhibitors and transcriptional activators, but uniform reactivation in cell clones is restricted by additional mechanisms. J. Virol. 2007, 81, 2592–2604. [Google Scholar] [CrossRef] [PubMed]
- Shalginskikh, N.; Poleshko, A.; Skalka, A.M.; Katz, R.A. Retroviral DNA methylation and epigenetic repression are mediated by the antiviral host protein Daxx. J. Virol. 2013, 87, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Hejnar, J.; Plachy, J.; Geryk, J.; Machon, O.; Trejbalova, K.; Guntaka, R.V.; Svoboda, J. Inhibition of the rous sarcoma virus long terminal repeat-driven transcription by in vitro methylation: Different sensitivity in permissive chicken cells versus mammalian cells. Virology 1999, 255, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Hejnar, J.; Svoboda, J.; Geryk, J.; Fincham, V.J.; Hak, R. High rate of morphological reversion in tumor cell line H-19 associated with permanent transcriptional suppression of the LTR, v-src, LTR provirus. Cell Growth Differ. 1994, 5, 277–285. [Google Scholar] [PubMed]
- Searle, S.; Gillespie, D.A.; Chiswell, D.J.; Wyke, J.A. Analysis of the variations in proviral cytosine methylation that accompany transformation and morphological reversion in a line of Rous sarcoma virus-infected Rat-1 cells. Nucleic Acids Res. 1984, 12, 5193–5210. [Google Scholar] [CrossRef] [PubMed]
- Jahner, D.; Stuhlmann, H.; Stewart, C.L.; Harbers, K.; Lohler, J.; Simon, I.; Jaenisch, R. De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 1982, 298, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Klug, C.A.; Cheshier, S.; Weissman, I.L. Inactivation of a GFP retrovirus occurs at multiple levels in long-term repopulating stem cells and their differentiated progeny. Blood 2000, 96, 894–901. [Google Scholar] [PubMed]
- Hoeben, R.C.; Migchielsen, A.A.; van der Jagt, R.C.; van Ormondt, H.; van der Eb, A.J. Inactivation of the Moloney murine leukemia virus long terminal repeat in murine fibroblast cell lines is associated with methylation and dependent on its chromosomal position. J. Virol. 1991, 65, 904–912. [Google Scholar] [PubMed]
- Plachy, J.; Kotab, J.; Divina, P.; Reinisova, M.; Senigl, F.; Hejnar, J. Proviruses selected for high and stable expression of transduced genes accumulate in broadly transcribed genome areas. J. Virol. 2010, 84, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Senigl, F.; Auxt, M.; Hejnar, J. Transcriptional provirus silencing as a crosstalk of de novo DNA methylation and epigenomic features at the integration site. Nucleic Acids Res. 2012, 40, 5298–5312. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, M.N.; Skipper, K.A.; Anakok, O. Optimizing retroviral gene expression for effective therapies. Hum. Gene Ther. 2013, 24, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Benabdellah, K.; Gutierrez-Guerrero, A.; Cobo, M.; Munoz, P.; Martin, F. A chimeric HS4-SAR insulator (IS2) that prevents silencing and enhances expression of lentiviral vectors in pluripotent stem cells. PLoS One 2014, 9, e84268. [Google Scholar] [CrossRef] [PubMed]
- Senigl, F.; Plachy, J.; Hejnar, J. The core element of a CpG island protects avian sarcoma and leukosis virus-derived vectors from transcriptional silencing. J. Virol. 2008, 82, 7818–7827. [Google Scholar] [CrossRef] [PubMed]
- Hejnar, J.; Hajkova, P.; Plachy, J.; Elleder, D.; Stepanets, V.; Svoboda, J. CpG island protects Rous sarcoma virus-derived vectors integrated into nonpermissive cells from DNA methylation and transcriptional suppression. Proc. Natl. Acad. Sci. USA 2001, 98, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Merten, O.W. State-of-the-art of the production of retroviral vectors. J. Gene Med. 2004, 6, S105–S124. [Google Scholar] [CrossRef] [PubMed]
- Coroadinha, A.S.; Gama-Norton, L.; Amaral, A.I.; Hauser, H.; Alves, P.M.; Cruz, P.E. Production of retroviral vectors: Review. Curr. Gene Ther. 2010, 10, 456–473. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Starkey, W.; Vile, R.G. A replication-competent retrovirus arising from a split-function packaging cell line was generated by recombination events between the vector, one of the packaging constructs, and endogenous retroviral sequences. J. Virol. 1998, 72, 2663–2670. [Google Scholar] [PubMed]
- U.S. Food and Drug Administration. Guidance for Industry: Guidance for Human Somatic Cell Therapy and Gene Therapy. Available online: http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm072987.htm (accessed on 1 December 2014).
- Ni, Y.; Sun, S.; Oparaocha, I.; Humeau, L.; Davis, B.; Cohen, R.; Binder, G.; Chang, Y.N.; Slepushkin, V.; Dropulic, B. Generation of a packaging cell line for prolonged large-scale production of high-titer HIV-1-based lentiviral vector. J. Gene Med. 2005, 7, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.; Jones-Trower, A.; Vanin, E.F.; Stambaugh, K.; Mueller, S.N.; Anderson, W.F.; McGarrity, G.J. Characterization of a replication-competent retrovirus resulting from recombination of packaging and vector sequences. Hum. Gene Ther. 1994, 5, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Von Werder, A.; Seidler, B.; Schmid, R.M.; Schneider, G.; Saur, D. Production of avian retroviruses and tissue-specific somatic retroviral gene transfer in vivo using the RCAS/TVA system. Nat. Protoc. 2012, 7, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Morgan, R.A.; Cornetta, K.; June, C.H.; Binder-Scholl, G.; Dudley, M.E.; Feldman, S.A.; Rosenberg, S.A.; Shurtleff, S.A.; Rooney, C.M.; et al. Replication-competent retroviruses in gene-modified T cells used in clinical trials: Is it time to revise the testing requirements? Mol. Ther. 2012, 20, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Takeuchi, Y.; Martin, F.; Cosset, F.L.; Mitrophanous, K.; Collins, M. Continuous high-titer HIV-1 vector production. Nat. Biotechnol. 2003, 21, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Cosset, F.L.; Legras, C.; Chebloune, Y.; Savatier, P.; Thoraval, P.; Thomas, J.L.; Samarut, J.; Nigon, V.M.; Verdier, G. A new avian leukosis virus-based packaging cell line that uses two separate transcomplementing helper genomes. J. Virol. 1990, 64, 1070–1078. [Google Scholar] [PubMed]
- Schaefer-Klein, J.; Givol, I.; Barsov, E.V.; Whitcomb, J.M.; VanBrocklin, M.; Foster, D.N.; Federspiel, M.J.; Hughes, S.H. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology 1998, 248, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Himly, M.; Foster, D.N.; Bottoli, I.; Iacovoni, J.S.; Vogt, P.K. The DF-1 chicken fibroblast cell line: Transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 1998, 248, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Loftus, S.K.; Larson, D.M.; Watkins-Chow, D.; Church, D.M.; Pavan, W.J. Generation of RCAS vectors useful for functional genomic analyses. DNA Res. 2001, 8, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Schambach, A.; Baum, C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr. Opin. Immunol. 2012, 24, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Hotta, A.; Saito, Y.; Kyogoku, K.; Kawabe, Y.; Nishijima, K.; Kamihira, M.; Iijima, S. Characterization of transient expression system for retroviral vector production. J. Biosci. Bioeng. 2006, 101, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, V.; Boson, B.; Salmon, P.; Gay, W.; Negre, D.; Le Grand, R.; Trono, D.; Cosset, F.L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 2002, 100, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Ghani, K.; Cottin, S.; Kamen, A.; Caruso, M. Generation of a high-titer packaging cell line for the production of retroviral vectors in suspension and serum-free media. Gene Ther. 2007, 14, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Le Doux, J.M.; Morgan, J.R.; Snow, R.G.; Yarmush, M.L. Proteoglycans secreted by packaging cell lines inhibit retrovirus infection. J. Virol. 1996, 70, 6468–6473. [Google Scholar] [PubMed]
- Suomalainen, M.; Garoff, H. Incorporation of homologous and heterologous proteins into the envelope of Moloney murine leukemia virus. J. Virol. 1994, 68, 4879–4889. [Google Scholar] [PubMed]
- Cosset, F.L.; Takeuchi, Y.; Battini, J.L.; Weiss, R.A.; Collins, M.K. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J. Virol. 1995, 69, 7430–7436. [Google Scholar] [PubMed]
- Wicke, D.C.; Meyer, J.; Buesche, G.; Heckl, D.; Kreipe, H.; Li, Z.; Welte, K.H.; Ballmaier, M.; Baum, C.; Modlich, U. Gene therapy of MPL deficiency: Challenging balance between leukemia and pancytopenia. Mol. Ther. 2010, 18, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, C.; Galla, M.; Dannhauser, P.N.; Maetzig, T.; Sodeik, B.; Schambach, A.; Baum, C. Pseudotype-independent nonspecific uptake of gammaretroviral and lentiviral particles in human cells. Hum. Gene Ther. 2012, 23, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Hauer, J.; Lim, A.; Picard, C.; Wang, G.P.; Berry, C.C.; Martinache, C.; Rieux-Laucat, F.; Latour, S.; Belohradsky, B.H.; et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2010, 363, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Adams, S.; Howe, S.J.; Al Ghonaium, A.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2011, 3, 97ra79. [Google Scholar] [PubMed]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [PubMed]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Aebersold, P.; Cornetta, K.; Kasid, A.; Morgan, R.A.; Moen, R.; Karson, E.M.; Lotze, M.T.; Yang, J.C.; Topalian, S.L.; et al. Gene transfer into humans—Immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J. Med. 1990, 323, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.P.; Xue, S.A.; Thomas, S.; Cesco-Gaspere, M.; Tranter, A.; Willcox, B.; Lee, S.P.; Steven, N.; Morris, E.C.; Stauss, H.J. Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther. 2008, 15, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Riviere, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra153. [Google Scholar] [CrossRef]
- Newrzela, S.; Al-Ghaili, N.; Heinrich, T.; Petkova, M.; Hartmann, S.; Rengstl, B.; Kumar, A.; Jack, H.M.; Gerdes, S.; Roeder, I.; et al. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia 2012, 26, 2499–2507. [Google Scholar] [CrossRef] [PubMed]
- Newrzela, S.; Cornils, K.; Li, Z.; Baum, C.; Brugman, M.H.; Hartmann, M.; Meyer, J.; Hartmann, S.; Hansmann, M.L.; Fehse, B.; et al. Resistance of mature T cells to oncogene transformation. Blood 2008, 112, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, T.; Rengstl, B.; Muik, A.; Petkova, M.; Schmid, F.; Wistinghausen, R.; Warner, K.; Crispatzu, G.; Hansmann, M.L.; Herling, M.; et al. Mature T-cell lymphomagenesis induced by retroviral insertional activation of janus kinase 1. Mol. Ther. 2013, 21, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suerth, J.D.; Labenski, V.; Schambach, A. Alpharetroviral Vectors: From a Cancer-Causing Agent to a Useful Tool for Human Gene Therapy. Viruses 2014, 6, 4811-4838. https://doi.org/10.3390/v6124811
Suerth JD, Labenski V, Schambach A. Alpharetroviral Vectors: From a Cancer-Causing Agent to a Useful Tool for Human Gene Therapy. Viruses. 2014; 6(12):4811-4838. https://doi.org/10.3390/v6124811
Chicago/Turabian StyleSuerth, Julia D., Verena Labenski, and Axel Schambach. 2014. "Alpharetroviral Vectors: From a Cancer-Causing Agent to a Useful Tool for Human Gene Therapy" Viruses 6, no. 12: 4811-4838. https://doi.org/10.3390/v6124811
APA StyleSuerth, J. D., Labenski, V., & Schambach, A. (2014). Alpharetroviral Vectors: From a Cancer-Causing Agent to a Useful Tool for Human Gene Therapy. Viruses, 6(12), 4811-4838. https://doi.org/10.3390/v6124811