Cholesterol Balance in Prion Diseases and Alzheimer’s Disease


Abstract
:1. Introduction
2. Cholesterol Metabolism
2.1. Synthesis and Uptake
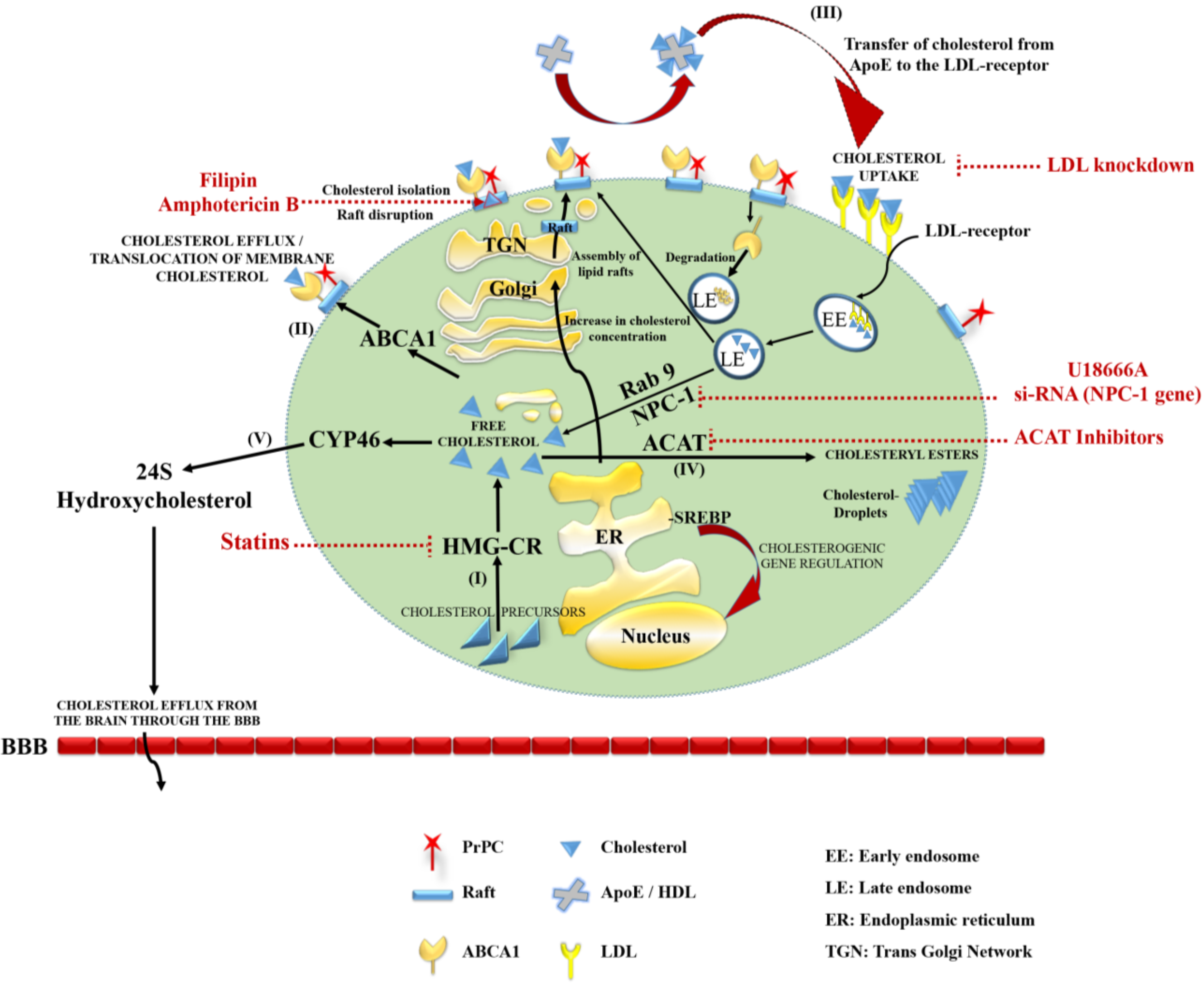
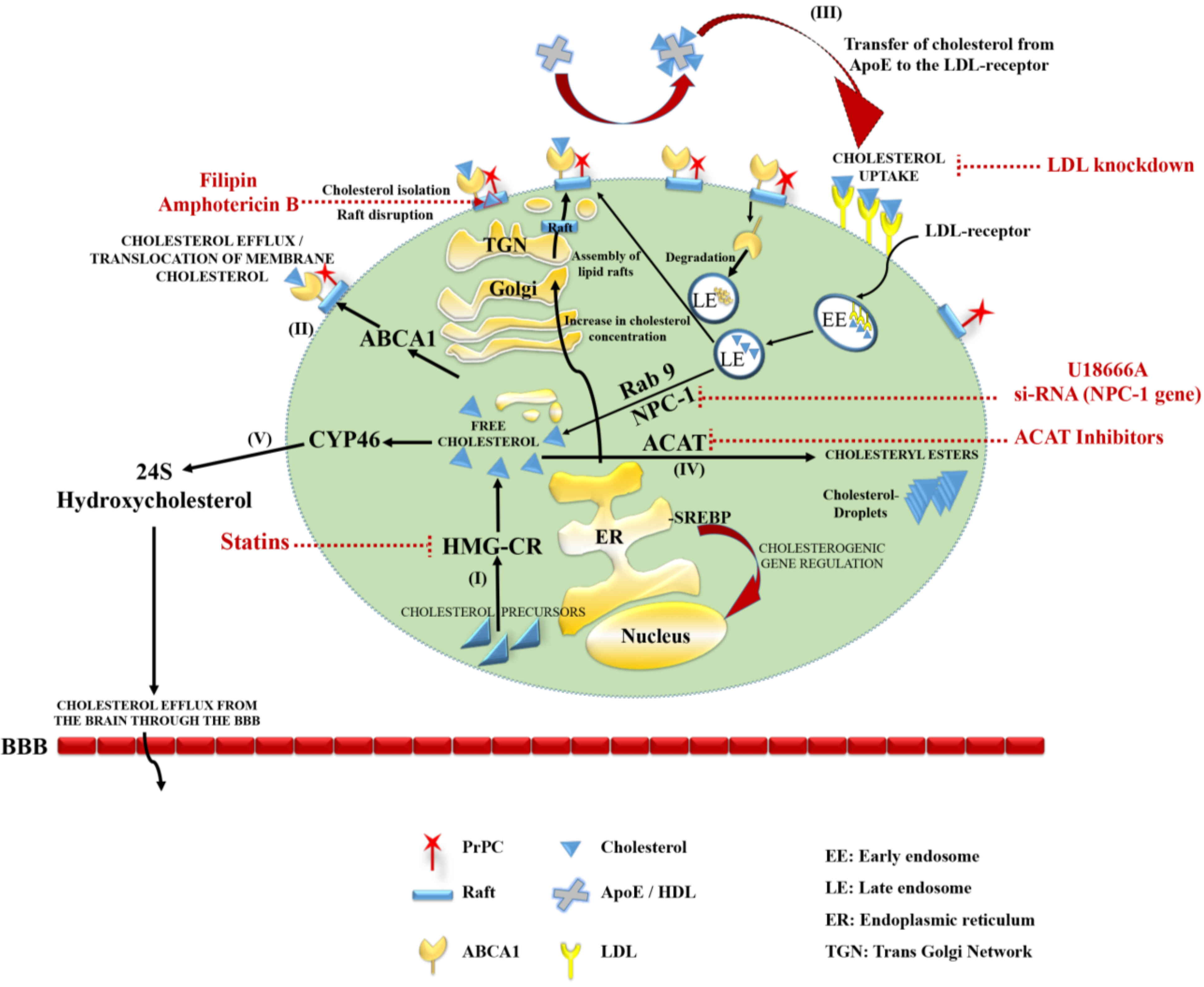
2.2. Storage and Elimination
2.3. Cellular Cholesterol Localization and Trafficking
2.4. Cholesterol in the Brain
2.4.1. Synthesis: Neurons and Astrocytes Interact in Cholesterol Homeostasis
2.4.2. Cholesterol Elimination from the Brain
3. Role of Cholesterol in the Formation of Microdomains
4. Cholesterol and Prions
4.1. Cholesterol-Enriched Lipid Rafts and Prion Protein Isoforms
4.2. How does Interference in Cholesterol Metabolism Influence PrPSc Propagation?
4.2.1. Inhibition of Cholesterol Synthesis
4.2.2. Cholesterol Esterification and Prion Formation
4.2.3. Cholesterol at the Plasma Membrane, PrP Isoforms and Prion Disease Progression
4.2.4. Manipulation of Cholesterol in the Endocytic Pathway
4.2.5. Cholesterol, a Potential Target for Treatment of Prion Diseases in Vivo

{kind=link}
| Compound | Model | Cholesterol level | Effect on PrPC/PrPSc or APP/Aβ level | References |
|---|---|---|---|---|
| Lovastatin | ScN2a cells | Depletion of cellular cholesterol | ↓PrPC degradation | [48] |
| HaB cells | ↓PrPSc accumulation | |||
| Squalestatin | ScN2a cells | Reduction of cellular cholesterol | ↓PrPSc accumulation | [51] |
| SMB cells | ||||
| ScGT1 cells | ||||
| Atorvastatin | N2a cells | Inhibition of cellular cholesterol | ↑PrPC level | [100] |
| Filipin | ScN2a cells | Sequestration of cholesterol and disruption of membrane structure | ↓PrPSc accumulation | [24] |
| Amphotericin B | ScN2a cells | Interaction with cholesterol and disruption of membrane structure | ↓PrPSc accumulation | [109] |
| ScGT1-7 cells | ||||
| ACAT inhibitors | N2a cells | Inhibition of cholesterol ester formation (cholesterol relocation) | ↓PrPSc accumulation | [102] |
| Simvastatin | C57BL/6 infected with ME7 (IC) | Cholesterol level unchanged | PrPC/PrPSc unchanged | [121] |
| Increased survival time | ||||
| Simvastatin | C57BL/6 infected with ME7 (IC) | Cholesterol level unchanged | PrPC/PrPSc unchanged | [121] |
| No significant effect | ||||
| Simvastatin | FVB/N infected with RML (IC/IP) | Cholesterol level unchanged | PrPC/PrPSc unchanged | [122] |
| Increased onset of symptoms | ||||
| Simvastatin | C57BL/6 infected with 139A (IC) | Cholesterol level unchanged | PrPC/PrPSc unchanged | [123] |
| Increased survival time | ||||
| Pravastatin | C57BL/6 mice infected with 139A (IC) | Cholesterol level unchanged | PrPC/PrPSc unchanged | [124] |
| Increased survival time | ||||
| Amphotericin B | Syrian hamster infected with 263 K (IC/IP) | Interaction with cholesterol and disruption of membrane structure | ↓PrPSc accumulation | [125,126] |
| Increased survival time | ||||
| Amphotericin B | Scrapie infected mice and hamsters | Interaction with cholesterol and disruption of membrane structure | ↓PrPSc accumulation | [127,128,129,130] |
| Simvastatin/Lovastatin | Rat Hippocampal neurons | Depletion of cellular cholesterol | ↓Aβ production | [131] |
| HEK 293 cells | ||||
| SHSY5Y cells | ||||
| ACAT inhibitors | CHO cells hippocampal primary neurons | inhibition of cholesterol ester formation (cholesterol relocation) | ↓Aβ production | [132] |
| ACAT inhibitors | transgenic APP-mice (London and Swedish mutation) | inhibition of cholesterol ester formation (cholesterol relocation) | ↓Amyloid plaques | [133] |
| ↓Aβ production | ||||
| Slightly improve of spatial learning | ||||
| Seladin 1 | Rat hippocampal neurons | Decrease cholesterol level | ↑Aβ production | [134] |
| CHO cells | ||||
| Statins (cholesterol-lowering drugs) | The Rotterdam study (6992 Non AD subjects) | Cholesterol-lowering | Reduced risk of late-onset AD | [135] |
4.3. How Does Prion Infection Interfere with Cholesterol Metabolism?
5. Cholesterol in Other Neurodegenerative Disorders
5.1. Cholesterol and AD
5.2. Cholesterol in the Brains of AD Patients
5.3. Cholesterol Reducing Agents in AD Models
5.4. Genes that Affect Cholesterol Metabolism and AD
6. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heikenwalder, M.; Miele, G. Progress and problems in the biology, diagnostics, and therapeutics of prion diseases. J. Clin. Investig. 2004, 114, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Balachandran, A.; Westaway, D. The expanding universe of prion diseases. PLoS Pathog. 2006, 2, e26. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; McBride, P.A.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar]
- Huang, Z.; Prusiner, S.B.; Cohen, F.E. Structures of prion proteins and conformational models for prion diseases. Curr. Top. Microbiol. Immunol. 1996, 207, 49–67. [Google Scholar] [PubMed]
- Lansbury, P.T., Jr.; Caughey, B. The chemistry of scrapie infection: Implications of the “ice 9” metaphor. Chem. Biol. 1995, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J.; Kocisko, D.A.; Lansbury, P.T., Jr. Scrapie infectivity correlates with converting activity, protease resistance, and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J. Virol. 1997, 71, 4107–4110. [Google Scholar] [PubMed]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Milhavet, O.; Lehmann, S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res. 2002, 38, 328–339. [Google Scholar] [CrossRef]
- Pauly, P.C.; Harris, D.A. Copper stimulates endocytosis of the prion protein. J. Biol. Chem. 1998, 273, 33107–33110. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Gains, M.; LeBlanc, A.C. Neuroprotective functions of prion protein. J. Neurosci. Res. 2004, 75, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Halliday, W.G.; Bell, J.; Johnston, A.R.; MacLeod, N.K.; Ingham, C.; Sayers, A.R.; Brown, D.A.; Fraser, J.R. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol. Appl. Neurobiol. 2000, 26, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Chesebro, B.; Race, R.; Wehrly, K.; Nishio, J.; Bloom, M.; Lechner, D.; Bergstrom, S.; Robbins, K.; Mayer, L.; Keith, J.M.; et al. Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 1985, 315, 331–333. [Google Scholar] [CrossRef]
- Robakis, N.K.; Sawh, P.R.; Wolfe, G.C.; Rubenstein, R.; Carp, R.I.; Innis, M.A. Isolation of a cDNA clone encoding the leader peptide of prion protein and expression of the homologous gene in various tissues. Proc. Natl. Acad. Sci. USA 1986, 83, 6377–6381. [Google Scholar] [CrossRef] [PubMed]
- Bendheim, P.E.; Brown, H.R.; Rudelli, R.D.; Scala, L.J.; Goller, N.L.; Wen, G.Y.; Kascsak, R.J.; Cashman, N.R.; Bolton, D.C. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 1992, 42, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, H.A.; Prusiner, S.B.; Stowring, L.E.; DeArmond, S.J. Scrapie prion proteins are synthesized in neurons. Am. J. Pathol. 1986, 122, 1–5. [Google Scholar] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Harmey, J.H.; Doyle, D.; Brown, V.; Rogers, M.S. The cellular isoform of the prion protein, PrPC, is associated with caveolae in mouse neuroblastoma (N2a) cells. Biochem. Biophys. Res Commun. 1995, 210, 753–759. [Google Scholar] [CrossRef]
- Vey, M.; Pilkuhn, S.; Wille, H.; Nixon, R.; DeArmond, S.J.; Smart, E.J.; Anderson, R.G.; Taraboulos, A.; Prusiner, S.B. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. USA 1996, 93, 14945–14949. [Google Scholar] [CrossRef] [PubMed]
- Silvius, J.R. Role of cholesterol in lipid raft formation: Lessons from lipid model systems. Biochim. Biophys. Acta 2003, 1610, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Vey, M.; Scott, M.; Pilkuhn, S.; Cohen, F.E.; Prusiner, S.B. COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc. Natl. Acad. Sci. USA 1997, 94, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Marella, M.; Lehmann, S.; Grassi, J.; Chabry, J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 2002, 277, 25457–25464. [Google Scholar] [CrossRef]
- Peters, P.J.; Mironov, A., Jr.; Peretz, D.; van Donselaar, E.; Leclerc, E.; Erpel, S.; DeArmond, S.J.; Burton, D.R.; Williamson, R.A.; Vey, M.; et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell Biol. 2003, 162, 703–717. [Google Scholar] [CrossRef]
- Shyng, S.L.; Heuser, J.E.; Harris, D.A. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J. Cell Biol. 1994, 125, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Watt, N.T.; Perera, W.S.; Hooper, N.M. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 2005, 118, 5141–5153. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Milhavet, O.; Mange, A. Trafficking of the cellular isoform of the prion protein. Biomed. Pharmacother. 1999, 53, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Race, R.E.; Ernst, D.; Buchmeier, M.J.; Chesebro, B. Prion protein biosynthesis in scrapie-infected and uninfected neuroblastoma cells. J. Virol. 1989, 63, 175–181. [Google Scholar] [PubMed]
- Shyng, S.L.; Huber, M.T.; Harris, D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993, 268, 15922–15928. [Google Scholar] [PubMed]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar] [PubMed]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Prusiner, S.B. Biosynthesis of the prion proteins in scrapie-infected cells in culture. Braz. J. Med. Biol. Res. 1994, 27, 303–307. [Google Scholar] [PubMed]
- Beranger, F.; Mange, A.; Goud, B.; Lehmann, S. Stimulation of PrPC retrograde transport toward the endoplasmic reticulum increases accumulation of PrPSc in prion-infected cells. J. Biol. Chem. 2002, 277, 38972–38977. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Goodsir, C.M.; Bruce, M.E.; McBride, P.A.; Scott, J.R.; Halliday, W.G. Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci. Lett. 1992, 147, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B. Scrapie associated PrP accumulation and its prevention: Insights from cell culture. Br. Med. Bull. 1993, 49, 860–872. [Google Scholar] [PubMed]
- McKinley, M.P.; Taraboulos, A.; Kenaga, L.; Serban, D.; Stieber, A.; DeArmond, S.J.; Prusiner, S.B.; Gonatas, N. Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab. Investig. 1991, 65, 622–630. [Google Scholar] [PubMed]
- Arnold, J.E.; Tipler, C.; Laszlo, L.; Hope, J.; Landon, M.; Mayer, R.J. The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J. Pathol. 1995, 176, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Veith, N.M.; Plattner, H.; Stuermer, C.A.; Schulz-Schaeffer, W.J.; Burkle, A. Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 2009, 88, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.J.; Parkyn, C.J.; Jen, A. Traffic of prion protein between different compartments on the neuronal surface, and the propagation of prion disease. FEBS Lett. 2006, 580, 5565–5571. [Google Scholar] [CrossRef] [PubMed]
- Godsave, S.F.; Wille, H.; Kujala, P.; Latawiec, D.; DeArmond, S.J.; Serban, A.; Prusiner, S.B.; Peters, P.J. Cryo-immunogold electron microscopy for prions: Toward identification of a conversion site. J. Neurosci. 2008, 28, 12489–12499. [Google Scholar] [CrossRef] [PubMed]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Raeber, A.J.; Borchelt, D.R.; Serban, D.; Prusiner, S.B. Synthesis and trafficking of prion proteins in cultured cells. Mol. Biol. Cell 1992, 3, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Gilch, S.; Winklhofer, K.F.; Groschup, M.H.; Nunziante, M.; Lucassen, R.; Spielhaupter, C.; Muranyi, W.; Riesner, D.; Tatzelt, J.; Schatzl, H.M. Intracellular re-routing of prion protein prevents propagation of PrPSc and delays onset of prion disease. EMBO J. 2001, 20, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Supattapone, S.; Nishina, K.; Rees, J.R. Pharmacological approaches to prion research. Biochem. Pharmacol. 2002, 63, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Geoghegan, J.C.; Nishina, K.; Kascsak, R.; Williamson, R.A.; Supattapone, S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. J. Biol. Chem. 2005, 280, 26873–26879. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.; Rabbanian, S.; Sutton, L.; Andre, R.; Arora, P.; Moonga, J.; Clarke, A.R.; Schiavo, G.; Jat, P.; Collinge, J.; et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2011, 2, 281. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Naslavsky, N.; Stein, R.; Yanai, A.; Friedlander, G.; Taraboulos, A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 1997, 272, 6324–6331. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.S.; Wehrly, K.; Dorward, D.W.; Chesebro, B.; Caughey, B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrPres (PrPSc) into contiguous membranes. EMBO J. 2002, 21, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Salmona, M.; Diomede, L.; Williams, A. Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J. Biol. Chem. 2004, 279, 14983–14990. [Google Scholar] [CrossRef] [PubMed]
- Gilch, S.; Kehler, C.; Schatzl, H.M. The prion protein requires cholesterol for cell surface localization. Mol. Cell. Neurosci. 2006, 31, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Models Mech. 2012, 5, 746–755. [Google Scholar] [CrossRef]
- Lange, Y.; Ye, J.; Rigney, M.; Steck, T.L. Regulation of endoplasmic reticulum cholesterol by plasma membrane cholesterol. J. Lipid Res. 1999, 40, 2264–2270. [Google Scholar] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Reid, P.C.; Sugii, S.; Ohgami, N.; Cruz, J.C.; Chang, C.C. Niemann-Pick type C disease and intracellular cholesterol trafficking. J. Biol. Chem. 2005, 280, 20917–20920. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Zha, X.; Tabas, I.; Maxfield, F.R. Cholesterol distribution in living cells: Fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys. J. 1998, 75, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and disease. J. Clin. Investig. 2002, 110, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, N.; Scott, D.W.; Castle, J.D.; Casanova, J.E.; Schwartz, M.A. Arf6 and microtubules in adhesion-dependent trafficking of lipid rafts. Nat. Cell Biol. 2007, 9, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Gupta, N. Tether and trap: Regulation of membrane-raft dynamics by actin-binding proteins. Nat. Rev. Immunol. 2007, 7, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Benson, E.P.; Saito, M.; Rosenberg, A. Metabolism of cholesterol and triacylglycerol in cultured chick neuronal cells, glial cells, and fibroblasts: Accumulation of esterified cholesterol in serum-free culture. J. Neurosci. Res. 1987, 18, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 2004, 279, 14009–14015. [Google Scholar] [CrossRef] [PubMed]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Brendza, R.P.; Heuser, J.E.; Kierson, M.; Cirrito, J.R.; Fryer, J.; Sullivan, P.M.; Fagan, A.M.; Han, X.; Holtzman, D.M. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human ApoE transgenic mice. Neurochem. Int. 2001, 39, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Lutjohann, D.; Breuer, O.; Ahlborg, G.; Nennesmo, I.; Siden, A.; Diczfalusy, U.; Bjorkhem, I. Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. USA 1996, 93, 9799–9804. [Google Scholar] [CrossRef] [PubMed]
- Bjorkhem, I.; Lutjohann, D.; Breuer, O.; Sakinis, A.; Wennmalm, A. Importance of a novel oxidative mechanism for elimination of brain cholesterol. Turnover of cholesterol and 24S-hydroxycholesterol in rat brain as measured with 18O2 techniques in vivo and in vitro. J. Biol. Chem. 1997, 272, 30178–30184. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Caccia, C. 24S-hydroxycholesterol in plasma: A marker of cholesterol turnover in neurodegenerative diseases. Biochimie 2013, 95, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.G.; Guileyardo, J.M.; Russell, D.W. CDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. USA 1999, 96, 7238–7243. [Google Scholar] [CrossRef] [PubMed]
- Abildayeva, K.; Jansen, P.J.; Hirsch-Reinshagen, V.; Bloks, V.W.; Bakker, A.H.; Ramaekers, F.C.; de Vente, J.; Groen, A.K.; Wellington, C.L.; Kuipers, F.; et al. 24S-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 2006, 281, 12799–12808. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W. Outsourcing in the brain: Do neurons depend on cholesterol delivery by astrocytes? BioEssays 2003, 25, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Shobab, L.A.; Hsiung, G.Y.; Feldman, H.H. Cholesterol in Alzheimer’s disease. Lancet Neurol. 2005, 4, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, L.D. Dynamic lipid heterogeneity and receptor events. Mol. Membr. Biol. 1995, 12, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Slotte, J.P. Sphingomyelin-cholesterol interactions in biological and model membranes. Chem. Phys. Lipids 1999, 102, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Honsho, M.; Ekroos, K.; Shevchenko, A.; Simons, K. Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. USA 2003, 100, 5795–5800. [Google Scholar] [CrossRef] [PubMed]
- Korlach, J.; Schwille, P.; Webb, W.W.; Feigenson, G.W. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- Kahya, N.; Scherfeld, D.; Bacia, K.; Poolman, B.; Schwille, P. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 2003, 278, 28109–28115. [Google Scholar] [CrossRef] [PubMed]
- Gaus, K.; Gratton, E.; Kable, E.P.; Jones, A.S.; Gelissen, I.; Kritharides, L.; Jessup, W. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 15554–15559. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.L.; Dawson, A.M. Free and esterified cholesterol in the cerebrospinal fluid of goats affected with experimental scrapie. Res. Vet. Sci. 1965, 6, 274–279. [Google Scholar] [PubMed]
- Baron, G.S.; Caughey, B. Effect of glycosylphosphatidylinositol anchor-dependent and independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J. Biol. Chem. 2003, 278, 14883–14892. [Google Scholar] [CrossRef] [PubMed]
- Nishina, K.; Deleault, N.R.; Lucassen, R.W.; Supattapone, S. In vitro prion protein conversion in detergent-solubilized membranes. Biochemistry 2004, 43, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.R.; Ratte, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.; Collinge, J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002, 21, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A. Cell biology. Prion toxicity: All sail and no anchor. Science 2005, 308, 1420–1421. [Google Scholar] [CrossRef] [PubMed]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rapp, J.; Martin-Lanneree, S.; Hirsch, T.Z.; Pradines, E.; Alleaume-Butaux, A.; Schneider, B.; Baudry, A.; Launay, J.M.; Mouillet-Richard, S. A PrPC-caveolin-Lyn complex negatively controls neuronal GSK3β and serotonin 1B receptor. Sci. Rep. 2014, 4, 4881. [Google Scholar] [CrossRef] [PubMed]
- Roffe, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Mutel, V.; Pietri, M.; Ermonval, M.; Mouillet-Richard, S.; Kellermann, O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and non neuronal cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13326–13331. [Google Scholar] [CrossRef] [PubMed]
- Pradines, E.; Loubet, D.; Mouillet-Richard, S.; Manivet, P.; Launay, J.M.; Kellermann, O.; Schneider, B. Cellular prion protein coupling to TACE-dependent TNF-alpha shedding controls neurotransmitter catabolism in neuronal cells. J. Neurochem. 2009, 110, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Campana, V.; Paladino, S.; Stornaiuolo, M.; Nitsch, L.; Zurzolo, C. PrPC association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell 2004, 15, 4031–4042. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Paladino, S.; Campana, V.; Grassi, J.; Nitsch, L.; Zurzolo, C. PrPC is sorted to the basolateral membrane of epithelial cells independently of its association with rafts. Traffic 2002, 3, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, T.; Fisher, S.; Olofsson, S.; Endo, T.; Groth, D.; Tarentino, A.; Borchelt, D.R.; Teplow, D.; Hood, L.; Burlingame, A.; et al. Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch. Biochem. Biophys. 1989, 274, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.M.; Stahl, N.; Prusiner, S.B. Purification and properties of the cellular prion protein from Syrian hamster brain. Protein Sci. 1992, 1, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Yasutaka, Y.; Nishioku, T.; Nakashima, A.; Futagami, K.; Yamauchi, A.; Kataoka, Y. Atorvastatin stimulates neuroblastoma cells to induce neurite outgrowth by increasing cellular prion protein expression. Neurosci. Lett. 2012, 531, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Tayebi, M.; Williams, A. Cholesterol esterification reduces the neurotoxicity of prions. Neuropharmacology 2008, 54, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Pani, A.; Norfo, C.; Abete, C.; Mulas, C.; Putzolu, M.; Laconi, S.; Orru, C.D.; Cannas, M.D.; Vascellari, S.; La Colla, P.; et al. Antiprion activity of cholesterol esterification modulators: A comparative study using ex vivo sheep fibroblasts and lymphocytes and mouse neuroblastoma cell lines. Antimicrob. Agents Chemother. 2007, 51, 4141–4147. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Banni, S.; Vacca, C.; Vetrugno, V.; Cardone, F.; Di Bari, M.A.; La Colla, P.; Pani, A. Accumulation and aberrant composition of cholesteryl esters in scrapie-infected N2a cells and C57BL/6 mouse brains. Lipids Health Dis. 2011, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Cerneus, D.P.; Ueffing, E.; Posthuma, G.; Strous, G.J.; van der Ende, A. Detergent insolubility of alkaline phosphatase during biosynthetic transport and endocytosis. Role of cholesterol. J. Biol. Chem. 1993, 268, 3150–3155. [Google Scholar] [PubMed]
- Scheiffele, P.; Roth, M.G.; Simons, K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997, 16, 5501–5508. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Hooper, N.M. The prion protein and lipid rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, P.; Haskins, N.J.; Howlett, D.R. Beta-cyclodextrin interacts with the Alzheimer amyloid beta-A4 peptide. FEBS Lett. 1994, 341, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Amyx, H.; Salazar, A.M.; Gajdusek, D.C.; Gibbs, C.J. Chemotherapeutic trial in experimental slow virus disease. Neurology 1984, 34, 149. [Google Scholar]
- Mange, A.; Nishida, N.; Milhavet, O.; McMahon, H.E.; Casanova, D.; Lehmann, S. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J. Virol. 2000, 74, 3135–3140. [Google Scholar] [CrossRef] [PubMed]
- Prior, M.; Lehmann, S.; Sy, M.S.; Molloy, B.; McMahon, H.E. Cyclodextrins inhibit replication of scrapie prion protein in cell culture. J. Virol. 2007, 81, 11195–11207. [Google Scholar] [CrossRef] [PubMed]
- Gilch, S.; Bach, C.; Lutzny, G.; Vorberg, I.; Schatzl, H.M. Inhibition of cholesterol recycling impairs cellular PrPSc propagation. Cell. Mol. Life Sci. 2009, 66, 3979–3991. [Google Scholar] [CrossRef] [PubMed]
- Klingenstein, R.; Lober, S.; Kujala, P.; Godsave, S.; Leliveld, S.R.; Gmeiner, P.; Peters, P.J.; Korth, C. Tricyclic antidepressants, quinacrine and a novel, synthetic chimera thereof clear prions by destabilizing detergent-resistant membrane compartments. J. Neurochem. 2006, 98, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Beuchat, M.H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118. [Google Scholar] [PubMed]
- Lange, Y.; Ye, J.; Rigney, M.; Steck, T. Cholesterol movement in Niemann-Pick type C cells and in cells treated with amphiphiles. J. Biol. Chem. 2000, 275, 17468–17475. [Google Scholar] [CrossRef] [PubMed]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schatzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schatzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Aguib, Y.; Schatzl, H.M. Autophagy, prion infection and their mutual interactions. Curr. Issues Mol. Biol. 2010, 12, 87–97. [Google Scholar] [PubMed]
- Ganley, I.G.; Pfeffer, S.R. Cholesterol accumulation sequesters Rab9 and disrupts late endosome function in NPC1-deficient cells. J. Biol. Chem. 2006, 281, 17890–17899. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Nakamura, Y.; Nishijima, M.; Yamakawa, Y. Prevention of prion propagation by dehydrocholesterol reductase inhibitors in cultured cells and a therapeutic trial in mice. Biol. Pharm.Bull. 2007, 30, 835–838. [Google Scholar] [CrossRef] [PubMed]
- Marzo, L.; Marijanovic, Z.; Browman, D.; Chamoun, Z.; Caputo, A.; Zurzolo, C. 4-hydroxytamoxifen leads to PrPSc clearance by conveying both PrPC and PrPSc to lysosomes independently of autophagy. J. Cell Sci. 2013, 126, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Kempster, S.; Bate, C.; Williams, A. Simvastatin treatment prolongs the survival of scrapie-infected mice. Neuroreport 2007, 18, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Haviv, Y.; Avrahami, D.; Ovadia, H.; Ben-Hur, T.; Gabizon, R.; Sharon, R. Induced neuroprotection independently from PrPSc accumulation in a mouse model for prion disease treated with simvastatin. Arch. Neurol. 2008, 65, 762–775. [Google Scholar] [PubMed]
- Mok, S.W.; Thelen, K.M.; Riemer, C.; Bamme, T.; Gultner, S.; Lutjohann, D.; Baier, M. Simvastatin prolongs survival times in prion infections of the central nervous system. Biochem. Biophys. Res. Commun. 2006, 348, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Vetrugno, V.; Di Bari, M.A.; Nonno, R.; Puopolo, M.; D’Agostino, C.; Pirisinu, L.; Pocchiari, M.; Agrimi, U. Oral pravastatin prolongs survival time of scrapie-infected mice. J. Gen. Virol. 2009, 90, 1775–1780. [Google Scholar] [CrossRef] [PubMed]
- Pocchiari, M.; Schmittinger, S.; Masullo, C. Amphotericin B delays the incubation period of scrapie in intracerebrally inoculated hamsters. J. Gen. Virol. 1987, 68, 219–223. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, D.; Kaczkowski, J.; Marsh, R.; Aiken, J. Amphotericin B delays both scrapie agent replication and PrPres accumulation early in infection. J. Virol. 1994, 68, 7534–7536. [Google Scholar] [PubMed]
- Demaimay, R.; Race, R.; Chesebro, B. Effectiveness of polyene antibiotics in treatment of transmissible spongiform encephalopathy in transgenic mice expressing Syrian hamster PrP only in neurons. J. Virol. 1999, 73, 3511–3513. [Google Scholar] [PubMed]
- Adjou, K.T.; Demaimay, R.; Lasmezas, C.I.; Seman, M.; Deslys, J.P.; Dormont, D. Differential effects of a new Amphotericin B derivative, MS-8209, on mouse BSE and scrapie: Implications for the mechanism of action of polyene antibiotics. Res. Virol. 1996, 147, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Demaimay, R.; Adjou, K.T.; Beringue, V.; Demart, S.; Lasmezas, C.I.; Deslys, J.P.; Seman, M.; Dormont, D. Late treatment with polyene antibiotics can prolong the survival time of scrapie-infected animals. J. Virol. 1997, 71, 9685–9689. [Google Scholar] [PubMed]
- Adjou, K.T.; Demaimay, R.; Lasmezas, C.; Deslys, J.P.; Seman, M.; Dormont, D. MS-8209, a new Amphotericin B derivative, provides enhanced efficacy in delaying hamster scrapie. Antimicrob. Agents Chemother. 1995, 39, 2810–2812. [Google Scholar] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides A-beta 42 and A-beta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.; Berezovska, O.; Hyman, B.T.; Chang, T.Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Hutter-Paier, B.; Huttunen, H.J.; Puglielli, L.; Eckman, C.B.; Kim, D.Y.; Hofmeister, A.; Moir, R.D.; Domnitz, S.B.; Frosch, M.P.; Windisch, M.; et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 2004, 44, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Abad-Rodriguez, J.; Ledesma, M.D.; Craessaerts, K.; Perga, S.; Medina, M.; Delacourte, A.; Dingwall, C.; De Strooper, B.; Dotti, C.G. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J. Cell Biol. 2004, 167, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Haag, M.D.; Hofman, A.; Koudstaal, P.J.; Stricker, B.H.; Breteler, M.M. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The rotterdam study. J. Neurol. Neurosurg. Psychiatry 2009, 80, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Hernandez-Romero, M.C.; Machado, A.; Cano, J. Zocor forte (simvastatin) has a neuroprotective effect against LPS striatal dopaminergic terminals injury, whereas against MPP+ does not. Eur. J. Pharmacol. 2009, 609, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.G.; Ingrosso, L.; Ladogana, A.; Masullo, C.; Pocchiari, M. Amphotericin B treatment dissociates in vivo replication of the scrapie agent from PrP accumulation. Nature 1992, 356, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Cronier, S.; Beringue, V.; Bellon, A.; Peyrin, J.M.; Laude, H. Prion strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J. Virol. 2007, 81, 13794–13800. [Google Scholar] [CrossRef] [PubMed]
- Bach, C.; Gilch, S.; Rost, R.; Greenwood, A.D.; Horsch, M.; Hajj, G.N.; Brodesser, S.; Facius, A.; Schadler, S.; Sandhoff, K.; et al. Prion-induced activation of cholesterogenic gene expression by SREBP2 in neuronal cells. J. Biol. Chem. 2009, 284, 31260–31269. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Rebus, S.; McKimmie, C.S.; Robertson, K.; Williams, A.; Fazakerley, J.K. Gene expression profiling of the preclinical scrapie-infected hippocampus. Biochem. Biophys. Res. Commun. 2005, 334, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Qiu, Y.; Hyun, W.; Nixon, R.; VanCleff, J.; Sanchez-Salazar, J.; Prusiner, S.B.; DeArmond, S.J. Decreased receptor-mediated calcium response in prion-infected cells correlates with decreased membrane fluidity and IP3 release. Neurology 1996, 47, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Tayebi, M.; Diomede, L.; Salmona, M.; Williams, A. Docosahexaenoic and eicosapentaenoic acids increase prion formation in neuronal cells. BMC Biol. 2008, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Riemer, C.; Neidhold, S.; Burwinkel, M.; Schwarz, A.; Schultz, J.; Kratzschmar, J.; Monning, U.; Baier, M. Gene expression profiling of scrapie-infected brain tissue. Biochem. Biophys. Res. Commun. 2004, 323, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Hummel, M.; Mitteregger, G.; Pace, C.; Windl, O.; Mansmann, U.; Kretzschmar, H.A. Transcriptome analysis reveals altered cholesterol metabolism during the neurodegeneration in mouse scrapie model. J. Neurochem. 2007, 102, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; McClain, D.; Young, R.; Carlson, G.A. Cholesterol transporter ATP-binding cassette A1 (ABCA1) is elevated in prion disease and affects PrPC and PrPSc concentrations in cultured cells. J. Gen. Virol. 2008, 89, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.L.; Guo, B.; Scicluna, B.; Coleman, B.M.; Lawson, V.A.; Ellett, L.; Meikle, P.J.; Bukrinsky, M.; Mukhamedova, N.; Sviridov, D.; et al. Prion infection impairs cholesterol metabolism in neuronal cells. J. Biol. Chem. 2014, 289, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.K.; Lin, D.S.; Connor, W.E. Cholestanol metabolism in patients with cerebrotendinous xanthomatosis: Absorption, turnover, and tissue deposition. J. Lipid Res. 2007, 48, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Bjorkhem, I.; Leitersdorf, E. Sterol 27-hydroxylase deficiency: A rare cause of xanthomas in normocholesterolemic humans. Trends Endocrinol. Metab. 2000, 11, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Bjorkhem, I.; Starck, L.; Andersson, U.; Lutjohann, D.; von Bahr, S.; Pikuleva, I.; Babiker, A.; Diczfalusy, U. Oxysterols in the circulation of patients with the Smith-Lemli-Opitz syndrome: Abnormal levels of 24S- and 27-hydroxycholesterol. J. Lipid Res. 2001, 42, 366–371. [Google Scholar] [PubMed]
- Porter, F.D. Smith-Lemli-Opitz syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum.Genet. 2008, 16, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Lipid imbalance in the neurological disorder, Niemann-Pick C disease. FEBS Lett. 2006, 580, 5518–5524. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Oltersdorf, T.; Lee, M.G.; Barbour, R.; Blomquist, C.; Davis, D.L.; Bryant, K.; Fritz, L.C.; Galasko, D.; Thal, L.J.; et al. Secretion of beta-amyloid precursor protein cleaved at the amino terminus of the beta-amyloid peptide. Nature 1993, 361, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Cutler, R.G.; Kelly, J.; Storie, K.; Pedersen, W.A.; Tammara, A.; Hatanpaa, K.; Troncoso, J.C.; Mattson, M.P. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and A-beta production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Bakovic, M. Lipid rafts in health and disease. Biol. Cell/Under Auspices Eur. Cell Biol. Organ. 2007, 99, 129–140. [Google Scholar] [CrossRef]
- Ma, D.W. Lipid mediators in membrane rafts are important determinants of human health and disease. Appl. Physiol. Nutr. Metab. 2007, 32, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Schengrund, C.L. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 2010, 82, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jicha, G.A.; Markesbery, W.R. Omega-3 fatty acids: Potential role in the management of early Alzheimer’s disease. Clin. Interv. Aging 2010, 5, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Farlow, M.R. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J. Lipid Res. 2005, 46, 949–968. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marin, R.; Ferrer, I.; Diaz, M. Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimer’s Dis. 2010, 19, 489–502. [Google Scholar]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lecart, S.; Saudou, F.; Duyckaerts, C.; Leveque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursor protein-BACE1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Schneider, A.; Schlechtingen, G.; Weidlich, S.; Ries, J.; Braxmeier, T.; Schwille, P.; Schulz, J.B.; Schroeder, C.; Simons, M.; et al. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 2008, 320, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zerbinatti, C.V.; Zhang, J.; Hoe, H.S.; Wang, B.; Cole, S.L.; Herz, J.; Muglia, L.; Bu, G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 2007, 56, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Grimm, H.S.; Patzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschape, J.A.; De Strooper, B.; Muller, U.; Shen, J.; et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Coma, M.; Pera, M.; Clarimon, J.; Sereno, L.; Agullo, J.M.; Molina-Porcel, L.; Gallardo, E.; Deng, A.; Berezovska, O.; et al. Mild cholesterol depletion reduces amyloid-beta production by impairing APP trafficking to the cell surface. J. Neurochem. 2009, 110, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Lemkul, J.A.; Bevan, D.R. Lipid composition influences the release of Alzheimer’s amyloid beta-peptide from membranes. Protein Sci. 2011, 20, 1530–1545. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Kirkland, S.; Hogan, D.B.; MacKnight, C.; Merry, H.; Verreault, R.; Wolfson, C.; McDowell, I. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch. Neurol. 2002, 59, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Barrett-Connor, E.; Lin, F.; Grady, D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch. Neurol. 2002, 59, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.; Blauw, G.J.; Murphy, M.B.; Bollen, E.L.; Buckley, B.M.; Cobbe, S.M.; Ford, I.; Gaw, A.; Hyland, M.; Jukema, J.W.; et al. Pravastatin in elderly individuals at risk of vascular disease (prosper): A randomised controlled trial. Lancet 2002, 360, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Higdon, R.; Kukull, W.A.; Peskind, E.; Van Valen Moore, K.; Tsuang, D.; van Belle, G.; McCormick, W.; Bowen, J.D.; Teri, L.; et al. Statin therapy and risk of dementia in the elderly: A community-based prospective cohort study. Neurology 2004, 63, 1624–1628. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Sparks, D.L.; Khachaturian, A.S.; Tschanz, J.; Norton, M.; Steinberg, M.; Welsh-Bohmer, K.A.; Breitner, J.C.; Cache County Study Investigators. Do statins reduce risk of incident dementia and Alzheimer disease? The cache county study. Arch. Gen. Psychiatry 2005, 62, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Kurinami, H.; Sato, N.; Shinohara, M.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Ogihara, T.; Morishita, R. Prevention of amyloid beta-induced memory impairment by fluvastatin, associated with the decrease in amyloid beta accumulation and oxidative stress in amyloid beta injection mouse model. Int. J. Mol. Med. 2008, 21, 531–537. [Google Scholar] [PubMed]
- Li, L.; Cao, D.; Kim, H.; Lester, R.; Fukuchi, K. Simvastatin enhances learning and memory independent of amyloid load in mice. Ann. Neurol. 2006, 60, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Petanceska, S.S.; DeRosa, S.; Olm, V.; Diaz, N.; Sharma, A.; Thomas-Bryant, T.; Duff, K.; Pappolla, M.; Refolo, L.M. Statin therapy for Alzheimer’s disease: Will it work? J. Mol. Neurosci. 2002, 19, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Shie, F.S.; Jin, L.W.; Cook, D.G.; Leverenz, J.B.; LeBoeuf, R.C. Diet-induced hypercholesterolemia enhances brain A-beta accumulation in transgenic mice. Neuroreport 2002, 13, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A gift from nature: The birth of the statins. Nat. Med. 2008, 14, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Stuve, O.; Youssef, S.; Steinman, L.; Zamvil, S.S. Statins as potential therapeutic agents in neuroinflammatory disorders. Curr. Opin. Neurol. 2003, 16, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.; Kritharides, L. The pleiotropic effects of HMG-CoA reductase inhibitors: Their role in osteoporosis and dementia. Drugs 2003, 63, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.D.; Geoghagen, N.S.; Friedhoff, L.T. Cholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid A-beta peptide. J. Alzheimer’s Dis. 2001, 3, 221–229. [Google Scholar]
- Paris, D.; Townsend, K.P.; Humphrey, J.; Obregon, D.F.; Yokota, K.; Mullan, M. Statins inhibit A-beta-neurotoxicity in vitro and A-beta-induced vasoconstriction and inflammation in rat aortae. Atherosclerosis 2002, 161, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, C.; Eckert, G.P.; Mueller, W.E. Statin effects on cholesterol micro-domains in brain plasma membranes. Biochem. Pharmacol. 2003, 65, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.M.; Wilkinson, B.L.; Golde, T.E.; Landreth, G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J. Biol. Chem. 2007, 282, 26832–26844. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Ellis, B.C.; Ingano, L.A.; Kovacs, D.M. Role of Acyl-coenzyme A: Cholesterol acyltransferase activity in the processing of the amyloid precursor protein. J. Mol. Neurosci. 2004, 24, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Puglielli, L.; Ellis, B.C.; MacKenzie Ingano, L.A.; Kovacs, D.M. Novel N-terminal cleavage of APP precludes A-beta generation in ACAT-defective AC29 cells. J. Mol. Neurosci. 2009, 37, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Van Dam, D.; Kulik, W.; De Deyn, P.P.; Stet, F.S.; Ahouansou, O.; Benraiss, A.; Delacourte, A.; Bougneres, P.; Aubourg, P.; et al. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol. Ther. 2010, 18, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Halford, R.W.; Russell, D.W. Reduction of cholesterol synthesis in the mouse brain does not affect amyloid formation in Alzheimer’s disease, but does extend lifespan. Proc. Natl. Acad. Sci. USA 2009, 106, 3502–3506. [Google Scholar] [CrossRef] [PubMed]
- Greeve, I.; Hermans-Borgmeyer, I.; Brellinger, C.; Kasper, D.; Gomez-Isla, T.; Behl, C.; Levkau, B.; Nitsch, R.M. The human DIMINUTO/DWARF1 homolog Seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J. Neurosci. 2000, 20, 7345–7352. [Google Scholar] [PubMed]
- Crameri, A.; Biondi, E.; Kuehnle, K.; Lutjohann, D.; Thelen, K.M.; Perga, S.; Dotti, C.G.; Nitsch, R.M.; Ledesma, M.D.; Mohajeri, M.H. The role of Seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and A-beta generation in vivo. EMBO J. 2006, 25, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, C.; Rosati, F.; Pensalfini, A.; Formigli, L.; Nosi, D.; Liguri, G.; Dichiara, F.; Morello, M.; Danza, G.; Pieraccini, G.; et al. Seladin-1/DHCR24 protects neuroblastoma cells against A-beta toxicity by increasing membrane cholesterol content. J. Cell. Mol. Med. 2008, 12, 1990–2002. [Google Scholar] [CrossRef] [PubMed]
- Subasinghe, S.; Unabia, S.; Barrow, C.J.; Mok, S.S.; Aguilar, M.I.; Small, D.H. Cholesterol is necessary both for the toxic effect of A-beta peptides on vascular smooth muscle cells and for A-beta binding to vascular smooth muscle cell membranes. J. Neurochem. 2003, 84, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Peri, A.; Serio, M. Neuroprotective effects of the Alzheimer’s disease-related gene Seladin-1. J. Mol. Endocrinol. 2008, 41, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [PubMed]
- Rapp, A.; Gmeiner, B.; Huttinger, M. Implication of ApoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 2006, 88, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Michikawa, M.; Fan, Q.W.; Isobe, I.; Yanagisawa, K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 2000, 74, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Igbavboa, U.; Hamanaka, H.; Kobayashi, M.; Fujita, S.C.; Wood, W.G.; Yanagisawa, K. Cholesterol is increased in the exofacial leaflet of synaptic plasma membranes of human apolipoprotein E4 knock-in mice. Neuroreport 2002, 13, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Naslund, J.; Thyberg, J.; Tjernberg, L.O.; Wernstedt, C.; Karlstrom, A.R.; Bogdanovic, N.; Gandy, S.E.; Lannfelt, L.; Terenius, L.; Nordstedt, C. Characterization of stable complexes involving apolipoprotein E and the amyloid beta peptide in Alzheimer’s disease brain. Neuron 1995, 15, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Tiraboschi, P.; Hansen, L.A.; Masliah, E.; Alford, M.; Thal, L.J.; Corey-Bloom, J. Impact of ApoE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 2004, 62, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Liu, X.; Bandy, D.; Yu, M.; Lee, W.; Ayutyanont, N.; Keppler, J.; Reeder, S.A.; Langbaum, J.B.; et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 6820–6825. [Google Scholar] [PubMed]
- Morris, J.C.; Roe, C.M.; Xiong, C.; Fagan, A.M.; Goate, A.M.; Holtzman, D.M.; Mintun, M.A. ApoE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 2010, 67, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Moir, R.D.; Rebeck, G.W.; Bush, A.I.; Argraves, W.S.; Tanzi, R.E.; Hyman, B.T.; Strickland, D.K. LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted beta-amyloid precursor protein and mediates its degradation. Cell 1995, 82, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Sato, N.; Kurinami, H.; Takeuchi, D.; Takeda, S.; Shimamura, M.; Yamashita, T.; Uchiyama, Y.; Rakugi, H.; Morishita, R. Reduction of brain beta-amyloid (A-beta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and A-beta clearance. J. Biol. Chem. 2010, 285, 22091–22102. [Google Scholar] [CrossRef] [PubMed]
- Pfrieger, F.W. Role of cholesterol in synapse formation and function. Biochim. Biophys. Acta 2003, 1610, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Claudepierre, T.; Pfrieger, F.W. New aspects of cholesterol in the central nervous system. Med. Sci. 2003, 19, 601–605. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannaoui, S.; Shim, S.Y.; Cheng, Y.C.; Corda, E.; Gilch, S. Cholesterol Balance in Prion Diseases and Alzheimer’s Disease. Viruses 2014, 6, 4505-4535. https://doi.org/10.3390/v6114505
Hannaoui S, Shim SY, Cheng YC, Corda E, Gilch S. Cholesterol Balance in Prion Diseases and Alzheimer’s Disease. Viruses. 2014; 6(11):4505-4535. https://doi.org/10.3390/v6114505
Chicago/Turabian StyleHannaoui, Samia, Su Yeon Shim, Yo Ching Cheng, Erica Corda, and Sabine Gilch. 2014. "Cholesterol Balance in Prion Diseases and Alzheimer’s Disease" Viruses 6, no. 11: 4505-4535. https://doi.org/10.3390/v6114505
APA StyleHannaoui, S., Shim, S. Y., Cheng, Y. C., Corda, E., & Gilch, S. (2014). Cholesterol Balance in Prion Diseases and Alzheimer’s Disease. Viruses, 6(11), 4505-4535. https://doi.org/10.3390/v6114505