The Innate Immune Playbook for Restricting West Nile Virus Infection

{kind=link}
Abstract
:1. Introduction
1.1. West Nile Virus is a Serious Public Health Concern
1.2. WNV Biology and Pathogenesis
2. The Innate Immune Players
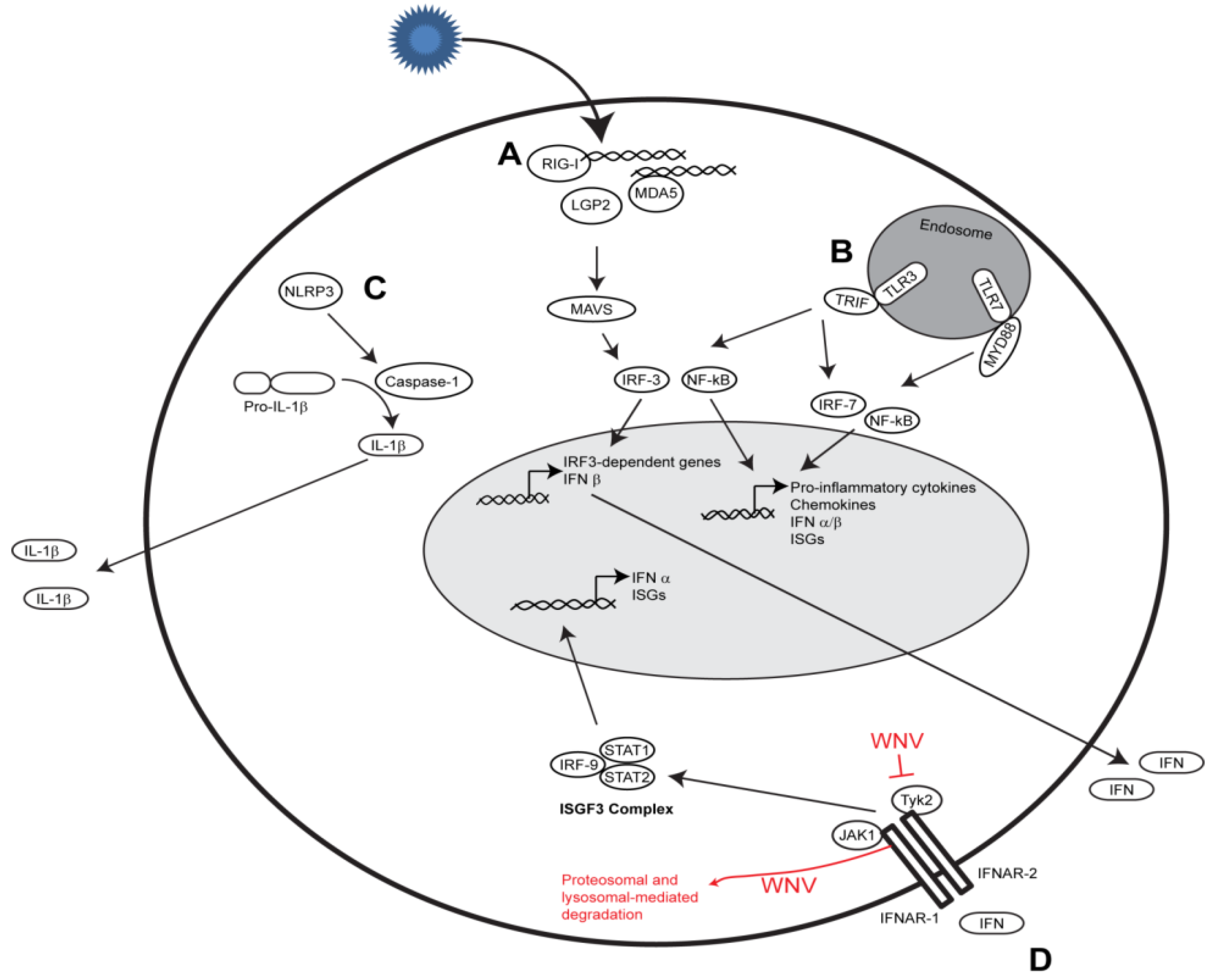
2.1. RIG-I-like Receptor Signaling
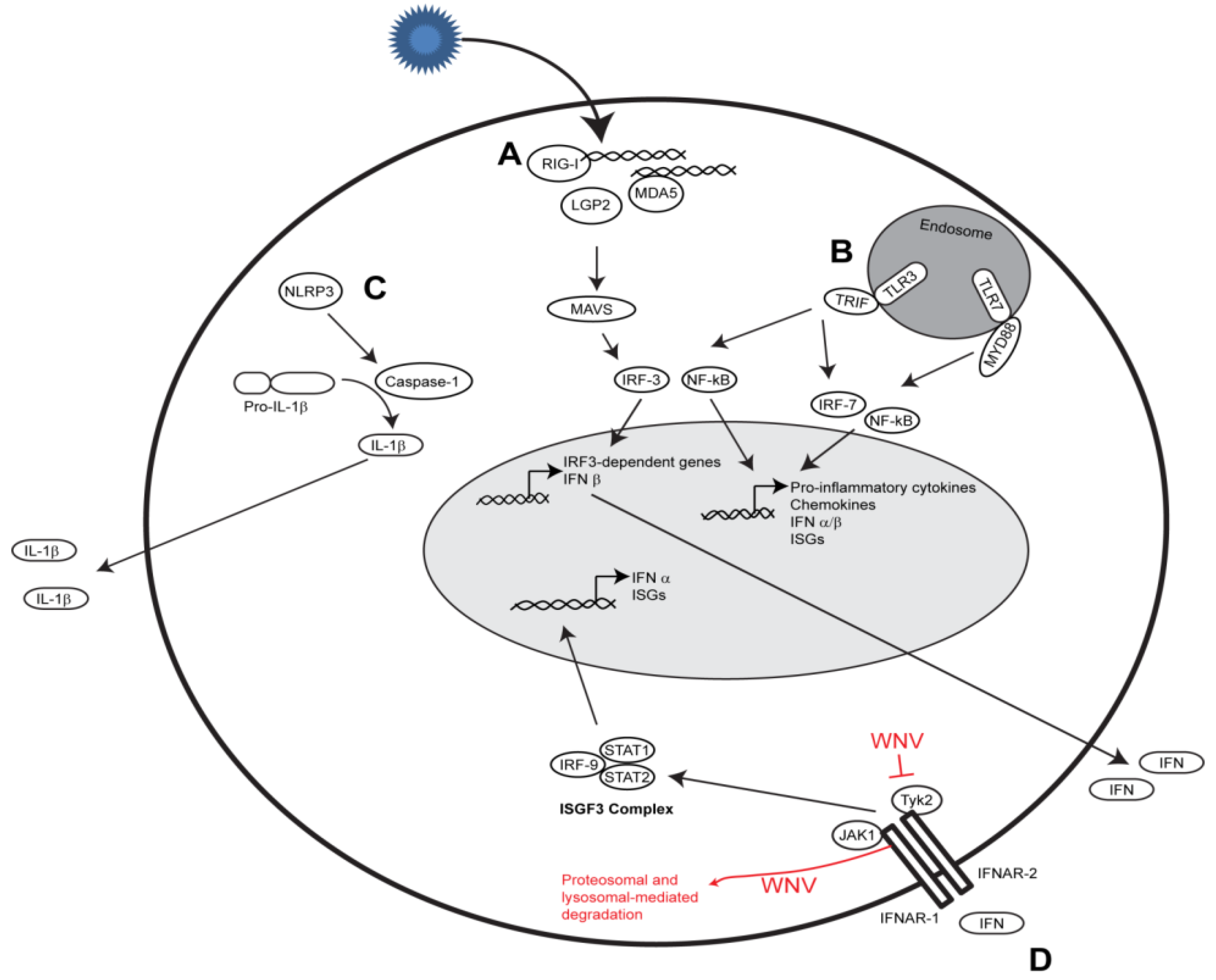
2.2. Toll-like Receptor Signaling
2.3. Nod-like Receptor Signaling
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Petersen, L.R.; Brault, A.C.; Nasci, R.S. West Nile virus: Review of the literature. JAMA 2013, 310, 308–315. [Google Scholar] [CrossRef]
- Petersen, L.R.; Carson, P.J.; Biggerstaff, B.J.; Custer, B.; Borchardt, S.M.; Busch, M.P. Estimated cumulative incidence of West Nile virus infection in US adults, 1999–2010. Epidemiol. Infect. 2012, 28, 1–5. [Google Scholar]
- Reiter, P. West Nile virus in Europe: Understanding the present to gauge the future. Euro Surveill. 2010, 15, e19508. [Google Scholar]
- Brinton, M.A. Host factors involved in West Nile virus replication. Ann. NY Acad. Sci. 2001, 951, 207–219. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar]
- Komuro, A.; Horvath, C.M. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol. 2006, 80, 12332–12342. [Google Scholar] [CrossRef]
- Venkataraman, T.; Valdes, M.; Elsby, R.; Kakuta, S.; Caceres, G.; Saijo, S.; Iwakura, Y.; Barber, G.N. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 2007, 178, 6444–6455. [Google Scholar]
- Suthar, M.S.; Ramos, H.J.; Brassil, M.M.; Netland, J.; Chappell, C.P.; Blahnik, G.; McMillan, A.; Diamond, M.S.; Clark, E.A.; Bevan, M.J.; et al. The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 2012, 37, 235–248. [Google Scholar]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M., Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar]
- Liu, H.M.; Loo, Y.M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The mitochondrial targeting chaperone 14-3-3epsilon regulates a RIG-I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef]
- Wies, E.; Wang, M.K.; Maharaj, N.P.; Chen, K.; Zhou, S.; Finberg, R.W.; Gack, M.U. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 2013, 38, 437–449. [Google Scholar]
- Gack, M.U.; Nistal-Villan, E.; Inn, K.S.; Garcia-Sastre, A.; Jung, J.U. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J. Virol. 2010, 84, 3220–3229. [Google Scholar]
- Daffis, S.; Samuel, M.A.; Keller, B.C.; Gale, M., Jr.; Diamond, M.S. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007, 3, e106. [Google Scholar]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Keller, B.C.; Gale, M., Jr.; Diamond, M.S. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J. Virol. 2008, 82, 8465–8475. [Google Scholar]
- Daffis, S.; Suthar, M.S.; Szretter, K.J.; Gale, M., Jr.; Diamond, M.S. Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 2009, 5, e1000607. [Google Scholar]
- Lazear, H.M.; Lancaster, A.; Wilkins, C.; Suthar, M.S.; Huang, A.; Vick, S.C.; Clepper, L.; Thackray, L.; Brassil, M.M.; Virgin, H.W.; et al. IRF-3, IRF-5, and IRF-7 coordinately regulate the Type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 2013, 9, e1003118. [Google Scholar]
- Suthar, M.S.; Ma, D.Y.; Thomas, S.; Lund, J.M.; Zhang, N.; Daffis, S.; Rudensky, A.Y.; Bevan, M.J.; Clark, E.A.; Kaja, M.K.; et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010, 6, e1000757. [Google Scholar] [CrossRef]
- Errett, J.S.; Suthar, M.S.; McMillan, A.; Diamond, M.S.; Gale, M., Jr. The essential, non-redundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J. Virol. 2013, 87, 11416–11425. [Google Scholar]
- Lazear, H.M.; Pinto, A.K.; Ramos, H.J.; Vick, S.C.; Shrestha, B.; Suthar, M.S.; Gale, M., Jr.; Diamond, M.S. The pattern recognition receptor MDA5 modulates CD8+ T cell-dependent clearance of West Nile virus from the central nervous system. J. Virol. 2013, 87, 11401–11415. [Google Scholar]
- Wang, P.; Arjona, A.; Zhang, Y.; Sultana, H.; Dai, J.; Yang, L.; LeBlanc, P.M.; Doiron, K.; Saleh, M.; Fikrig, E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat. Immunol. 2010, 11, 912–919. [Google Scholar]
- Fredericksen, B.L.; Gale, M., Jr. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 2006, 80, 2913–2923. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M., Jr. Establishment and maintenance of the innate antiviral response to West Nile virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar]
- Shipley, J.G.; Vandergaast, R.; Deng, L.; Mariuzza, R.A.; Fredericksen, B.L. Identification of multiple RIG-I-specific pathogen associated molecular patterns within the West Nile virus genome and antigenome. Virology 2012, 432, 232–238. [Google Scholar]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar]
- Hoenen, A.; Liu, W.; Kochs, G.; Khromykh, A.A.; Mackenzie, J.M. West Nile virus-induced cytoplasmic membrane structures provide partial protection against the interferon-induced antiviral MxA protein. J. Gen. Virol. 2007, 88, 3013–3017. [Google Scholar]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; e Sousa, C.R. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M., Jr.; Diamond, M.S. Toll-like receptor 3 has a protective role against West Nile virus infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar]
- Szretter, K.J.; Daffis, S.; Patel, J.; Suthar, M.S.; Klein, R.S.; Gale, M., Jr.; Diamond, M.S. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol. 2010, 84, 12125–12138. [Google Scholar]
- Welte, T.; Reagan, K.; Fang, H.; Machain-Williams, C.; Zheng, X.; Mendell, N.; Chang, G.J.; Wu, P.; Blair, C.D.; Wang, T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J. Gen. Virol. 2009, 90, 2660–2668. [Google Scholar]
- Town, T.; Bai, F.; Wang, T.; Kaplan, A.T.; Qian, F.; Montgomery, R.R.; Anderson, J.F.; Flavell, R.A.; Fikrig, E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 2009, 30, 242–253. [Google Scholar]
- Xia, J.; Winkelmann, E.R.; Gorder, S.R.; Mason, P.W.; Milligan, G.N. TLR3- and MyD88-dependent signaling differentially influence the development of West Nile virus-specific B cell responses in mice following immunization with the single-cycle flavivirus (SCFV) RepliVAX WN. J. Virol. 2013, 87, 12090–12101. [Google Scholar]
- Wilson, J.R.; de Sessions, P.F.; Leon, M.A.; Scholle, F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar]
- Baronti, C.; Sire, J.; de Lamballerie, X.; Querat, G. Nonstructural NS1 proteins of several mosquito-borne Flavivirus do not inhibit TLR3 signaling. Virology 2010, 404, 319–330. [Google Scholar]
- Lamkanfi, M.; Dixit, V.M. Modulation of inflammasome pathways by bacterial and viral pathogens. J. Immunol. 2011, 187, 597–602. [Google Scholar] [CrossRef]
- Ramos, H.J.; Lanteri, M.C.; Blahnik, G.; Negash, A.; Suthar, M.S.; Brassil, M.M.; Sodhi, K.; Treuting, P.M.; Busch, M.P.; Norris, P.J.; et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012, 8, e1003039. [Google Scholar]
- Durrant, D.M.; Robinette, M.L.; Klein, R.S. IL-1R1 is required for dendritic cell-mediated T cell reactivation within the CNS during West Nile virus encephalitis. J. Exp. Med. 2013, 210, 503–516. [Google Scholar] [CrossRef]
- Kumar, M.; Roe, K.; Orillo, B.; Muruve, D.A.; Nerurkar, V.R.; Gale, M., Jr.; Verma, S. Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J. Virol. 2013, 87, 3655–3667. [Google Scholar]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Invest. Dermatol. 2001, 117, 702–709. [Google Scholar]
- Horvath, C.M. The Jak-STAT pathway stimulated by interferon alpha or interferon beta. Sci. STKE 2004, 2004, tr10. [Google Scholar]
- Porterfield, J. A simple plaque inhibition test for antiviral agents: Application to assay of interferon. Lancet 1959, 274, 326–327. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Type I IFN protects against lethal West Nile Virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef]
- Suthar, M.S.; Brassil, M.M.; Blahnik, G.; McMillan, A.; Ramos, H.J.; Proll, S.C.; Belisle, S.E.; Katze, M.G.; Gale, M., Jr. A systems biology approach reveals that tissue tropism to West Nile virus is regulated by antiviral genes and innate immune cellular processes. PLoS Pathog. 2013, 9, e1003168. [Google Scholar] [CrossRef]
- Lazear, H.M.; Pinto, A.K.; Vogt, M.R.; Gale, M., Jr.; Diamond, M.S. Beta interferon controls West Nile virus infection and pathogenesis in mice. J. Virol. 2011, 85, 7186–7194. [Google Scholar]
- Purtha, W.E.; Chachu, K.A.; Virgin, H.W.; Diamond, M.S. Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J. Virol. 2008, 82, 10964–10974. [Google Scholar]
- Pinto, A.K.; Daffis, S.; Brien, J.D.; Gainey, M.D.; Yokoyama, W.M.; Sheehan, K.C.; Murphy, K.M.; Schreiber, R.D.; Diamond, M.S. A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS Pathog. 2011, 7, e1002407. [Google Scholar]
- Ma, D.; Jiang, D.; Qing, M.; Weidner, J.M.; Qu, X.; Guo, H.; Chang, J.; Gu, B.; Shi, P.Y.; Block, T.M.; et al. Antiviral effect of interferon lambda against West Nile virus. Antiviral Res. 2009, 83, 53–60. [Google Scholar] [CrossRef]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M., Jr. Resistance to alpha/beta interferon is a determinant of West Nile virus replication fitness and virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar]
- Evans, J.D.; Crown, R.A.; Sohn, J.A.; Seeger, C. West Nile virus infection induces depletion of IFNAR1 protein levels. Viral Immunol. 2011, 24, 253–263. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Khromykh, A.A.; Parton, R.G. Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2007, 2, 229–239. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 2013, 87, 2206–2214. [Google Scholar] [CrossRef]
- Ambrose, R.L.; Mackenzie, J.M. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 2011, 85, 2723–2732. [Google Scholar] [CrossRef]
- Suthar, M.S.; Brassil, M.M.; Blahnik, G.; Gale, M., Jr. Infectious clones of novel lineage 1 and lineage 2 West Nile virus strains WNV-TX02 and WNV-Madagascar. J. Virol. 2012, 86, 7704–7709. [Google Scholar]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.Y.; Randall, R.; Khromykh, A.A. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar]
- Evans, J.D.; Seeger, C. Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J. Virol. 2007, 81, 11809–11816. [Google Scholar] [CrossRef]
- Munoz-Jordan, J.L.; Laurent-Rolle, M.; Ashour, J.; Martinez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; Garcia-Sastre, A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.F.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar]
- Schuessler, A.; Funk, A.; Lazear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J. Virol. 2012, 86, 5708–5718. [Google Scholar]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M., Jr.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar]
- Jiang, D.; Weidner, J.M.; Qing, M.; Pan, X.B.; Guo, H.; Xu, C.; Zhang, X.; Birk, A.; Chang, J.; Shi, P.Y.; et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J. Virol. 2010, 84, 8332–8341. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar]
- Li, J.; Ding, S.C.; Cho, H.; Chung, B.C.; Gale, M., Jr.; Chanda, S.K.; Diamond, M.S. A short hairpin RNA screen of interferon-stimulated genes identifies a novel negative regulator of the cellular antiviral response. MBio 2013, 4, e00385–e00313. [Google Scholar]
- Qian, F.; Chung, L.; Zheng, W.; Bruno, V.; Alexander, R.P.; Wang, Z.; Wang, X.; Kurscheid, S.; Zhao, H.; Fikrig, E.; et al. Identification of genes critical for resistance to infection by West Nile virus using RNA-Seq analysis. Viruses 2013, 5, 1664–1681. [Google Scholar] [CrossRef]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 2001, re2. [Google Scholar] [CrossRef]
- Samuel, M.A.; Whitby, K.; Keller, B.C.; Marri, A.; Barchet, W.; Williams, B.R.; Silverman, R.H.; Gale, M., Jr.; Diamond, M.S. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006, 80, 7009–7019. [Google Scholar] [CrossRef]
- Scherbik, S.V.; Paranjape, J.M.; Stockman, B.M.; Silverman, R.H.; Brinton, M.A. RNase L plays a role in the antiviral response to West Nile virus. J. Virol. 2006, 80, 2987–2999. [Google Scholar]
- Gilfoy, F.D.; Mason, P.W. West Nile virus-induced IFN production is mediated by the double-stranded RNA-dependent protein kinase, PKR. J. Virol. 2007, 81, 11148–11158. [Google Scholar] [CrossRef]
- Elbahesh, H.; Scherbik, S.V.; Brinton, M.A. West Nile virus infection does not induce PKR activation in rodent cells. Virology 2011, 421, 51–60. [Google Scholar] [CrossRef]
- Fensterl, V.; Sen, G.C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef]
- Perwitasari, O.; Cho, H.; Diamond, M.S.; Gale, M., Jr. Inhibitor of kappaB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J. Biol. Chem. 2011, 286, 44412–44423. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2'-O-methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar]
- Wacher, C.; Muller, M.; Hofer, M.J.; Getts, D.R.; Zabaras, R.; Ousman, S.S.; Terenzi, F.; Sen, G.C.; King, N.J.; Campbell, I.L. Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J. Virol. 2007, 81, 860–871. [Google Scholar]
- Szretter, K.J.; Daniels, B.P.; Cho, H.; Gainey, M.D.; Yokoyama, W.M.; Gale, M., Jr.; Virgin, H.W.; Klein, R.S.; Sen, G.C.; Diamond, M.S. 2'-O-methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012, 8, e1002698. [Google Scholar] [CrossRef]
- Cho, H.; Shrestha, B.; Sen, G.C.; Diamond, M.S. A role for Ifit2 in restricting West Nile virus infection in the brain. J. Virol. 2013, 87, 8363–8371. [Google Scholar]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar]
- Wilkins, C.; Woodward, J.; Lau, D.T.; Barnes, A.; Joyce, M.; McFarlane, N.; McKeating, J.A.; Tyrrell, D.L.; Gale, M., Jr. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013, 57, 461–469. [Google Scholar] [CrossRef]
- Seo, J.Y.; Yaneva, R.; Cresswell, P. Viperin: A multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 2011, 10, 534–539. [Google Scholar] [CrossRef]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar]
- Saitoh, T.; Satoh, T.; Yamamoto, N.; Uematsu, S.; Takeuchi, O.; Kawai, T.; Akira, S. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 2011, 34, 352–363. [Google Scholar]
- Helbig, K.J.; Carr, J.M.; Calvert, J.K.; Wati, S.; Clarke, J.N.; Eyre, N.S.; Narayana, S.K.; Fiches, G.N.; McCartney, E.M.; Beard, M.R. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLoS Negl. Trop. Dis. 2013, 7, e2178. [Google Scholar] [CrossRef]
- Hinson, E.R.; Cresswell, P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc. Natl. Acad. Sci. USA 2009, 106, 20452–20457. [Google Scholar] [CrossRef]
- Perelygin, A.A.; Scherbik, S.V.; Zhulin, I.B.; Stockman, B.M.; Li, Y.; Brinton, M.A. Positional cloning of the murine flavivirus resistance gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9322–9327. [Google Scholar]
- Zhou, A.; Paranjape, J.; Brown, T.L.; Nie, H.; Naik, S.; Dong, B.; Chang, A.; Trapp, B.; Fairchild, R.; Colmenares, C.; et al. Interferon action and apoptosis are defective in mice devoid of 2',5'-oligoadenylate-dependent RNase L. EMBO J. 1997, 16, 6355–6363. [Google Scholar] [CrossRef]
- Malathi, K.; Saito, T.; Crochet, N.; Barton, D.J.; Gale, M., Jr.; Silverman, R.H. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 2010, 16, 2108–2119. [Google Scholar]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar]
- Mashimo, T.; Lucas, M.; Simon-Chazottes, D.; Frenkiel, M.P.; Montagutelli, X.; Ceccaldi, P.E.; Deubel, V.; Guenet, J.L.; Despres, P. A nonsense mutation in the gene encoding 2'-5'-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11311–11316. [Google Scholar]
- Elbahesh, H.; Jha, B.K.; Silverman, R.H.; Scherbik, S.V.; Brinton, M.A. The Flvr-encoded murine oligoadenylate synthetase 1b (Oas1b) suppresses 2–5A synthesis in intact cells. Virology 2011, 409, 262–270. [Google Scholar] [CrossRef]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; Huynh, L.; Ward, J.M.; Johnson, B.; Johnson, H.; Pape, J.; Foster, G.A.; Krysztof, D.; et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009, 5, e1000321. [Google Scholar] [CrossRef]
- Bigham, A.W.; Buckingham, K.J.; Husain, S.; Emond, M.J.; Bofferding, K.M.; Gildersleeve, H.; Rutherford, A.; Astakhova, N.M.; Perelygin, A.A.; Busch, M.P.; et al. Host genetic risk factors for West Nile virus infection and disease progression. PLoS One 2011, 6, e24745. [Google Scholar] [CrossRef]
- Kajaste-Rudnitski, A.; Mashimo, T.; Frenkiel, M.P.; Guenet, J.L.; Lucas, M.; Despres, P. The 2',5'-oligoadenylate synthetase 1b is a potent inhibitor of West Nile virus replication inside infected cells. J. Biol. Chem. 2006, 281, 4624–4637. [Google Scholar]
- Courtney, S.C.; Di, H.; Stockman, B.M.; Liu, H.; Scherbik, S.V.; Brinton, M.A. Identification of novel host cell binding partners of oas1b, the protein conferring resistance to flavivirus-induced disease in mice. J. Virol. 2012, 86, 7953–7963. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quicke, K.M.; Suthar, M.S. The Innate Immune Playbook for Restricting West Nile Virus Infection. Viruses 2013, 5, 2643-2658. https://doi.org/10.3390/v5112643
Quicke KM, Suthar MS. The Innate Immune Playbook for Restricting West Nile Virus Infection. Viruses. 2013; 5(11):2643-2658. https://doi.org/10.3390/v5112643
Chicago/Turabian StyleQuicke, Kendra M., and Mehul S. Suthar. 2013. "The Innate Immune Playbook for Restricting West Nile Virus Infection" Viruses 5, no. 11: 2643-2658. https://doi.org/10.3390/v5112643
APA StyleQuicke, K. M., & Suthar, M. S. (2013). The Innate Immune Playbook for Restricting West Nile Virus Infection. Viruses, 5(11), 2643-2658. https://doi.org/10.3390/v5112643