Viral and Cellular Requirements for the Nuclear Entry of Retroviral Preintegration Nucleoprotein Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
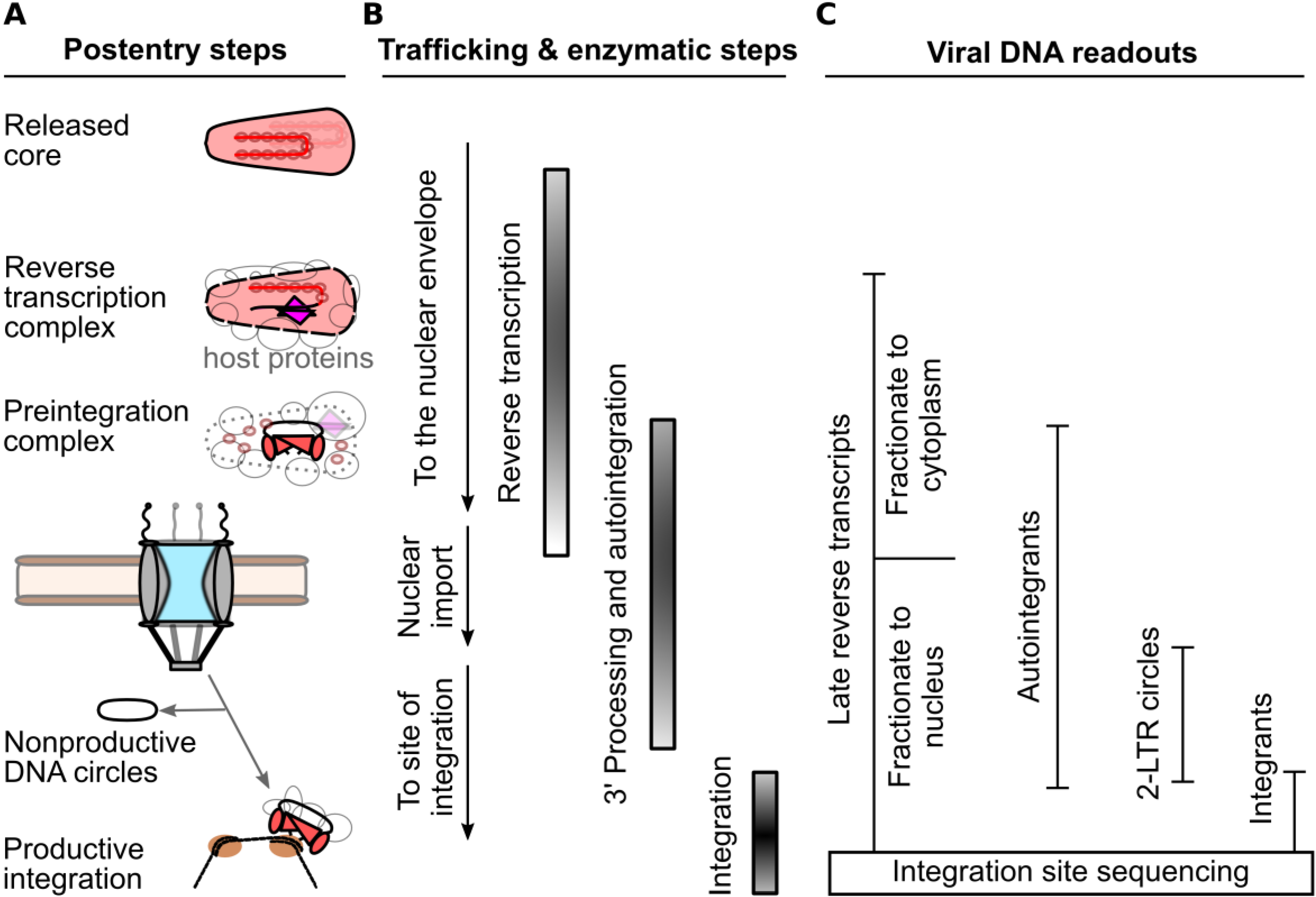
2. Measurements of the HIV-1 Nucleoprotein Substrate for Nuclear Import
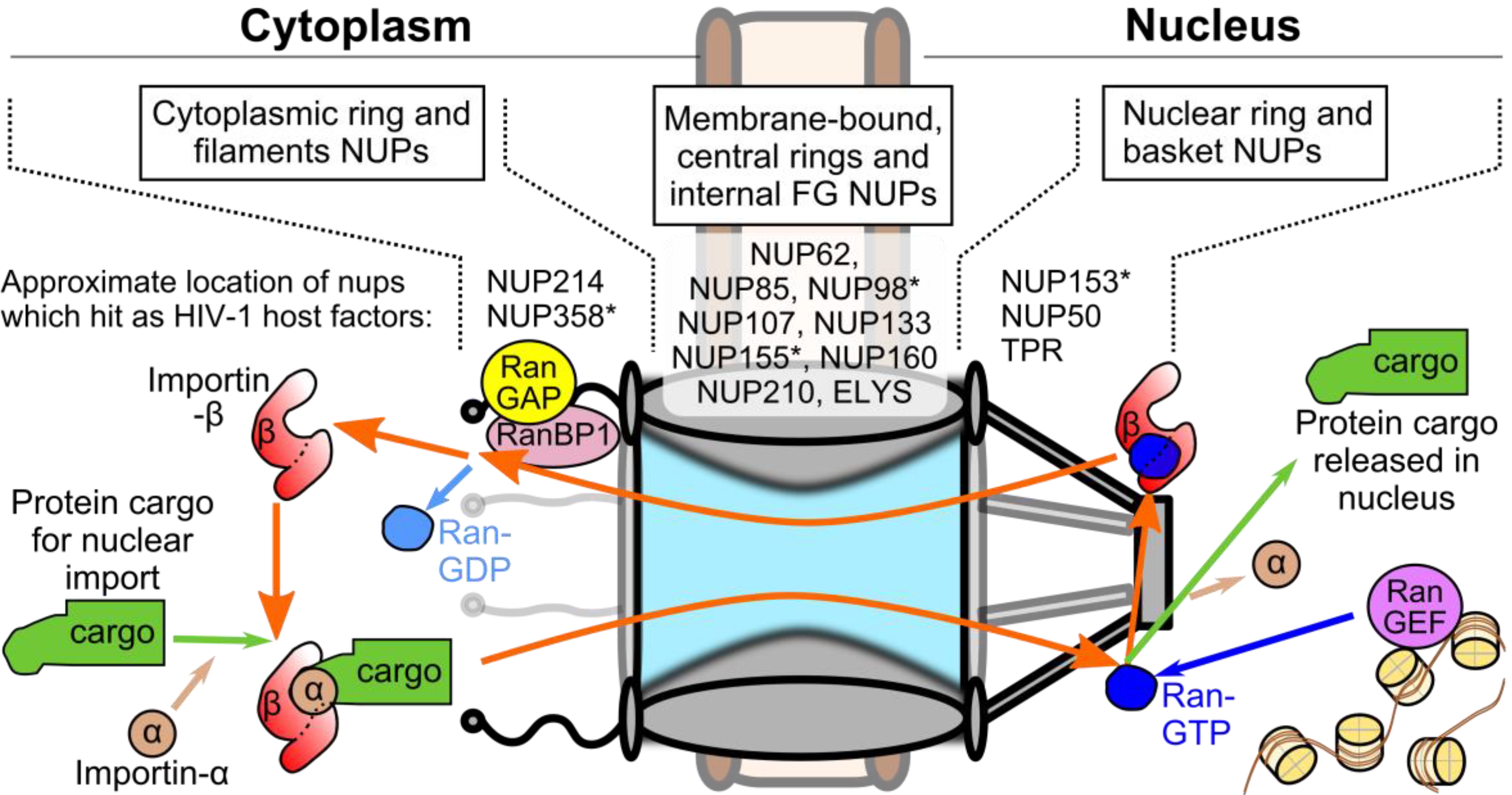
3. Viral and Cellular Elements Implicated in HIV-1 PIC Nuclear Import
4. CA Functionally Determines Requirements for Nuclear Trafficking
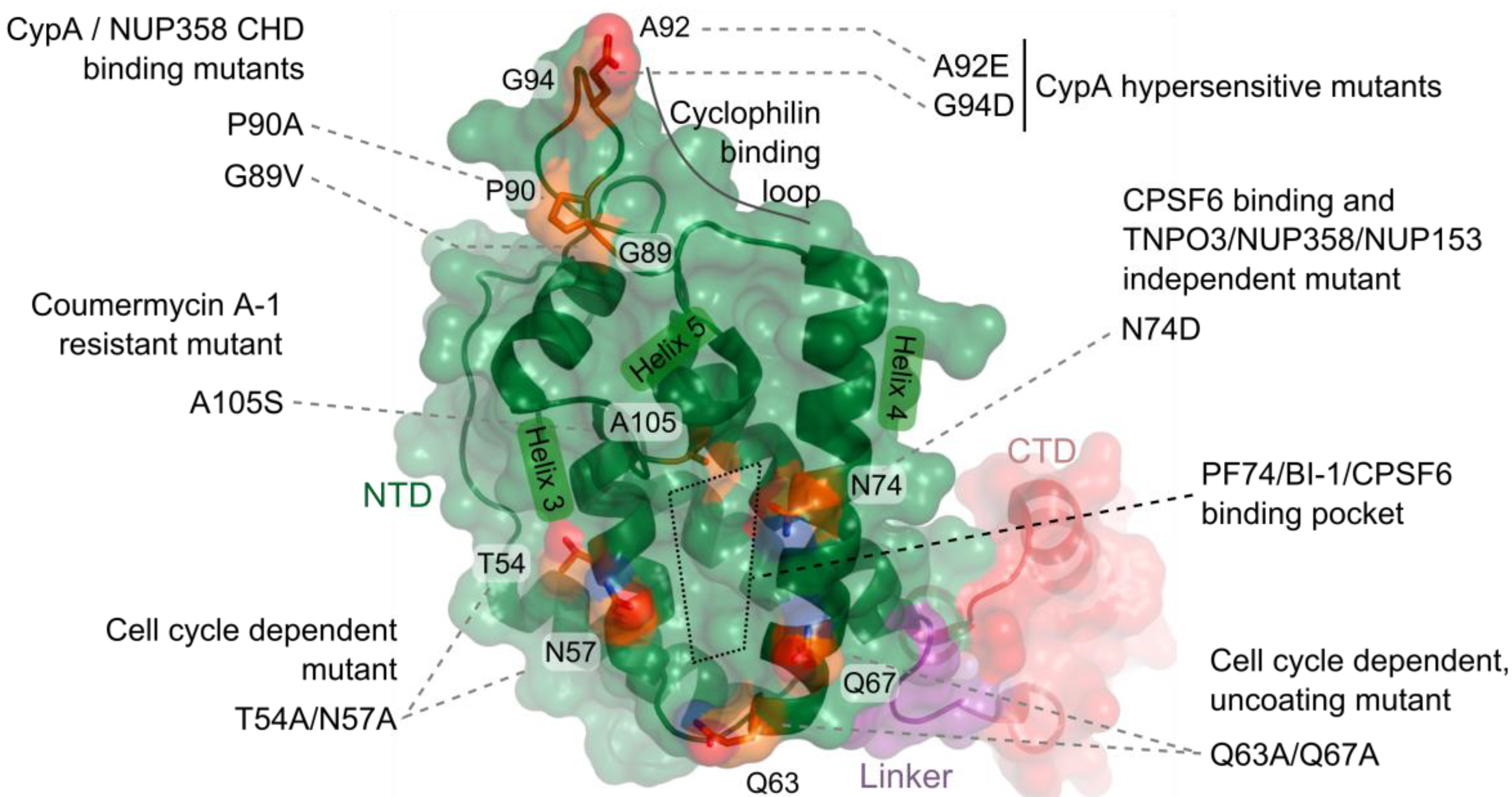
5. CA-Associated Host Proteins that Promote HIV-1 Nuclear Import
5.1. Potential Roles of TNPO3 in HIV-1 Infection
5.2. Viral Interactions with NUP153 and NUP358
5.3. Interdependence of CA-determined Host Factors during Infection
6. Effects of Nuclear Transport Proteins on Integration Site Selection
7. Model of CA and Nuclear Transport Factors during HIV-1 Nuclear Entry
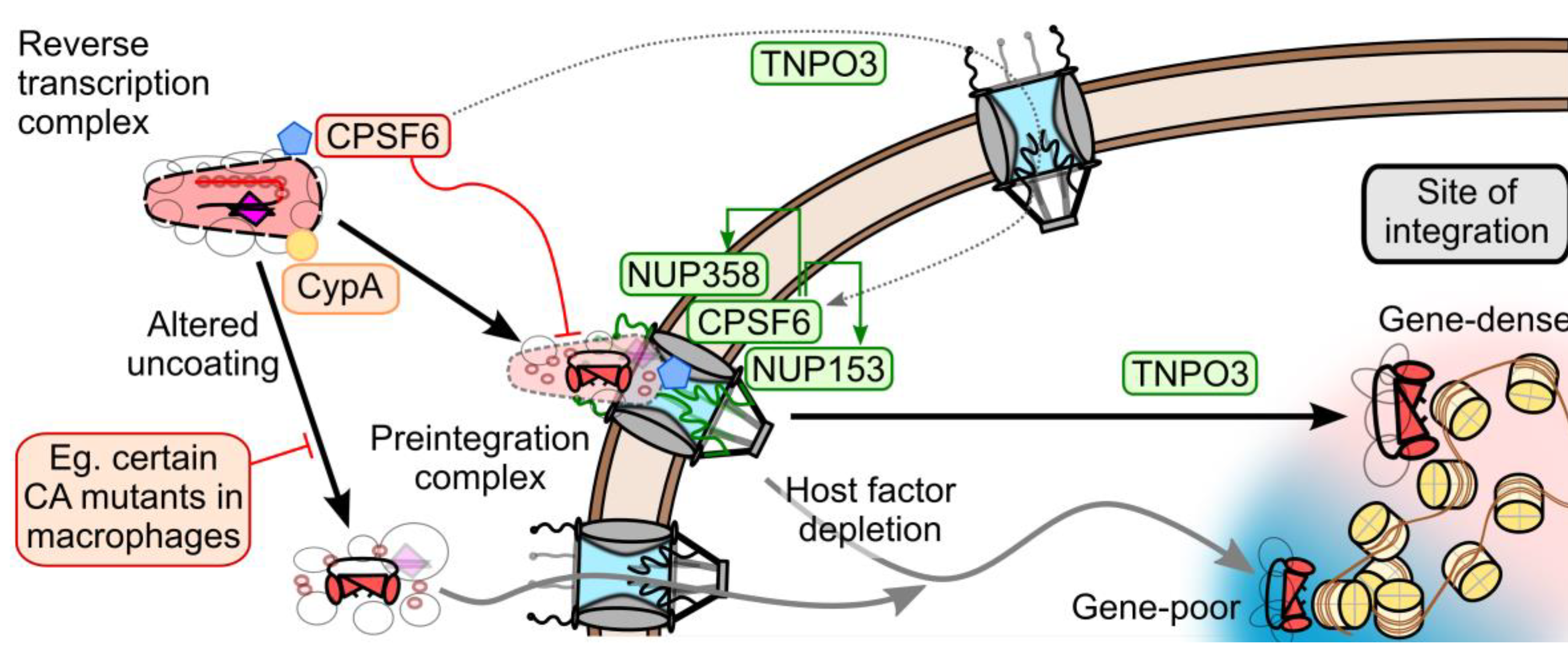
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of Murine Leukemia Virus DNA Depends on Mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar]
- Gartner, S.; Markovits, P.; Markovitz, D.M.; Kaplan, M.H.; Gallo, R.C.; Popovic, M. The Role of Mononuclear Phagocytes in HTLV–III/LAV Infection. Science 1986, 233, 215–219. [Google Scholar]
- Hoelz, A.; Debler, E.W.; Blobel, G. The Structure of the Nuclear Pore Complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef]
- Rout, M.P.; Aitchison, J.D.; Suprapto, A.; Hjertaas, K.; Zhao, Y.; Chait, B.T. The Yeast Nuclear Pore Complex: Composition, Architecture, and Transport Mechanism. J. Cell Biol. 2000, 148, 635–651. [Google Scholar] [CrossRef]
- Cronshaw, J.M.; Krutchinsky, A.N.; Zhang, W.; Chait, B.T.; Matunis, M.J. Proteomic Analysis of the Mammalian Nuclear Pore Complex. J. Cell Biol. 2002, 158, 915–927. [Google Scholar] [CrossRef]
- Rout, M.P.; Blobel, G. Isolation of the Yeast Nuclear Pore Complex. J. Cell Biol. 1993, 123, 771–783. [Google Scholar] [CrossRef]
- Reichelt, R.; Holzenburg, A.; Buhle, E.L., Jr.; Jarnik, M.; Engel, A.; Aebi, U. Correlation between Structure and Mass Distribution of the Nuclear Pore Complex and of Distinct Pore Complex Components. J. Cell Biol. 1990, 110, 883–894. [Google Scholar] [CrossRef]
- Pante, N.; Kann, M. Nuclear Pore Complex Is Able to Transport Macromolecules with Diameters of About 39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef]
- Conti, E.; Muller, C.W.; Stewart, M. Karyopherin Flexibility in Nucleocytoplasmic Transport. Curr. Opin. Struct. Biol. 2006, 16, 237–244. [Google Scholar] [CrossRef]
- Terry, L.J.; Wente, S.R. Flexible Gates: Dynamic Topologies and Functions for FG Nucleoporins in Nucleocytoplasmic Transport. Eukaryot. Cell 2009, 8, 1814–1827. [Google Scholar] [CrossRef]
- Nemergut, M.E.; Mizzen, C.A.; Stukenberg, T.; Allis, C.D.; Macara, I.G. Chromatin Docking and Exchange Activity Enhancement of RCC1 by Histones H2A and H2B. Science 2001, 292, 1540–1543. [Google Scholar] [CrossRef]
- Saitoh, H.; Pu, R.; Cavenagh, M.; Dasso, M. RanBP2 Associates with Ubc9p and a Modified Form of RanGAP1. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 3736–3741. [Google Scholar] [CrossRef]
- Radu, A.; Moore, M.S.; Blobel, G. The Peptide Repeat Domain of Nucleoporin NUP98 Functions as a Docking Site in Transport across the Nuclear Pore Complex. Cell 1995, 81, 215–222. [Google Scholar] [CrossRef]
- Bischoff, F.R.; Klebe, C.; Kretschmer, J.; Wittinghofer, A.; Ponstingl, H. RanGAP1 Induces GTPase Activity of Nuclear Ras–Related Ran. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 2587–2591. [Google Scholar] [CrossRef]
- Kehlenbach, R.H.; Dickmanns, A.; Kehlenbach, A.; Guan, T.; Gerace, L. A Role for RanBP1 in the Release of Crm1 from the Nuclear Pore Complex in a Terminal Step of Nuclear Export. J. Cell Biol. 1999, 145, 645–657. [Google Scholar] [CrossRef]
- Koyama, M.; Matsuura, Y. An Allosteric Mechanism to Displace Nuclear Export Cargo from Crm1 and RanGTP by RanBP1. EMBO J. 2010, 29, 2002–2013. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of Host Proteins Required for HIV Infection through a Functional Genomic Screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Konig, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global Analysis of Host–Pathogen Interactions That Regulate Early–Stage HIV–1 Replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome–Scale RNAi Screen for Host Factors Required for HIV Replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Yeung, M.L.; Houzet, L.; Yedavalli, V.S.; Jeang, K.T. A Genome–Wide Short Hairpin RNA Screening of Jurkat T–Cells for Human Proteins Contributing to Productive HIV–1 Replication. J. Biol. Chem. 2009, 284, 19463–19473. [Google Scholar] [CrossRef]
- Berthet–Colominas, C.; Monaco, S.; Novelli, A.; Sibai, G.; Mallet, F.; Cusack, S. Head–to–Tail Dimers and Interdomain Flexibility Revealed by the Crystal Structure of HIV–1 Capsid Protein (p24) Complexed with a Monoclonal Antibody Fab. EMBO J. 1999, 18, 1124–1136. [Google Scholar] [CrossRef]
- Ganser, B.K.; Li, S.; Klishko, V.Y.; Finch, J.T.; Sundquist, W.I. Assembly and Analysis of Conical Models for the HIV–1 Core. Science 1999, 283, 80–83. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser–Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X–Ray Structures of the Hexameric Building Block of the HIV Capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser–Pornillos, B.K.; Yeager, M. Atomic–Level Modelling of the HIV Capsid. Nature 2011, 469, 424–427. [Google Scholar] [CrossRef]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV–1 Capsid Structure by Cryo–Electron Microscopy and All–Atom Molecular Dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef]
- Fassati, A.; Goff, S.P. Characterization of Intracellular Reverse Transcription Complexes of Human Immunodeficiency Virus Type 1. J. Virol. 2001, 75, 3626–3635. [Google Scholar]
- Hulme, A.E.; Perez, O.; Hope, T.J. Complementary Assays Reveal a Relationship between HIV–1 Uncoating and Reverse Transcription. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9975–9980. [Google Scholar]
- Arfi, V.; Lienard, J.; Nguyen, X.N.; Berger, G.; Rigal, D.; Darlix, J.L.; Cimarelli, A. Characterization of the Behavior of Functional Viral Genomes During the Early Steps of Human Immunodeficiency Virus Type 1 Infection. J. Virol. 2009, 83, 7524–7535. [Google Scholar]
- Yu, Z.; Dobro, M.J.; Woodward, C.L.; Levandovsky, A.; Danielson, C.M.; Sandrin, V.; Shi, J.; Aiken, C.; Zandi, R.; Hope, T.J.; et al. Unclosed HIV–1 Capsids Suggest a Curled Sheet Model of Assembly. J. Mol. Biol. 2013, 425, 112–123. [Google Scholar]
- Pereira, C.F.; Rossy, J.; Owen, D.M.; Mak, J.; Gaus, K. HIV Taken by Storm: Super–Resolution Fluorescence Microscopy of a Viral Infection. Virol. J. 2012, 9, 84. [Google Scholar] [CrossRef]
- Lelek, M.; Di Nunzio, F.; Henriques, R.; Charneau, P.; Arhel, N.; Zimmer, C. Superresolution Imaging of HIV in Infected Cells with FlAsH–PALM. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 8564–8569. [Google Scholar]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the Intracellular Behavior of HIV in Living Cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef]
- Zack, J.A.; Arrigo, S.J.; Weitsman, S.R.; Go, A.S.; Haislip, A.; Chen, I.S. HIV–1 Entry into Quiescent Primary Lymphocytes: Molecular Analysis Reveals a Labile, Latent Viral Structure. Cell 1990, 61, 213–222. [Google Scholar] [CrossRef]
- Butler, S.L.; Hansen, M.S.; Bushman, F.D. A Quantitative Assay for HIV DNA Integration In Vivo. Nat. Med. 2001, 7, 631–634. [Google Scholar] [CrossRef]
- Sherman, P.A.; Fyfe, J.A. Human Immunodeficiency Virus Integration Protein Expressed in Escherichia Coli Possesses Selective DNA Cleaving Activity. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 5119–5123. [Google Scholar] [CrossRef]
- Bushman, F.D.; Craigie, R. Activities of Human Immunodeficiency Virus (HIV) Integration Protein In Vitro: Specific Cleavage and Integration of HIV DNA. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 1339–1343. [Google Scholar] [CrossRef]
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV–1 DNA Integration: Mechanism of Viral DNA Cleavage and DNA Strand Transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human Immunodeficiency Virus Type 1 Preintegration Complexes: Studies of Organization and Composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar]
- Thomas, J.A.; Ott, D.E.; Gorelick, R.J. Efficiency of Human Immunodeficiency Virus Type 1 Postentry Infection Processes: Evidence against Disproportionate Numbers of Defective Virions. J. Virol. 2007, 81, 4367–4370. [Google Scholar] [CrossRef]
- Iordanskiy, S.; Berro, R.; Altieri, M.; Kashanchi, F.; Bukrinsky, M. Intracytoplasmic Maturation of the Human Immunodeficiency Virus Type 1 Reverse Transcription Complexes Determines Their Capacity to Integrate into Chromatin. Retrovirology 2006, 3, 4. [Google Scholar] [CrossRef]
- Yamashita, M.; Perez, O.; Hope, T.J.; Emerman, M. Evidence for Direct Involvement of the Capsid Protein in HIV Infection of Nondividing Cells. PLoS Pathog. 2007, 3, 1502–1510. [Google Scholar]
- Thomas, J.A.; Gagliardi, T.D.; Alvord, W.G.; Lubomirski, M.; Bosche, W.J.; Gorelick, R.J. Human Immunodeficiency Virus Type 1 Nucleocapsid Zinc–Finger Mutations Cause Defects in Reverse Transcription and Integration. Virology 2006, 353, 41–51. [Google Scholar] [CrossRef]
- Kilzer, J.M.; Stracker, T.; Beitzel, B.; Meek, K.; Weitzman, M.; Bushman, F.D. Roles of Host Cell Factors in Circularization of Retroviral DNA. Virology 2003, 314, 460–467. [Google Scholar] [CrossRef]
- Miller, M.D.; Wang, B.; Bushman, F.D. Human Immunodeficiency Virus Type 1 Preintegration Complexes Containing Discontinuous Plus Strands Are Competent to Integrate In Vitro. J. Virol. 1995, 69, 3938–3944. [Google Scholar]
- Munir, S.; Thierry, S.; Subra, F.; Deprez, E.; Delelis, O. Quantitative Analysis of the Time–Course of Viral DNA Forms During the HIV–1 Life Cycle. Retrovirology 2013, 10, 87. [Google Scholar] [CrossRef]
- Li, L.; Olvera, J.M.; Yoder, K.E.; Mitchell, R.S.; Butler, S.L.; Lieber, M.; Martin, S.L.; Bushman, F.D. Role of the Non–Homologous DNA End Joining Pathway in the Early Steps of Retroviral Infection. EMBO J. 2001, 20, 3272–3281. [Google Scholar] [CrossRef]
- Li, Y.; Kappes, J.C.; Conway, J.A.; Price, R.W.; Shaw, G.M.; Hahn, B.H. Molecular Characterization of Human Immunodeficiency Virus Type 1 Cloned Directly from Uncultured Human Brain Tissue: Identification of Replication–Competent and –Defective Viral Genomes. J. Virol. 1991, 65, 3973–3985. [Google Scholar]
- Farnet, C.M.; Haseltine, W.A. Circularization of Human Immunodeficiency Virus Type 1 DNA In Vitro. J. Virol. 1991, 65, 6942–6952. [Google Scholar]
- Yan, N.; Cherepanov, P.; Daigle, J.E.; Engelman, A.; Lieberman, J. The SET Complex Acts as a Barrier to Autointegration of HIV–1. PLoS Pathog.. 2009, 5, e1000327. [Google Scholar] [CrossRef]
- De Iaco, A.; Santoni, F.; Vannier, A.; Guipponi, M.; Antonarakis, S.; Luban, J. TNPO3 Protects HIV–1 Replication from CPSF6–Mediated Capsid Stabilization in the Host Cell Cytoplasm. Retrovirology 2013, 10, 20. [Google Scholar] [CrossRef]
- Brussel, A.; Sonigo, P. Analysis of Early Human Immunodeficiency Virus Type 1 DNA Synthesis by Use of a New Sensitive Assay for Quantifying Integrated Provirus. J. Virol. 2003, 77, 10119–10124. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV–1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences. PLoS Biol. 2004, 2, E234. [Google Scholar] [CrossRef]
- Christ, F.; Thys, W.; De Rijck, J.; Gijsbers, R.; Albanese, A.; Arosio, D.; Emiliani, S.; Rain, J.C.; Benarous, R.; Cereseto, A.; et al. Transportin–SR2 Imports HIV into the Nucleus. Curr. Biol. 2008, 18, 1192–1202. [Google Scholar] [CrossRef]
- Di Primio, C.; Quercioli, V.; Allouch, A.; Gijsbers, R.; Christ, F.; Debyser, Z.; Arosio, D.; Cereseto, A. Single–Cell Imaging of HIV–1 Provirus (SCIP). Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5636–5641. [Google Scholar] [CrossRef]
- Li, M.; Mizuuchi, M.; Burke, T.R., Jr.; Craigie, R. Retroviral DNA Integration: Reaction Pathway and Critical Intermediates. EMBO J. 2006, 25, 1295–1304. [Google Scholar] [CrossRef]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral Intasome Assembly and Inhibition of DNA Strand Transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Sharova, N.; McDonald, T.L.; Pushkarskaya, T.; Tarpley, W.G.; Stevenson, M. Association of Integrase, Matrix, and Reverse Transcriptase Antigens of Human Immunodeficiency Virus Type 1 with Viral Nucleic Acids Following Acute Infection. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6125–6129. [Google Scholar] [CrossRef]
- Gallay, P.; Swingler, S.; Song, J.; Bushman, F.; Trono, D. HIV Nuclear Import Is Governed by the Phosphotyrosine–Mediated Binding of Matrix to the Core Domain of Integrase. Cell 1995, 83, 569–576. [Google Scholar] [CrossRef]
- Gallay, P.; Hope, T.; Chin, D.; Trono, D. HIV–1 Infection of Nondividing Cells through the Recognition of Integrase by the Importin/Karyopherin Pathway. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 9825–9830. [Google Scholar] [CrossRef]
- Heinzinger, N.K.; Bukinsky, M.I.; Haggerty, S.A.; Ragland, A.M.; Kewalramani, V.; Lee, M.A.; Gendelman, H.E.; Ratner, L.; Stevenson, M.; Emerman, M. The Vpr Protein of Human Immunodeficiency Virus Type 1 Influences Nuclear Localization of Viral Nucleic Acids in Nondividing Host Cells. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 7311–7315. [Google Scholar] [CrossRef]
- Karageorgos, L.; Li, P.; Burrell, C. Characterization of HIV Replication Complexes Early after Cell–to–Cell Infection. AIDS Res. Hum. Retrov. 1993, 9, 817–823. [Google Scholar] [CrossRef]
- Arhel, N.J.; Souquere–Besse, S.; Munier, S.; Souque, P.; Guadagnini, S.; Rutherford, S.; Prevost, M.C.; Allen, T.D.; Charneau, P. HIV–1 DNA Flap Formation Promotes Uncoating of the Pre–Integration Complex at the Nuclear Pore. EMBO J. 2007, 26, 3025–3037. [Google Scholar] [CrossRef]
- Meehan, A.M.; Saenz, D.T.; Morrison, J.; Hu, C.; Peretz, M.; Poeschla, E.M. LEDGF Dominant Interference Proteins Demonstrate Prenuclear Exposure of HIV–1 Integrase and Synergize with LEDGF Depletion to Destroy Viral Infectivity. J. Virol. 2011, 85, 3570–3583. [Google Scholar] [CrossRef]
- Zhou, L.; Sokolskaja, E.; Jolly, C.; James, W.; Cowley, S.A.; Fassati, A. Transportin 3 Promotes a Nuclear Maturation Step Required for Efficient HIV–1 Integration. PLoS Pathog. 2011, 7, e1002194. [Google Scholar] [CrossRef]
- Di Nunzio, F.; Danckaert, A.; Fricke, T.; Perez, P.; Fernandez, J.; Perret, E.; Roux, P.; Shorte, S.; Charneau, P.; Diaz–Griffero, F.; et al. Human Nucleoporins Promote HIV–1 Docking at the Nuclear Pore, Nuclear Import and Integration. PLoS One 2012, 7, e46037. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Haggerty, S.; Dempsey, M.P.; Sharova, N.; Adzhubel, A.; Spitz, L.; Lewis, P.; Goldfarb, D.; Emerman, M.; Stevenson, M. A Nuclear Localization Signal within HIV–1 Matrix Protein That Governs Infection of Non–Dividing Cells. Nature 1993, 365, 666–669. [Google Scholar] [CrossRef]
- Haffar, O.K.; Popov, S.; Dubrovsky, L.; Agostini, I.; Tang, H.; Pushkarsky, T.; Nadler, S.G.; Bukrinsky, M. Two Nuclear Localization Signals in the HIV–1 Matrix Protein Regulate Nuclear Import of the HIV–1 Pre–Integration Complex. J. Mol. Biol. 2000, 299, 359–368. [Google Scholar] [CrossRef]
- Bouyac–Bertoia, M.; Dvorin, J.D.; Fouchier, R.A.; Jenkins, Y.; Meyer, B.E.; Wu, L.I.; Emerman, M.; Malim, M.H. HIV–1 Infection Requires a Functional Integrase NLS. Mol. Cell. 2001, 7, 1025–1035. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Meyer, B.E.; Simon, J.H.; Fischer, U.; Albright, A.V.; Gonzalez–Scarano, F.; Malim, M.H. Interaction of the Human Immunodeficiency Virus Type 1 Vpr Protein with the Nuclear Pore Complex. J. Virol. 1998, 72, 6004–6013. [Google Scholar]
- Vodicka, M.A.; Koepp, D.M.; Silver, P.A.; Emerman, M. HIV–1 Vpr Interacts with the Nuclear Transport Pathway to Promote Macrophage Infection. Genes Dev. 1998, 12, 175–185. [Google Scholar] [CrossRef]
- Popov, S.; Rexach, M.; Ratner, L.; Blobel, G.; Bukrinsky, M. Viral Protein R Regulates Docking of the HIV–1 Preintegration Complex to the Nuclear Pore Complex. J. Biol. Chem. 1998, 273, 13347–13352. [Google Scholar]
- Popov, S.; Rexach, M.; Zybarth, G.; Reiling, N.; Lee, M.A.; Ratner, L.; Lane, C.M.; Moore, M.S.; Blobel, G.; Bukrinsky, M. Viral Protein R Regulates Nuclear Import of the HIV–1 Pre–Integration Complex. EMBO J. 1998, 17, 909–917. [Google Scholar] [CrossRef]
- Jenkins, Y.; McEntee, M.; Weis, K.; Greene, W.C. Characterization of HIV–1 Vpr Nuclear Import: Analysis of Signals and Pathways. J. Cell Biol. 1998, 143, 875–885. [Google Scholar] [CrossRef]
- Ao, Z.; Danappa Jayappa, K.; Wang, B.; Zheng, Y.; Kung, S.; Rassart, E.; Depping, R.; Kohler, M.; Cohen, E.A.; Yao, X. Importin Alpha3 Interacts with HIV–1 Integrase and Contributes to HIV–1 Nuclear Import and Replication. J. Virol. 2010, 84, 8650–8663. [Google Scholar] [CrossRef]
- Fassati, A.; Gorlich, D.; Harrison, I.; Zaytseva, L.; Mingot, J.M. Nuclear Import of HIV–1 Intracellular Reverse Transcription Complexes Is Mediated by Importin 7. EMBO J. 2003, 22, 3675–3685. [Google Scholar] [CrossRef]
- Ao, Z.; Huang, G.; Yao, H.; Xu, Z.; Labine, M.; Cochrane, A.W.; Yao, X. Interaction of Human Immunodeficiency Virus Type 1 Integrase with Cellular Nuclear Import Receptor Importin 7 and Its Impact on Viral Replication. J. Biol. Chem. 2007, 282, 13456–13467. [Google Scholar] [CrossRef]
- Larue, R.; Gupta, K.; Wuensch, C.; Shkriabai, N.; Kessl, J.J.; Danhart, E.; Feng, L.; Taltynov, O.; Christ, F.; Van Duyne, G.D.; et al. Interaction of the HIV–1 Intasome with Transportin 3 Protein (TNPO3 or TRN–SR2). J. Biol. Chem. 2012, 287, 34044–34058. [Google Scholar] [CrossRef]
- Zielske, S.P.; Stevenson, M. Importin 7 May Be Dispensable for Human Immunodeficiency Virus Type 1 and Simian Immunodeficiency Virus Infection of Primary Macrophages. J. Virol. 2005, 79, 11541–11546. [Google Scholar] [CrossRef]
- Krishnan, L.; Matreyek, K.A.; Oztop, I.; Lee, K.; Tipper, C.H.; Li, X.; Dar, M.J.; Kewalramani, V.N.; Engelman, A. The Requirement for Cellular Transportin 3 (TNPO3 or TRN–SR2) During Infection Maps to Human Immunodeficiency Virus Type 1 Capsid and Not Integrase. J. Virol. 2010, 84, 397–406. [Google Scholar] [CrossRef]
- Cribier, A.; Segeral, E.; Delelis, O.; Parissi, V.; Simon, A.; Ruff, M.; Benarous, R.; Emiliani, S. Mutations Affecting Interaction of Integrase with TNPO3 Do Not Prevent HIV–1 cDNA Nuclear Import. Retrovirology 2011, 8, 104. [Google Scholar] [CrossRef]
- Woodward, C.L.; Prakobwanakit, S.; Mosessian, S.; Chow, S.A. Integrase Interacts with Nucleoporin NUP153 to Mediate the Nuclear Import of Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 6522–6533. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Mousnier, A.; Rustum, C.; Stutz, F.; Hallberg, E.; Dargemont, C.; Benichou, S. Docking of HIV–1 Vpr to the Nuclear Envelope Is Mediated by the Interaction with the Nucleoporin HCG1. J. Biol. Chem. 2002, 277, 45091–45098. [Google Scholar]
- Zennou, V.; Petit, C.; Guetard, D.; Nerhbass, U.; Montagnier, L.; Charneau, P. HIV–1 Genome Nuclear Import Is Mediated by a Central DNA Flap. Cell 2000, 101, 173–185. [Google Scholar] [CrossRef]
- Follenzi, A.; Ailles, L.E.; Bakovic, S.; Geuna, M.; Naldini, L. Gene Transfer by Lentiviral Vectors Is Limited by Nuclear Translocation and Rescued by HIV–1 Pol Sequences. Nat. Genet. 2000, 25, 217–222. [Google Scholar] [CrossRef]
- Limon, A.; Nakajima, N.; Lu, R.; Ghory, H.Z.; Engelman, A. Wild–Type Levels of Nuclear Localization and Human Immunodeficiency Virus Type 1 Replication in the Absence of the Central DNA Flap. J. Virol. 2002, 76, 12078–12086. [Google Scholar] [CrossRef]
- Dvorin, J.D.; Bell, P.; Maul, G.G.; Yamashita, M.; Emerman, M.; Malim, M.H. Reassessment of the Roles of Integrase and the Central DNA Flap in Human Immunodeficiency Virus Type 1 Nuclear Import. J. Virol. 2002, 76, 12087–12096. [Google Scholar] [CrossRef]
- Marsden, M.D.; Zack, J.A. Human Immunodeficiency Virus Bearing a Disrupted Central DNA Flap Is Pathogenic In Vivo. J. Virol. 2007, 81, 6146–6150. [Google Scholar] [CrossRef]
- De Rijck, J.; Debyser, Z. The Central DNA Flap of the Human Immunodeficiency Virus Type 1 Is Important for Viral Replication. Biochem. Biophys. Res. Commun. 2006, 349, 1100–1110. [Google Scholar] [CrossRef]
- Riviere, L.; Darlix, J.L.; Cimarelli, A. Analysis of the Viral Elements Required in the Nuclear Import of HIV–1 DNA. J. Virol. 2010, 84, 729–739. [Google Scholar] [CrossRef]
- Ao, Z.; Yao, X.; Cohen, E.A. Assessment of the Role of the Central DNA Flap in Human Immunodeficiency Virus Type 1 Replication by Using a Single–Cycle Replication System. J. Virol. 2004, 78, 3170–3177. [Google Scholar] [CrossRef]
- Skasko, M.; Kim, B. Compensatory Role of Human Immunodeficiency Virus Central Polypurine Tract Sequence in Kinetically Disrupted Reverse Transcription. J. Virol. 2008, 82, 7716–7720. [Google Scholar] [CrossRef]
- Hu, C.; Saenz, D.T.; Fadel, H.J.; Walker, W.; Peretz, M.; Poeschla, E.M. The HIV–1 Central Polypurine Tract Functions as a Second Line of Defense against APOBEC3G/F. J. Virol. 2010, 84, 11981–11993. [Google Scholar] [CrossRef]
- Wurtzer, S.; Goubard, A.; Mammano, F.; Saragosti, S.; Lecossier, D.; Hance, A.J.; Clavel, F. Functional Central Polypurine Tract Provides Downstream Protection of the Human Immunodeficiency Virus Type 1 Genome from Editing by APOBEC3G and APOBEC3B. J. Virol. 2006, 80, 3679–3683. [Google Scholar] [CrossRef]
- Suspene, R.; Rusniok, C.; Vartanian, J.P.; Wain–Hobson, S. Twin Gradients in APOBEC3 Edited HIV–1 DNA Reflect the Dynamics of Lentiviral Replication. Nucleic Acids Res. 2006, 34, 4677–4684. [Google Scholar] [CrossRef]
- Poeschla, E. The Importance of Becoming Double–Stranded: Innate Immunity and the Kinetic Model of HIV–1 Central Plus Strand Synthesis. Virology 2013, 441, 1–11. [Google Scholar] [CrossRef]
- Limon, A.; Devroe, E.; Lu, R.; Ghory, H.Z.; Silver, P.A.; Engelman, A. Nuclear Localization of Human Immunodeficiency Virus Type 1 Preintegration Complexes (PICS): V165A and R166A Are Pleiotropic Integrase Mutants Primarily Defective for Integration, Not PIC Nuclear Import. J. Virol. 2002, 76, 10598–10607. [Google Scholar] [CrossRef]
- Fouchier, R.A.; Meyer, B.E.; Simon, J.H.; Fischer, U.; Malim, M.H. HIV–1 Infection of Non–Dividing Cells: Evidence That the Amino–Terminal Basic Region of the Viral Matrix Protein Is Important for Gag Processing but Not for Post–Entry Nuclear Import. EMBO J. 1997, 16, 4531–4539. [Google Scholar] [CrossRef]
- Freed, E.O.; Englund, G.; Martin, M.A. Role of the Basic Domain of Human Immunodeficiency Virus Type 1 Matrix in Macrophage Infection. J. Virol. 1995, 69, 3949–3954. [Google Scholar]
- Katz, R.A.; Greger, J.G.; Boimel, P.; Skalka, A.M. Human Immunodeficiency Virus Type 1 DNA Nuclear Import and Integration Are Mitosis Independent in Cycling Cells. J. Virol. 2003, 77, 13412–13417. [Google Scholar] [CrossRef]
- Yamashita, M.; Emerman, M. Capsid Is a Dominant Determinant of Retrovirus Infectivity in Nondividing Cells. J. Virol. 2004, 78, 5670–5678. [Google Scholar] [CrossRef]
- Qi, M.; Yang, R.; Aiken, C. Cyclophilin A–Dependent Restriction of Human Immunodeficiency Virus Type 1 Capsid Mutants for Infection of Nondividing Cells. J. Virol. 2008, 82, 12001–12008. [Google Scholar] [CrossRef]
- Ylinen, L.M.; Schaller, T.; Price, A.; Fletcher, A.J.; Noursadeghi, M.; James, L.C.; Towers, G.J. Cyclophilin A Levels Dictate Infection Efficiency of Human Immunodeficiency Virus Type 1 Capsid Escape Mutants A92E and G94D. J. Virol. 2009, 83, 2044–2047. [Google Scholar] [CrossRef]
- Dismuke, D.J.; Aiken, C. Evidence for a Functional Link between Uncoating of the Human Immunodeficiency Virus Type 1 Core and Nuclear Import of the Viral Preintegration Complex. J. Virol. 2006, 80, 3712–3720. [Google Scholar] [CrossRef]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a Human Immunodeficiency Virus Type 1 Core of Optimal Stability Is Crucial for Viral Replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The Cytoplasmic Body Component Trim5alpha Restricts HIV–1 Infection in Old World Monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz–Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific Recognition and Accelerated Uncoating of Retroviral Capsids by the Trim5alpha Restriction Factor. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome Inhibitors Uncouple Rhesus Trim5alpha Restriction of HIV–1 Reverse Transcription and Infection. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7465–7470. [Google Scholar]
- Kutluay, S.B.; Perez–Caballero, D.; Bieniasz, P.D. Fates of Retroviral Core Components During Unrestricted and Trim5–Restricted Infection. PLoS Pathog. 2013, 9, e1003214. [Google Scholar] [CrossRef]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome Inhibition Reveals That a Functional Preintegration Complex Intermediate Can Be Generated During Restriction by Diverse Trim5 Proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef]
- Danielson, C.M.; Cianci, G.C.; Hope, T.J. Recruitment and Dynamics of Proteasome Association with rhTrim5alpha Cytoplasmic Complexes During HIV–1 Infection. Traffic 2012, 13, 1206–1217. [Google Scholar] [CrossRef]
- Yap, M.W.; Dodding, M.P.; Stoye, J.P. Trim–Cyclophilin A Fusion Proteins Can Restrict Human Immunodeficiency Virus Type 1 Infection at Two Distinct Phases in the Viral Life Cycle. J. Virol. 2006, 80, 4061–4067. [Google Scholar] [CrossRef]
- Hori, T.; Takeuchi, H.; Saito, H.; Sakuma, R.; Inagaki, Y.; Yamaoka, S. A Carboxy–Terminally Truncated Human CPSF6 Lacking Residues Encoded by Exon 6 Inhibits HIV–1 cDNA Synthesis and Promotes Capsid Disassembly. J. Virol. 2013. [Google Scholar]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV–1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef]
- Lee, K.; Mulky, A.; Yuen, W.; Martin, T.D.; Meyerson, N.R.; Choi, L.; Yu, H.; Sawyer, S.L.; Kewalramani, V.N. HIV–1 Capsid–Targeting Domain of Cleavage and Polyadenylation Specificity Factor 6. J. Virol. 2012, 86, 3851–3860. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.; Schulz, R.; Malim, M.H. Human MX2 Is an Interferon–Induced Post–Entry Inhibitor of HIV–1 Infection. Nature 2013. [Google Scholar] [CrossRef]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The Interferon–Inducible MXB Protein Inhibits HIV–1 Infection. Cell Host Microbe 2013. [Google Scholar] [CrossRef]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV Capsid Is a Tractable Target for Small Molecule Therapeutic Intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef]
- Shi, J.; Zhou, J.; Shah, V.B.; Aiken, C.; Whitby, K. Small–Molecule Inhibition of Human Immunodeficiency Virus Type 1 Infection by Virus Capsid Destabilization. J. Virol. 2011, 85, 542–549. [Google Scholar] [CrossRef]
- Lamorte, L.; Titolo, S.; Lemke, C.T.; Goudreau, N.; Mercier, J.F.; Wardrop, E.; Shah, V.B.; von Schwedler, U.K.; Langelier, C.; Banik, S.S.; et al. Discovery of Novel Small Molecule HIV–1 Replication Inhibitors That Stabilize Capsid Complexes. Antimicrob. Agents Chemother. 2013, 57, 4622–4631. [Google Scholar] [CrossRef]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 Defines a Conserved Capsid Interface That Modulates HIV–1 Replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yücel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 Phenylalanine–Glycine Motifs Engage a Common Binding Pocket within the HIV–1 Capsid Protein to Mediate Lentiviral Infectivity. PLoS Pathog. 2013. In Press. [Google Scholar]
- Vozzolo, L.; Loh, B.; Gane, P.J.; Tribak, M.; Zhou, L.; Anderson, I.; Nyakatura, E.; Jenner, R.G.; Selwood, D.; Fassati, A. Gyrase B Inhibitor Impairs HIV–1 Replication by Targeting HSP90 and the Capsid Protein. J. Biol. Chem. 2010, 285, 39314–39328. [Google Scholar] [CrossRef]
- Fassati, A.; Goff, S.P. Characterization of Intracellular Reverse Transcription Complexes of Moloney Murine Leukemia Virus. J. Virol. 1999, 73, 8919–8925. [Google Scholar]
- Yuan, B.; Li, X.; Goff, S.P. Mutations Altering the Moloney Murine Leukemia Virus p12 Gag Protein Affect Virion Production and Early Events of the Virus Life Cycle. EMBO J. 1999, 18, 4700–4710. [Google Scholar] [CrossRef]
- Prizan–Ravid, A.; Elis, E.; Laham–Karam, N.; Selig, S.; Ehrlich, M.; Bacharach, E. The Gag Cleavage Product, p12, Is a Functional Constituent of the Murine Leukemia Virus Pre–Integration Complex. PLoS Pathog. 2010, 6, e1001183. [Google Scholar] [CrossRef]
- Schneider, W.M.; Brzezinski, J.D.; Aiyer, S.; Malani, N.; Gyuricza, M.; Bushman, F.D.; Roth, M.J. Viral DNA Tethering Domains Complement Replication–Defective Mutations in the p12 Protein of MuLV Gag. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9487–9492. [Google Scholar]
- Elis, E.; Ehrlich, M.; Prizan–Ravid, A.; Laham–Karam, N.; Bacharach, E. P12 Tethers the Murine Leukemia Virus Pre–Integration Complex to Mitotic Chromosomes. PLoS Pathog. 2012, 8, e1003103. [Google Scholar] [CrossRef]
- Best, S.; Le Tissier, P.; Towers, G.; Stoye, J.P. Positional Cloning of the Mouse Retrovirus Restriction Gene Fv1. Nature 1996, 382, 826–829. [Google Scholar] [CrossRef]
- Hopkins, N.; Schindler, J.; Hynes, R. Six–NB–Tropic Murine Leukemia Viruses Derived from a B–Tropic Virus of Balb/C Have Altered p30. J. Virol. 1977, 21, 309–318. [Google Scholar]
- Rommelaere, J.; Donis–Keller, H.; Hopkins, N. RNA Sequencing Provides Evidence for Allelism of Determinants of the N–, B– or NB–Tropism of Murine Leukemia Viruses. Cell 1979, 16, 43–50. [Google Scholar] [CrossRef]
- Hilditch, L.; Matadeen, R.; Goldstone, D.C.; Rosenthal, P.B.; Taylor, I.A.; Stoye, J.P. Ordered Assembly of Murine Leukemia Virus Capsid Protein on Lipid Nanotubes Directs Specific Binding by the Restriction Factor, Fv1. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5771–5776. [Google Scholar]
- Pryciak, P.M.; Varmus, H.E. Fv–1 Restriction and Its Effects on Murine Leukemia Virus Integration In Vivo and In Vitro. J. Virol. 1992, 66, 5959–5966. [Google Scholar]
- Yang, W.K.; Kiggans, J.O.; Yang, D.M.; Ou, C.Y.; Tennant, R.W.; Brown, A.; Bassin, R.H. Synthesis and Circularization of N– and B–Tropic Retroviral DNA Fv–1 Permissive and Restrictive Mouse Cells. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 2994–2998. [Google Scholar]
- Jolicoeur, P.; Rassart, E. Effect of Fv–1 Gene Product on Synthesis of Linear and Supercoiled Viral DNA in Cells Infected with Murine Leukemia Virus. J. Virol. 1980, 33, 183–195. [Google Scholar]
- Schaller, T.; Ylinen, L.M.; Webb, B.L.; Singh, S.; Towers, G.J. Fusion of Cyclophilin A to Fv1 Enables Cyclosporine–Sensitive Restriction of Human and Feline Immunodeficiency Viruses. J. Virol. 2007, 81, 10055–10063. [Google Scholar] [CrossRef]
- Bushman, F.D.; Malani, N.; Fernandes, J.; D'Orso, I.; Cagney, G.; Diamond, T.L.; Zhou, H.; Hazuda, D.J.; Espeseth, A.S.; Konig, R.; et al. Host Cell Factors in HIV Replication: Meta–Analysis of Genome–Wide Studies. PLoS Pathog. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Zhang, R.; Mehla, R.; Chauhan, A. Perturbation of Host Nuclear Membrane Component RanBP2 Impairs the Nuclear Import of Human Immunodeficiency Virus 1 Preintegration Complex (DNA). PLoS One 2010, 5, e15620. [Google Scholar] [CrossRef]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hue, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV–1 Capsid–Cyclophilin Interactions Determine Nuclear Import Pathway, Integration Targeting and Replication Efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Engelman, A. The Requirement for Nucleoporin NUP153 During Human Immunodeficiency Virus Type 1 Infection Is Determined by the Viral Capsid. J. Virol. 2011, 85, 7818–7827. [Google Scholar] [CrossRef]
- Logue, E.C.; Taylor, K.T.; Goff, P.H.; Landau, N.R. The Cargo–Binding Domain of Transportin 3 Is Required for Lentivirus Nuclear Import. J. Virol. 2011, 85, 12950–12961. [Google Scholar] [CrossRef]
- De Iaco, A.; Luban, J. Inhibition of HIV–1 Infection by TNPO3 Depletion Is Determined by Capsid and Detectable after Viral cDNA Enters the Nucleus. Retrovirology 2011, 8, 98. [Google Scholar] [CrossRef]
- Valle–Casuso, J.C.; Di Nunzio, F.; Yang, Y.; Reszka, N.; Lienlaf, M.; Arhel, N.; Perez, P.; Brass, A.L.; Diaz–Griffero, F. TNPO3 Is Required for HIV–1 Replication after Nuclear Import but Prior to Integration and Binds the HIV–1 Core. J. Virol. 2012, 86, 5931–5936. [Google Scholar] [CrossRef]
- Hutten, S.; Kehlenbach, R.H. NUP214 Is Required for Crm1–Dependent Nuclear Protein Export In Vivo. Mol. Cell. Biol. 2006, 26, 6772–6785. [Google Scholar] [CrossRef]
- Walther, T.C.; Alves, A.; Pickersgill, H.; Loiodice, I.; Hetzer, M.; Galy, V.; Hulsmann, B.B.; Kocher, T.; Wilm, M.; Allen, T.; et al. The Conserved NUP107–160 Complex Is Critical for Nuclear Pore Complex Assembly. Cell 2003, 113, 195–206. [Google Scholar] [CrossRef]
- Hase, M.E.; Cordes, V.C. Direct Interaction with NUP153 Mediates Binding of Tpr to the Periphery of the Nuclear Pore Complex. Mol. Biol. Cell 2003, 14, 1923–1940. [Google Scholar] [CrossRef]
- Arnaoutov, A.; Azuma, Y.; Ribbeck, K.; Joseph, J.; Boyarchuk, Y.; Karpova, T.; McNally, J.; Dasso, M. Crm1 Is a Mitotic Effector of Ran–GTP in Somatic Cells. Nat Cell Biol 2005, 7, 626–632. [Google Scholar] [CrossRef]
- Dawlaty, M.M.; Malureanu, L.; Jeganathan, K.B.; Kao, E.; Sustmann, C.; Tahk, S.; Shuai, K.; Grosschedl, R.; van Deursen, J.M. Resolution of Sister Centromeres Requires RanBP2–Mediated Sumoylation of Topoisomerase IIalpha. Cell 2008, 133, 103–115. [Google Scholar] [CrossRef]
- Zuccolo, M.; Alves, A.; Galy, V.; Bolhy, S.; Formstecher, E.; Racine, V.; Sibarita, J.B.; Fukagawa, T.; Shiekhattar, R.; Yen, T.; et al. The Human NUP107–160 Nuclear Pore Subcomplex Contributes to Proper Kinetochore Functions. EMBO J. 2007, 26, 1853–1864. [Google Scholar] [CrossRef]
- Thys, W.; De Houwer, S.; Demeulemeester, J.; Taltynov, O.; Vancraenenbroeck, R.; Gerard, M.; De Rijck, J.; Gijsbers, R.; Christ, F.; Debyser, Z. Interplay between HIV Entry and Transportin–SR2 Dependency. Retrovirology 2011, 8, 7. [Google Scholar] [CrossRef]
- Fricke, T.; Valle–Casuso, J.C.; White, T.E.; Brandariz–Nunez, A.; Bosche, W.J.; Reszka, N.; Gorelick, R.; Diaz–Griffero, F. The Ability of TNPO3–Depleted Cells to Inhibit HIV–1 Infection Requires CPSF6. Retrovirology 2013, 10, 46. [Google Scholar] [CrossRef]
- De Houwer, S.; Demeulemeester, J.; Thys, W.; Taltynov, O.; Zmajkovicova, K.; Christ, F.; Debyser, Z. Identification of Residues in the C–Terminal Domain of HIV–1 Integrase That Mediate Binding to the Transportin–SR2 Protein. J. Biol. Chem. 2012, 287, 34059–34068. [Google Scholar]
- Taltynov, O.; Demeulemeester, J.; Christ, F.; De Houwer, S.; Tsirkone, V.G.; Gerard, M.; Weeks, S.D.; Strelkov, S.V.; Debyser, Z. Interaction of Transportin–SR2 with Ras–Related Nuclear Protein (Ran) GTPase. J. Biol. Chem. 2013, 288, 25603–25613. [Google Scholar] [CrossRef]
- Shah, V.B.; Shi, J.; Hout, D.R.; Oztop, I.; Krishnan, L.; Ahn, J.; Shotwell, M.S.; Engelman, A.; Aiken, C. The Host Proteins Transportin SR2/TNPO3 and Cyclophilin A Exert Opposing Effects on HIV–1 Uncoating. J. Virol. 2013, 87, 422–432. [Google Scholar] [CrossRef]
- Kataoka, N.; Bachorik, J.L.; Dreyfuss, G. Transportin–SR, a Nuclear Import Receptor for SR Proteins. J. Cell Biol. 1999, 145, 1145–1152. [Google Scholar] [CrossRef]
- Sukegawa, J.; Blobel, G. A Nuclear Pore Complex Protein That Contains Zinc Finger Motifs, Binds DNA, and Faces the Nucleoplasm. Cell 1993, 72, 29–38. [Google Scholar] [CrossRef]
- Enarson, P.; Enarson, M.; Bastos, R.; Burke, B. Amino–Terminal Sequences That Direct Nucleoporin NUP153 to the Inner Surface of the Nuclear Envelope. Chromosoma 1998, 107, 228–236. [Google Scholar]
- Lim, R.Y.; Huang, N.P.; Koser, J.; Deng, J.; Lau, K.H.; Schwarz–Herion, K.; Fahrenkrog, B.; Aebi, U. Flexible Phenylalanine–Glycine Nucleoporins as Entropic Barriers to Nucleocytoplasmic Transport. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 9512–9517. [Google Scholar]
- Cardarelli, F.; Lanzano, L.; Gratton, E. Capturing Directed Molecular Motion in the Nuclear Pore Complex of Live Cells. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9863–9868. [Google Scholar] [CrossRef]
- Fahrenkrog, B.; Maco, B.; Fager, A.M.; Koser, J.; Sauder, U.; Ullman, K.S.; Aebi, U. Domain–Specific Antibodies Reveal Multiple–Site Topology of NUP153 within the Nuclear Pore Complex. J. Struct. Biol. 2002, 140, 254–267. [Google Scholar] [CrossRef]
- Paulillo, S.M.; Phillips, E.M.; Koser, J.; Sauder, U.; Ullman, K.S.; Powers, M.A.; Fahrenkrog, B. Nucleoporin Domain Topology Is Linked to the Transport Status of the Nuclear Pore Complex. J. Mol. Biol. 2005, 351, 784–798. [Google Scholar] [CrossRef]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle–Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P.; et al. NUP153 and NUP98 Bind the HIV–1 Core and Contribute to the Early Steps of HIV–1 Replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef]
- Yokoyama, N.; Hayashi, N.; Seki, T.; Pante, N.; Ohba, T.; Nishii, K.; Kuma, K.; Hayashida, T.; Miyata, T.; Aebi, U.; et al. A Giant Nucleopore Protein That Binds Ran/TC4. Nature 1995, 376, 184–188. [Google Scholar] [CrossRef]
- Wu, J.; Matunis, M.J.; Kraemer, D.; Blobel, G.; Coutavas, E. Nup358, a Cytoplasmically Exposed Nucleoporin with Peptide Repeats, Ran–GTP Binding Sites, Zinc Fingers, a Cyclophilin A Homologous Domain, and a Leucine–Rich Region. J. Biol. Chem. 1995, 270, 14209–14213. [Google Scholar] [CrossRef]
- Wilken, N.; Senecal, J.L.; Scheer, U.; Dabauvalle, M.C. Localization of the Ran–GTP Binding Protein RanBP2 at the Cytoplasmic Side of the Nuclear Pore Complex. Eur J. Cell Biol. 1995, 68, 211–219. [Google Scholar]
- Lin, D.H.; Zimmermann, S.; Stuwe, T.; Stuwe, E.; Hoelz, A. Structural and Functional Analysis of the C–Terminal Domain of NUP358/RanBP2. J. Mol. Biol. 2013, 425, 1318–1329. [Google Scholar] [CrossRef]
- Bichel, K.; Price, A.J.; Schaller, T.; Towers, G.J.; Freund, S.M.; James, L.C. HIV–1 Capsid Undergoes Coupled Binding and Isomerization by the Nuclear Pore Protein NUP358. Retrovirology 2013, 10, 81. [Google Scholar] [CrossRef]
- Yoo, S.; Myszka, D.G.; Yeh, C.; McMurray, M.; Hill, C.P.; Sundquist, W.I. Molecular Recognition in the HIV–1 Capsid/Cyclophilin A Complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef]
- Ferreira, P.A.; Nakayama, T.A.; Travis, G.H. Interconversion of Red Opsin Isoforms by the Cyclophilin–Related Chaperone Protein Ran–Binding Protein 2. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 1556–1561. [Google Scholar] [CrossRef]
- Mamede, J.I.; Sitbon, M.; Battini, J.L.; Courgnaud, V. Heterogeneous Susceptibility of Circulating SIV Isolate Capsids to HIV–Interacting Factors. Retrovirology 2013, 10, 77. [Google Scholar] [CrossRef]
- Yaseen, N.R.; Blobel, G. Two Distinct Classes of Ran–Binding Sites on the Nucleoporin Nup–358. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5516–5521. [Google Scholar] [CrossRef]
- Higa, M.M.; Alam, S.L.; Sundquist, W.I.; Ullman, K.S. Molecular Characterization of the Ran–Binding Zinc Finger Domain of NUP153. J. Biol. Chem. 2007, 282, 17090–17100. [Google Scholar] [CrossRef]
- Prunuske, A.J.; Liu, J.; Elgort, S.; Joseph, J.; Dasso, M.; Ullman, K.S. Nuclear Envelope Breakdown Is Coordinated by Both NUP358/RanBP2 and NUP153, Two Nucleoporins with Zinc Finger Modules. Mol. Biol. Cell 2006, 17, 760–769. [Google Scholar]
- Li, Y.; Kar, A.K.; Sodroski, J. Target Cell Type–Dependent Modulation of Human Immunodeficiency Virus Type 1 Capsid Disassembly by Cyclophilin A. J. Virol. 2009, 83, 10951–10962. [Google Scholar] [CrossRef]
- Keckesova, Z.; Ylinen, L.M.; Towers, G.J. Cyclophilin A Renders Human Immunodeficiency Virus Type 1 Sensitive to Old World Monkey but Not Human Trim5 Alpha Antiviral Activity. J. Virol. 2006, 80, 4683–4690. [Google Scholar] [CrossRef]
- Holman, A.G.; Coffin, J.M. Symmetrical Base Preferences Surrounding HIV–1, Avian Sarcoma/Leukosis Virus, and Murine Leukemia Virus Integration Sites. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 6103–6107. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M.; Munroe, D.J. Weak Palindromic Consensus Sequences Are a Common Feature Found at the Integration Target Sites of Many Retroviruses. J. Virol. 2005, 79, 5211–5214. [Google Scholar]
- Pryciak, P.M.; Varmus, H.E. Nucleosomes, DNA–Binding Proteins, and DNA Sequence Modulate Retroviral Integration Target Site Selection. Cell 1992, 69, 769–780. [Google Scholar] [CrossRef]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV Integration Site Selection: Analysis by Massively Parallel Pyrosequencing Reveals Association with Epigenetic Modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A Role for LEDGF/p75 in Targeting HIV DNA Integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 Functions Downstream from Preintegration Complex Formation to Effect Gene–Specific HIV–1 Integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in Lentiviral Infectivity and Integration Targeting. PLoS One 2007, 2, e1340. [Google Scholar] [CrossRef]
- Ocwieja, K.E.; Brady, T.L.; Ronen, K.; Huegel, A.; Roth, S.L.; Schaller, T.; James, L.C.; Towers, G.J.; Young, J.A.; Chanda, S.K.; et al. HIV Integration Targeting: A Pathway Involving Transportin–3 and the Nuclear Pore Protein RanBP2. PLoS Pathog. 2011, 7, e1001313. [Google Scholar] [CrossRef]
- Koh, Y.; Wu, X.; Ferris, A.L.; Matreyek, K.A.; Smith, S.J.; Lee, K.; KewalRamani, V.N.; Hughes, S.H.; Engelman, A. Differential Effects of Human Immunodeficiency Virus Type 1 Capsid and Cellular Factors Nucleoporin 153 and LEDGF/p75 on the Efficiency and Specificity of Viral DNA Integration. J. Virol. 2013, 87, 648–658. [Google Scholar] [CrossRef]
- Vaquerizas, J.M.; Suyama, R.; Kind, J.; Miura, K.; Luscombe, N.M.; Akhtar, A. Nuclear Pore Proteins NUP153 and Megator Define Transcriptionally Active Regions in the Drosophila Genome. PLoS Genet. 2010, 6, e1000846. [Google Scholar] [CrossRef]
- Joseph, J.; Dasso, M. The Nucleoporin NUP358 Associates with and Regulates Interphase Microtubules. FEBS Lett. 2008, 582, 190–196. [Google Scholar] [CrossRef]
- Ambrose, Z.; Lee, K.; Ndjomou, J.; Xu, H.; Oztop, I.; Matous, J.; Takemura, T.; Unutmaz, D.; Engelman, A.; Hughes, S.H.; et al. Human Immunodeficiency Virus Type 1 Capsid Mutation N74D Alters Cyclophilin A Dependence and Impairs Macrophage Infection. J. Virol. 2012, 86, 4708–4714. [Google Scholar] [CrossRef]
- Saini, M.; Potash, M.J. Novel Activities of Cyclophilin A and Cyclosporin A During HIV–1 Infection of Primary Lymphocytes and Macrophages. J. Immunol. 2006, 177, 443–449. [Google Scholar]
- Goldstone, D.C.; Yap, M.W.; Robertson, L.E.; Haire, L.F.; Taylor, W.R.; Katzourakis, A.; Stoye, J.P.; Taylor, I.A. Structural and Functional Analysis of Prehistoric Lentiviruses Uncovers an Ancient Molecular Interface. Cell Host Microbe 2010, 8, 248–259. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Matreyek, K.A.; Engelman, A. Viral and Cellular Requirements for the Nuclear Entry of Retroviral Preintegration Nucleoprotein Complexes. Viruses 2013, 5, 2483-2511. https://doi.org/10.3390/v5102483
Matreyek KA, Engelman A. Viral and Cellular Requirements for the Nuclear Entry of Retroviral Preintegration Nucleoprotein Complexes. Viruses. 2013; 5(10):2483-2511. https://doi.org/10.3390/v5102483
Chicago/Turabian StyleMatreyek, Kenneth A., and Alan Engelman. 2013. "Viral and Cellular Requirements for the Nuclear Entry of Retroviral Preintegration Nucleoprotein Complexes" Viruses 5, no. 10: 2483-2511. https://doi.org/10.3390/v5102483
APA StyleMatreyek, K. A., & Engelman, A. (2013). Viral and Cellular Requirements for the Nuclear Entry of Retroviral Preintegration Nucleoprotein Complexes. Viruses, 5(10), 2483-2511. https://doi.org/10.3390/v5102483