Base Composition and Translational Selection are Insufficient to Explain Codon Usage Bias in Plant Viruses

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base Composition | |||
| Match | No Match | ||
| RSCU | Match | Both? | Translational Selection |
| No Match | Base Composition | Undetermined | |
2. Results
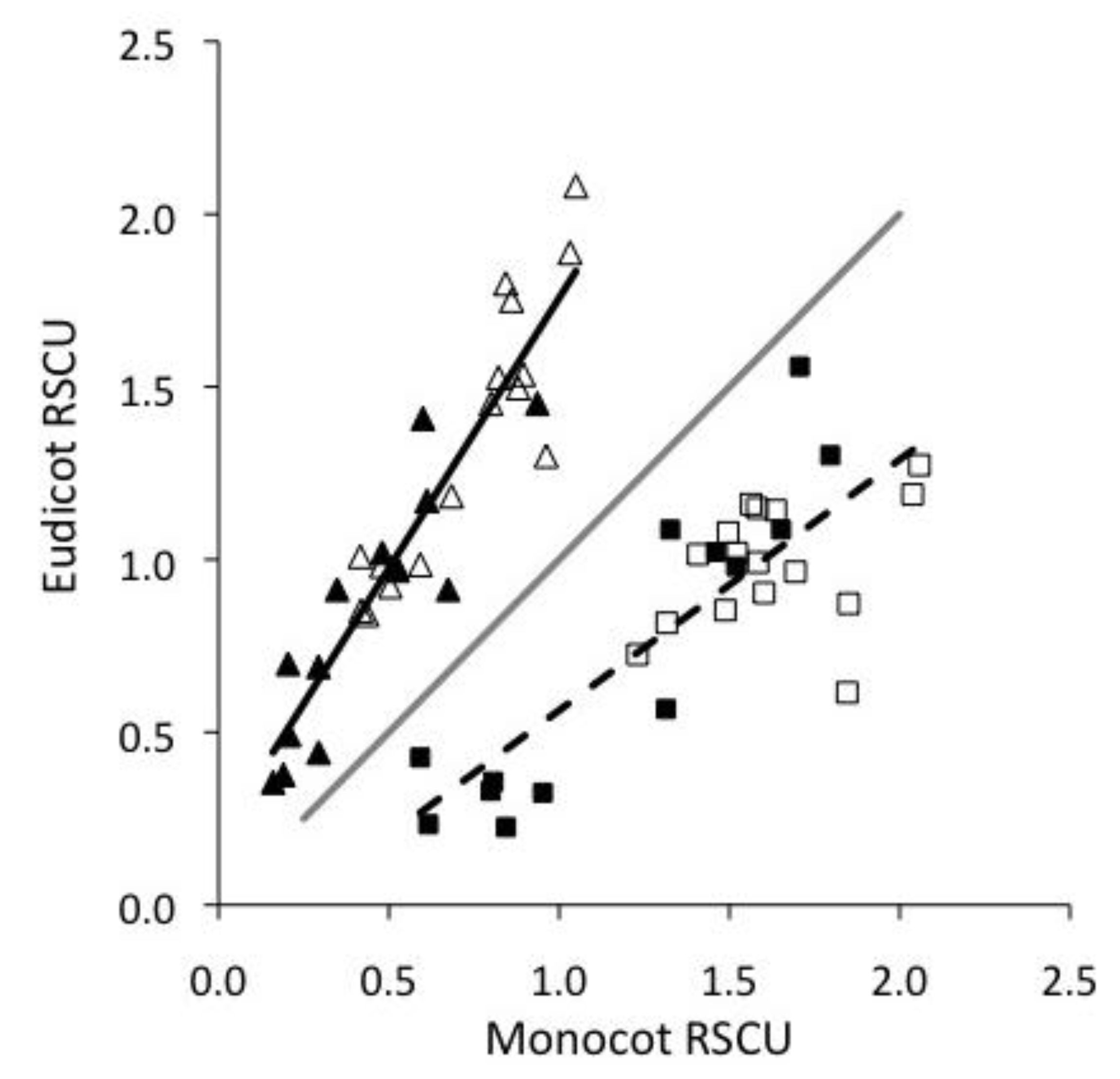
2.1. Monocots and Eudicots exhibit divergent CUB
| Monocots | Eudicots | ||||||
| tac | aac | ccc | tac | aac | |||
| ctc | acc | gac | |||||
| NNC | atc | gcc | tgc | ||||
| tcc | tac | cgc | |||||
| agc | cac | ggc | |||||
| NNG | ttg | aag | cag | ttg | aag | cag | |
| gag | agg | gag | |||||
| NNT | ctt | tct | gct | ||||
| gtt | gat | cgt | |||||
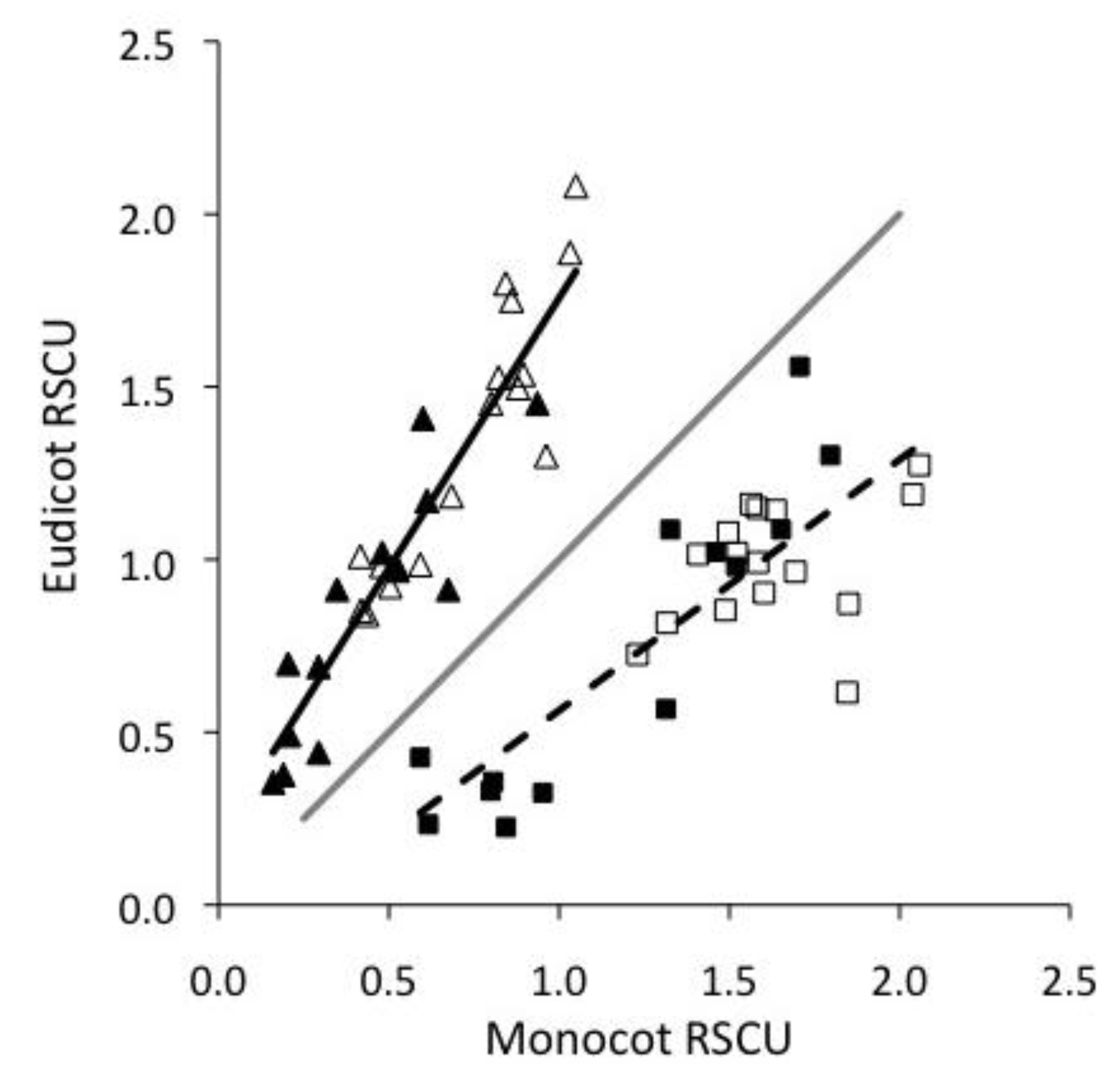
2.2. Base composition does not explain most CUB in plant viruses
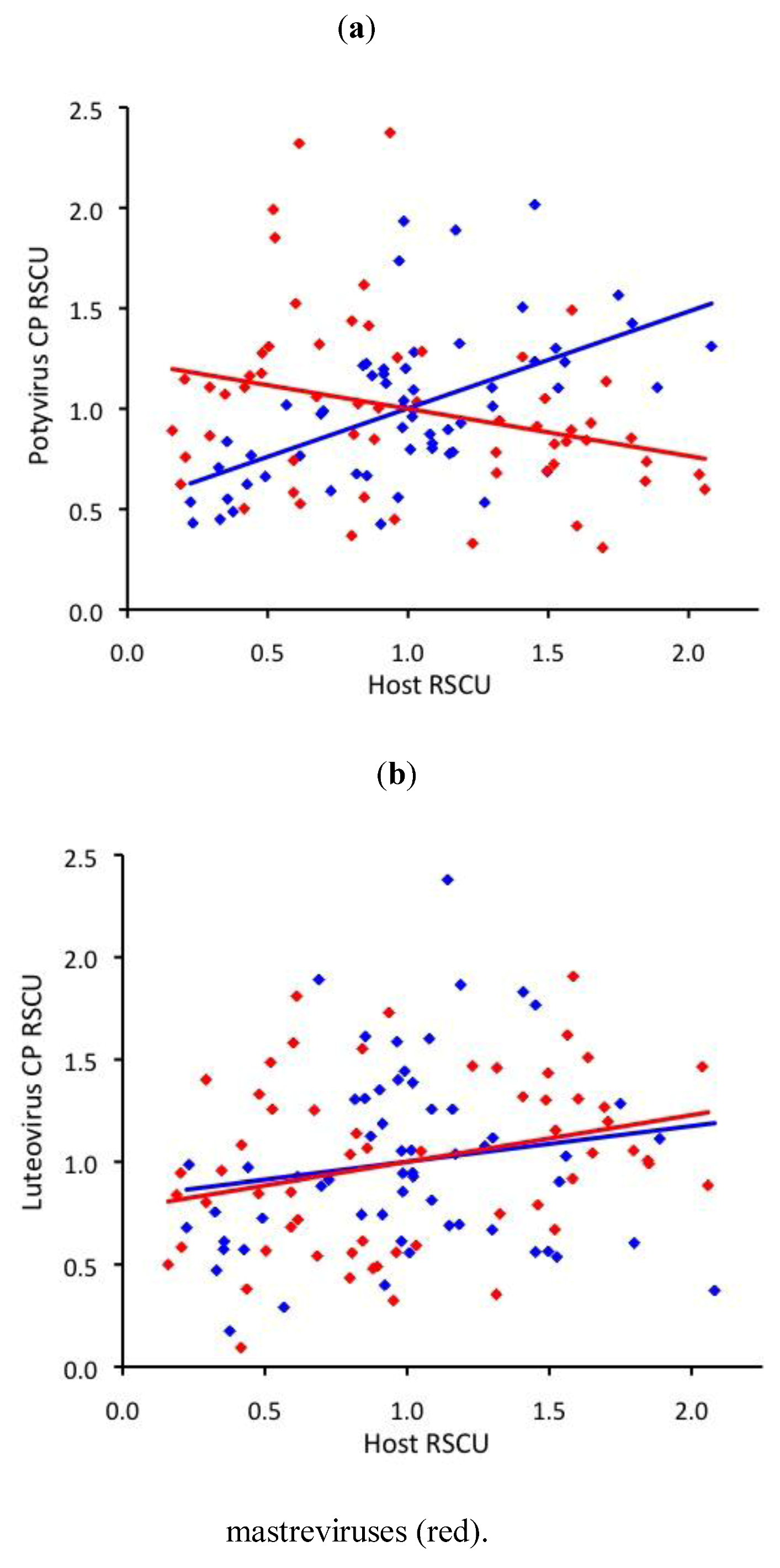
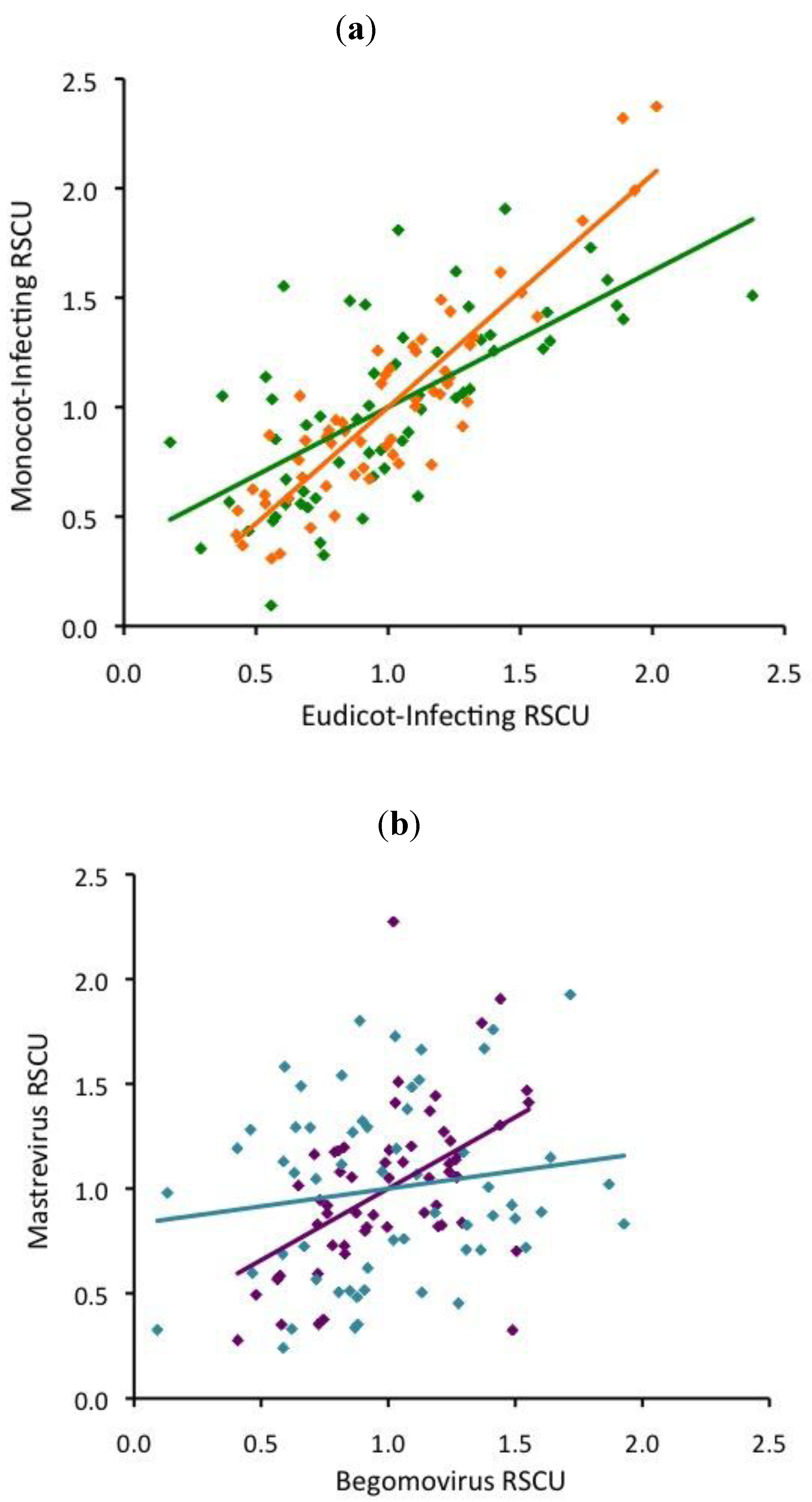
2.3. RNA virus CUB is independent of host use
| Potyviruses | Luteoviruses | ||||||||||||||
| Host | monocot | eudicot | monocot | eudicot | |||||||||||
| NNA | tca | cca | gca | tca | cca | gca | aga | ||||||||
| aga | aaa | aca | aga | gaa | gga | ||||||||||
| caa | |||||||||||||||
| NNC | tgc | cac | tgc | ttc | gac | tgc | ttc | gtc | |||||||
| tac | atc | ctc | |||||||||||||
| NNT | tat | aat | gat | tat | aat | gat | |||||||||
| ttt | ctt | gtt | ttt | ||||||||||||
| NNG | ttg | agg | |||||||||||||
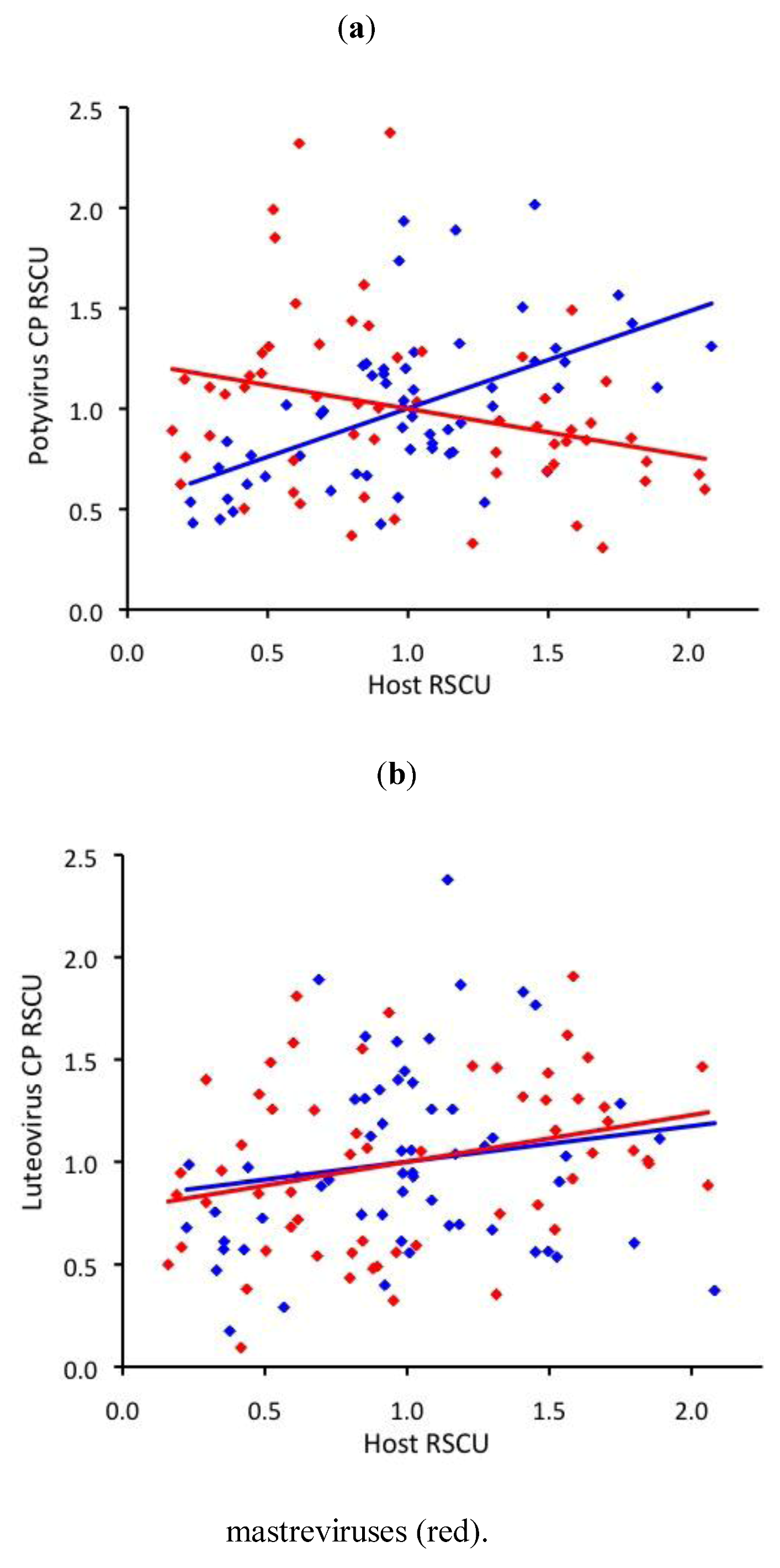
2.4. ssDNA virus RSCU does not indicate strong translational selection
| Mastreviruses | Begomoviruses | ||||||||||||||
| ORF | Rep | CP | Rep | CP | |||||||||||
| NNA | aaa | aaa | caa | gga | |||||||||||
| aca | cca | gaa | |||||||||||||
| aga | |||||||||||||||
| NNC | tac | ttc | gcc | gac | ttc | ctc | tgc | ttc | ccc | ||||||
| NNT | cat | cgt | agt | cat | aat | gat | cat | cgt | aat | ||||||
| gat | act | tgt | |||||||||||||
| att | ggt | gtt | |||||||||||||
| tat | |||||||||||||||
| NNG | ttg | agg | ttg | agg | gag | ttg | ttg | agg | gag | ||||||
| aag | cag | ctg | aag | ||||||||||||
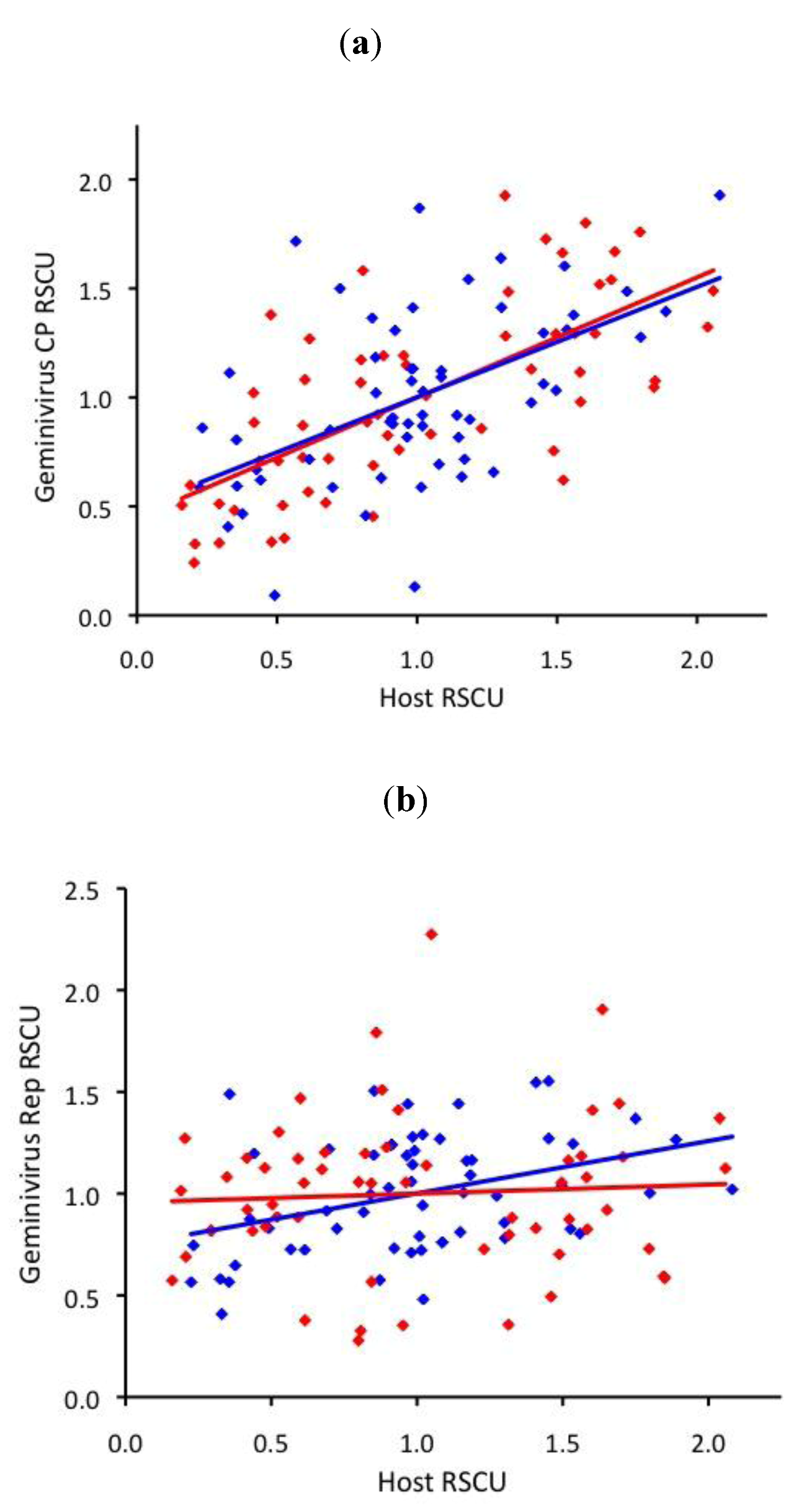
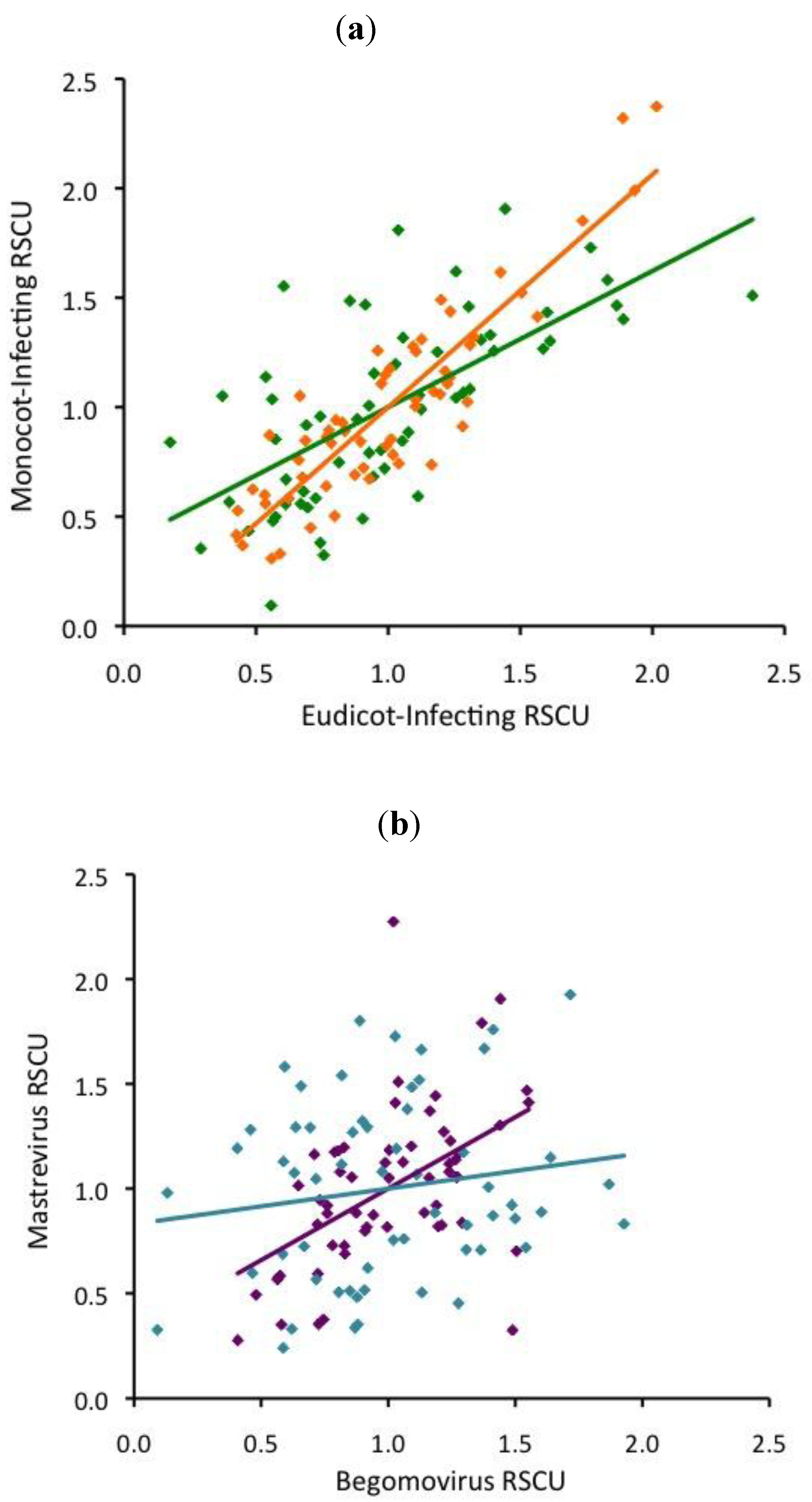
3. Discussion
3.1. Neither base composition nor translational selection explains our results
3.2. Possible alternative explanation for CUB in plant viruses
4. Methods
4.1. Host codon usage bias
4.2. Plant virus datasets
4.3. Base composition as a null hypothesis
4.4. Plant virus codon usage biases
4.5. Comparison with host CUB
5. Conclusions
Acknowledgments
Conflict of Interest
Supplementary Files
References
- Shields, D.C.; Sharp, P.M.; Higgins, D.G.; Wright, F. "Silent" sites in drosophila genes are not neutral: Evidence of selection among synonymous codons. Mol. Biol. Evol. 1988, 5, 704–716. [Google Scholar]
- Aota, S.; Ikemura, T. Diversity in g+c content at the third position of codons in vertebrate genes and its cause. Nucleic Acids Res. 1986, 14, 6345. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Hall, B.D. Codon selection in yeast. J. Biol. Chem. 1982, 257, 3026–3031. [Google Scholar]
- Sharp, P.M.; Li, W.-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Karlin, S.; Mrázek, J.; Campbell, a.M. Codon usages in different gene classes of the escherichia coli genome. Mol. Microbiol. 1998, 29, 1341–1355. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef]
- Hiraoka, Y.; Kawamata, K.; Haraguchi, T.; Chikashige, Y. Codon usage bias is correlated with gene expression levels in the fission yeast schizosaccharomyces pombe. Genes Cells 2009, 14, 499–509. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Danchin, A.; Iqbal, M.; Marsili, M.; Vergassola, M. Codon usage domains over bacterial chromosomes. PLoS Comput. Biol. 2006, 2, e37. [Google Scholar] [CrossRef]
- Lobry, J.R.; Sueoka, N. Asymmetric directional mutation pressures in bacteria. Genome Biol. 2002, 3. [Google Scholar]
- Camiolo, S.; Farina, L.; Porceddu, A. The relation of codon bias to tissue-specific gene expression in arabidopsis thaliana. Genetics 2012, 192, 641–649. [Google Scholar] [CrossRef]
- Ikemura, T. Correlation between the abundance of escherichia coli transfer rnas and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the e. Coli translational system. J. Mol. Biol. 1981, 151, 389–409. [Google Scholar] [CrossRef]
- Ikemura, T. Correlation between the abundance of yeast transfer rnas and the occurrence of the respective codons in protein genes: Differences in synonymous codon choice patterns of yeast and escherichia coli with reference to the abundance of isoaccepting transfer rnas. J. Mol. Biol. 1982, 158, 573–597. [Google Scholar] [CrossRef]
- Curran, J.F.; Yarus, M. Rates of aminoacyl-trna selection at 29 sense codons in vivo. J. Mol. Biol. 1989, 209, 65–77. [Google Scholar]
- Pedersen, S. Escherichia coli ribosomes translate in vivo with variable rate. The EMBO Journal 1984, 3, 2895–2898. [Google Scholar]
- Cai, M.-S.; Cheng, A.-C.; Wang, M.-S.; Zhao, L.-C.; Zhu, D.-K.; Luo, Q.-H.; Liu, F.; Chen, X.-Y. Characterization of synonymous codon usage bias in the duck plague virus ul35 gene. Intervirology 2009, 52, 266–278. [Google Scholar] [CrossRef]
- Jia, R.; Cheng, A.; Wang, M.; Xin, H.; Guo, Y.; Zhu, D.; Qi, X.; Zhao, L.; Ge, H.; Chen, X. Analysis of synonymous codon usage in the ul24 gene of duck enteritis virus. Virus Genes 2008, 38, 96–103. [Google Scholar]
- Au, K.G.; Welsh, K.; Modrich, P. Initiation of methyl-directed mismatch repair. J. Biol. Chem. 1992, 267, 12142–12148. [Google Scholar]
- Krieg, A.M.; Yi, A.-K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. Cpg motifs in bacterial DNA trigger direct b-cell activation. Nature 1995, 374, 546–549. [Google Scholar]
- Bull, J.J.; Molineux, I.J.; Wilke, C.O. Slow fitness recovery in a codon-modified viral genome. Mol. Biol. Evol. 2012, 29, 2997–3004. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Cardinale, D.J.; Duffy, S. Single-stranded genomic architecture constrains optimal codon usage. Bacteriophage 2011, 1, 219–224. [Google Scholar]
- Wang, L.; Roossinck, M.J. Comparative analysis of expressed sequences reveals a conserved pattern of optimal codon usage in plants. Plant Mol. Biol. 2006, 61, 699–710. [Google Scholar] [CrossRef]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus taxonomy: Classification and nomenclature of viruses: Ninth report of the international committee on the taxonomy of viruses; Elsevier Academic press: San Diego, CA, USA, 2011. [Google Scholar]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains of archaea, bacteria, and eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar]
- Rojas, M.R.; Zerbini, F.M.; Allison, R.F.; Robert, L.; Robert, L.; Gilbertson; Lucas, W.J. Capsid protein and helper component-proteinase function as potyvirus cell-to-cell movement proteins. Virology 1997, 237, 283–295. [Google Scholar] [CrossRef]
- Chare, E.R. Selection pressures in the capsid genes of plant RNA viruses reflect mode of transmission. J. Gen. Virol. 2004, 85, 3149–3157. [Google Scholar] [CrossRef]
- Krupovic, M.; Ravantti, J.J.; Bamford, D.H. Geminiviruses: A tale of a plasmid becoming a virus. BMC Evol. Biol. 2009, 9, 112. [Google Scholar] [CrossRef]
- Martin, D.P.; Biagini, P.; Lefeuvre, P.; Golden, M.; Roumagnac, P.; Varsani, A. Recombination in eukaryotic single stranded DNA viruses. Viruses 2011, 3, 1699–1738. [Google Scholar]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef]
- Varsani, A.; Shepherd, D.N.; Dent, K.; Monjane, A.L.; Rybicki, E.P.; Martin, D.P. A highly divergent south african geminivirus species illuminates the ancient evolutionary history of this family. Virol. J. 2009, 6, 36. [Google Scholar] [CrossRef]
- Gutierrez, C. Geminivirus DNA replication. Cell. Mol. Life Sci. 1999, 56, 313–329. [Google Scholar] [CrossRef]
- Fuglsang, A. Estimating the "effective number of codons": The Wright way of determining codon homozygosity leads to superior estimates. Genetics 2005, 172, 1301–1307. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F. Codon usage bias amongst plant viruses. Arch. Virol. 2004, 149, 113–135. [Google Scholar]
- Jiang, Y.; Deng, F.; Wang, H.; Hu, Z. An extensive analysis on the global codon usage pattern of baculoviruses. Arch. Virol. 2008, 153, 2273–2282. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Chen, A.Y.H. Codon usage bias and recombination events for neuraminidase and hemagglutinin genes in chinese isolates of influenza a virus subtype h9n2. Arch. Virol. 2010, 155, 685–693. [Google Scholar] [CrossRef]
- Fu, M. Codon usage bias in herpesvirus. Arch. Virol. 2010, 155, 391–396. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Zhou, J.-H.; Chen, H.-T.; Ma, L.-N.; Ding, Y.-Z.; Liu, W.-Q.; Liu, Y.-S. Analysis of codon usage in bovine viral diarrhea virus. Arch. Virol. 2010, 156, 153–160. [Google Scholar]
- Wang, M.; Liu, Y.-S.; Zhou, J.-H.; Chen, H.-T.; Ma, L.-N.; Ding, Y.-Z.; Liu, W.-Q.; Gu, Y.-X.; Zhang, J. Analysis of codon usage in newcastle disease virus. Virus Genes 2011, 42, 245–253. [Google Scholar]
- Xu, X.-Z.; Liu, Q.-P.; Fan, L.-J.; Cui, X.-F.; Zhou, X.-P. Analysis of synonymous codon usage and evolution of begomoviruses. Journal of Zhejiang University SCIENCE B 2008, 9, 667–674. [Google Scholar] [CrossRef]
- Gray, S.M.; Banerjee, N. Mechanisms of arthropod transmission of plant and animal viruses. Microbiol. Mol. Biol. Rev. 1999, 63, 128–148. [Google Scholar]
- Power, A.G. Insect transmission of plant viruses: A constraint on virus variability. Current Opinions in Plant Biology 2000, 3. [Google Scholar]
- Andret-Link, P.; Fuchs, M. Transmission specificity of plant viruses by vectors. Journal of Plant Pathology 2005, 87, 153–165. [Google Scholar]
- Sanjuan, R.; Agudelo-Romero, P.; Elena, S.F. Upper-limit mutation rate estimation for a plant RNA virus. Biol. Lett. 2009, 5, 394–396. [Google Scholar]
- Gibbs, A.J.; Ohshima, K.; Phillips, M.J.; Gibbs, M.J. The prehistory of potyviruses: Their initial radiation was during the dawn of agriculture. PLoS ONE 2008, 3, e2523. [Google Scholar]
- Pagan, I.; Holmes, E.C. Long-term evolution of the luteoviridae: Time scale and mode of virus speciation. J. Virol. 2010, 84, 6177–6187. [Google Scholar] [CrossRef]
- Duffy, S.; Holmes, E.C. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J. Virol. 2008, 82, 957–965. [Google Scholar]
- Duffy, S.; Holmes, E.C. Validation of high rates of nucleotide substitution in geminiviruses: Phylogenetic evidence from east african cassava mosaic viruses. J. Gen. Virol. 2009, 90, 1539–1547. [Google Scholar]
- Harkins, G.W.; Martin, D.P.; Duffy, S.; Monjane, A.L.; Shepherd, D.N.; Windram, O.P.; Owor, B.E.; Donaldson, L.; van Antwerpen, T.; Sayed, R.A.; et al. Dating the origins of the maize-adapted strain of maize streak virus, msv-a. J. Gen. Virol. 2009, 90, 3066–3074. [Google Scholar] [CrossRef]
- Lefeuvre, P.; Harkins, G.W.; Lett, J.-M.; Briddon, R.W.; Chase, M.W.; Moury, B.; Martin, D.P. Evolutionary time-scale of the begomoviruses: Evidence from integrated sequences in the nicotiana genome. PLoS ONE 2011, 6, e19193. [Google Scholar]
- Hofacker, I.L.; Stadler, P.F.; Stocsits, R.R. Conserved rna secondary structures in viral genomes: A survey. Bioinformatics 2004, 20, 1495–1499. [Google Scholar]
- Hofacker, I.L.; Fekete, M.; Flamm, C.; Huynen, M.A.; Rauscher, S.; Stolorz, P.E.; Stadler, P.F. Automatic detection of conserved rna structure elements in complete rna virus genomes. Nucleic Acids Res. 1998, 26, 3825–3836. [Google Scholar]
- Grantham, R.; Gautier, C.; Guoy, M.; Mercier, R.; Pave, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, r49–r62. [Google Scholar]
- Frederico, L.A.; Kunkel, T.A.; Shaw, B.R. A sensitive genetic assay for the detection of cytosine deamination: Determination of rate constants and the activation energy. Biochemistry (Mosc.) 1990, 29, 2532–2537. [Google Scholar] [CrossRef]
- Liu, X.-S.; Zhang, Y.-G.; Fang, Y.-Z.; Wang, Y.-L. Patterns and influencing factor of synonymous codon usage in porcine circovirus. Virol. J. 2012, 9, 68. [Google Scholar] [CrossRef]
- Shen, C.K.; Ikoku, A.; Hearst, J.E. A specific DNA orientation in the filamentous bacteriophage fd as probed by psoralen crosslinking and electron microscopy. J. Mol. Biol. 1979, 127, 163–175. [Google Scholar] [CrossRef]
- Incardona, N.L.; Prescott, B.; Sargent, D.; Lamba, O.P.; Thomas, G.J. Phage phi x174 probed by laser raman spectroscopy: Evidence for capsid-imposed constraint on DNA secondary structure. Biochemistry (Mosc.) 1987, 26, 1532–1538. [Google Scholar] [CrossRef]
- Benevides, J.M.; Stow, P.L.; Ilag, L.L.; Incardona, N.L.; Thomas, G.J. Differences in secondary structure between packaged and unpackaged single-stranded DNA of bacteriophage phi x174 determined by raman spectroscopy: A model for phi x174 DNA packaging. Biochemistry (Mosc.) 1991, 30, 4855–4863. [Google Scholar] [CrossRef]
- Welsh, L.C.; Marvin, D.A.; Perham, R.N. Analysis of x-ray diffraction from fibres of pf1 inovirus (filamentous bacteriophage) shows that the DNA in the virion is not highly ordered. J. Mol. Biol. 1998, 284, 1265–1271. [Google Scholar] [CrossRef]
- Wen, Z.Q.; Armstrong, A.; Thomas, G.J., Jr. Demonstration by ultraviolet resonance raman spectroscopy of differences in DNA organization and interactions in filamentous viruses pf1 and fd. Biochemistry (Mosc.) 1999, 38, 3148–3156. [Google Scholar]
- Tsuboi, M.; Tsunoda, M.; Overman, S.A.; Benevides, J.M.; Thomas, G.J. A structural model for the single-stranded DNA genome of filamentous bacteriophage pf1. Biochemistry (Mosc.) 2010, 49, 1737–1743. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, A.Y.; Cheng, F.; Guan, W.; Johnson, F.B.; Qiu, J. Molecular characterization of infectious clones of the minute virus of canines reveals unique features of bocaviruses. J. Virol. 2009, 83, 3956–3967. [Google Scholar]
- Mol, C.D.; Parikh, S.S.; Putname, C.D.; Lo, T.P.; Tainer, J.A. DNA repair machanisms for the recognition and removal of damaged DNA bases. Annual Review of Biophysics and Biomolecular Structures 1999, 28, 101–128. [Google Scholar] [CrossRef]
- McClelland, M. Selection against dam methylation sites in the genomes of DNA of enterobacteriophages. J. Mol. Evol. 1985, 21, 317–322. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Davidson, N.O.; Cho, S.-J.; Malim, M.H. Cytidine deamination of retroviral DNA by diverse apobec proteins. Curr. Biol. 2004, 14, 1392–1396. [Google Scholar] [CrossRef]
- Hadfield, J.; Thomas, J.E.; Schwinghamer, M.W.; Kraberger, S.; Stainton, D.; Dayaram, A.; Parry, J.N.; Pande, D.; Martin, D.P.; Varsani, A. Molecular characterisation of dicot-infecting mastreviruses from Australia. Virus Res. 2012, 166, 13–22. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. Caical: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cardinale, D.J.; DeRosa, K.; Duffy, S. Base Composition and Translational Selection are Insufficient to Explain Codon Usage Bias in Plant Viruses. Viruses 2013, 5, 162-181. https://doi.org/10.3390/v5010162
Cardinale DJ, DeRosa K, Duffy S. Base Composition and Translational Selection are Insufficient to Explain Codon Usage Bias in Plant Viruses. Viruses. 2013; 5(1):162-181. https://doi.org/10.3390/v5010162
Chicago/Turabian StyleCardinale, Daniel J., Kate DeRosa, and Siobain Duffy. 2013. "Base Composition and Translational Selection are Insufficient to Explain Codon Usage Bias in Plant Viruses" Viruses 5, no. 1: 162-181. https://doi.org/10.3390/v5010162
APA StyleCardinale, D. J., DeRosa, K., & Duffy, S. (2013). Base Composition and Translational Selection are Insufficient to Explain Codon Usage Bias in Plant Viruses. Viruses, 5(1), 162-181. https://doi.org/10.3390/v5010162