Mouse Models for Filovirus Infections


Abstract
:1. Introduction
2. Pathogenesis of Filovirus Disease in Humans and NHPs
3. Filovirus Infection of Mice
3.1. Wild-Type Filovirus Infections of Wild-Type Mice
3.2. Adaptation of Filoviruses to Virulence for Mice through Sequential Passage
3.2.1. EBOV
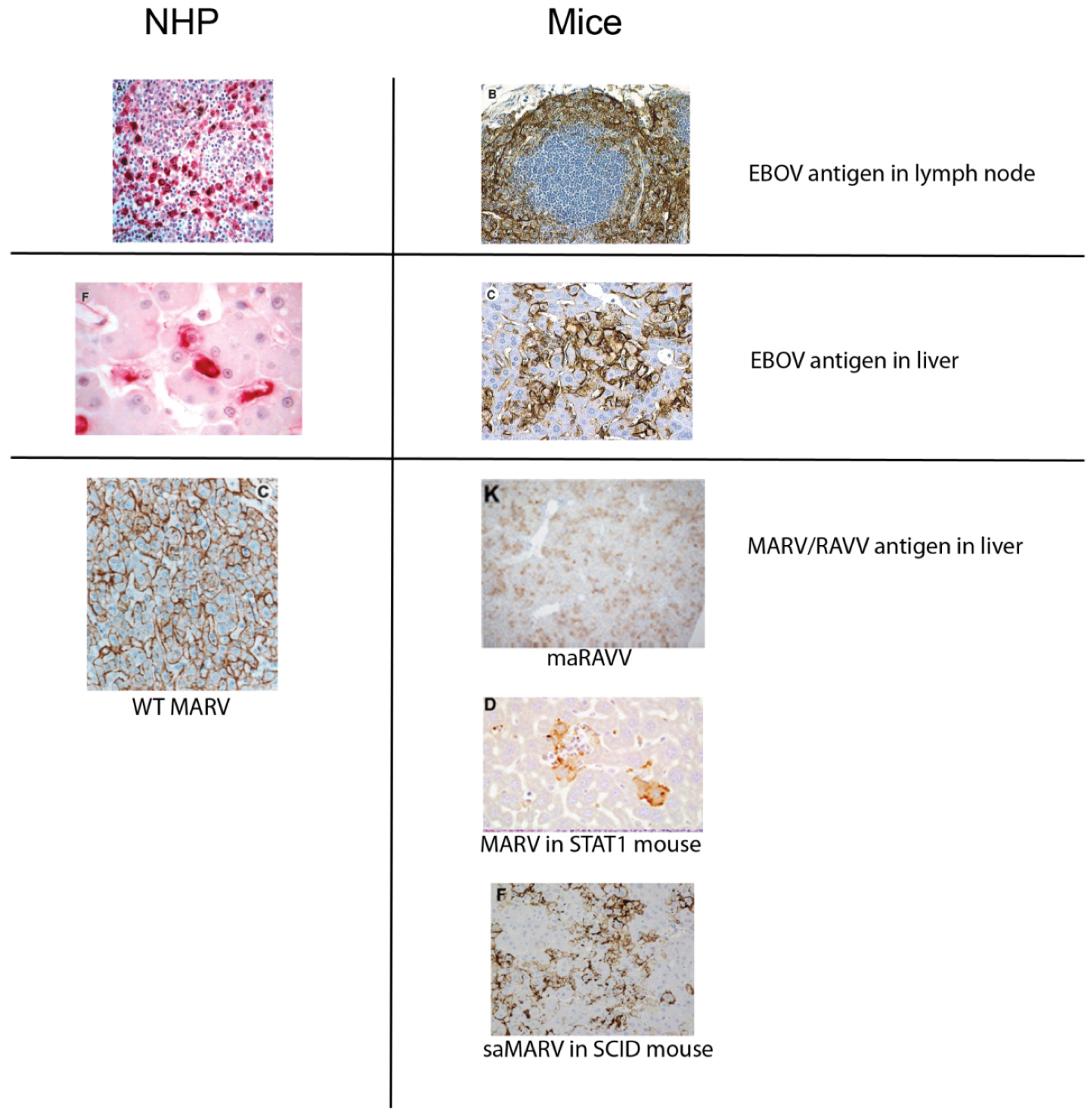
3.2.2. Marburgviruses
3.3. Wild-Type Filovirus Infection of Immunodeficient Mice
3.3.2. Mice with Defective Adaptive Immune Responses
4. Studies of Host Responses to Filovirus Infection in Mice
4.1. Type I IFN Responses
4.2. Determinants of Filovirus Virulence
4.3. Basis of Inherent Resistance to Infection
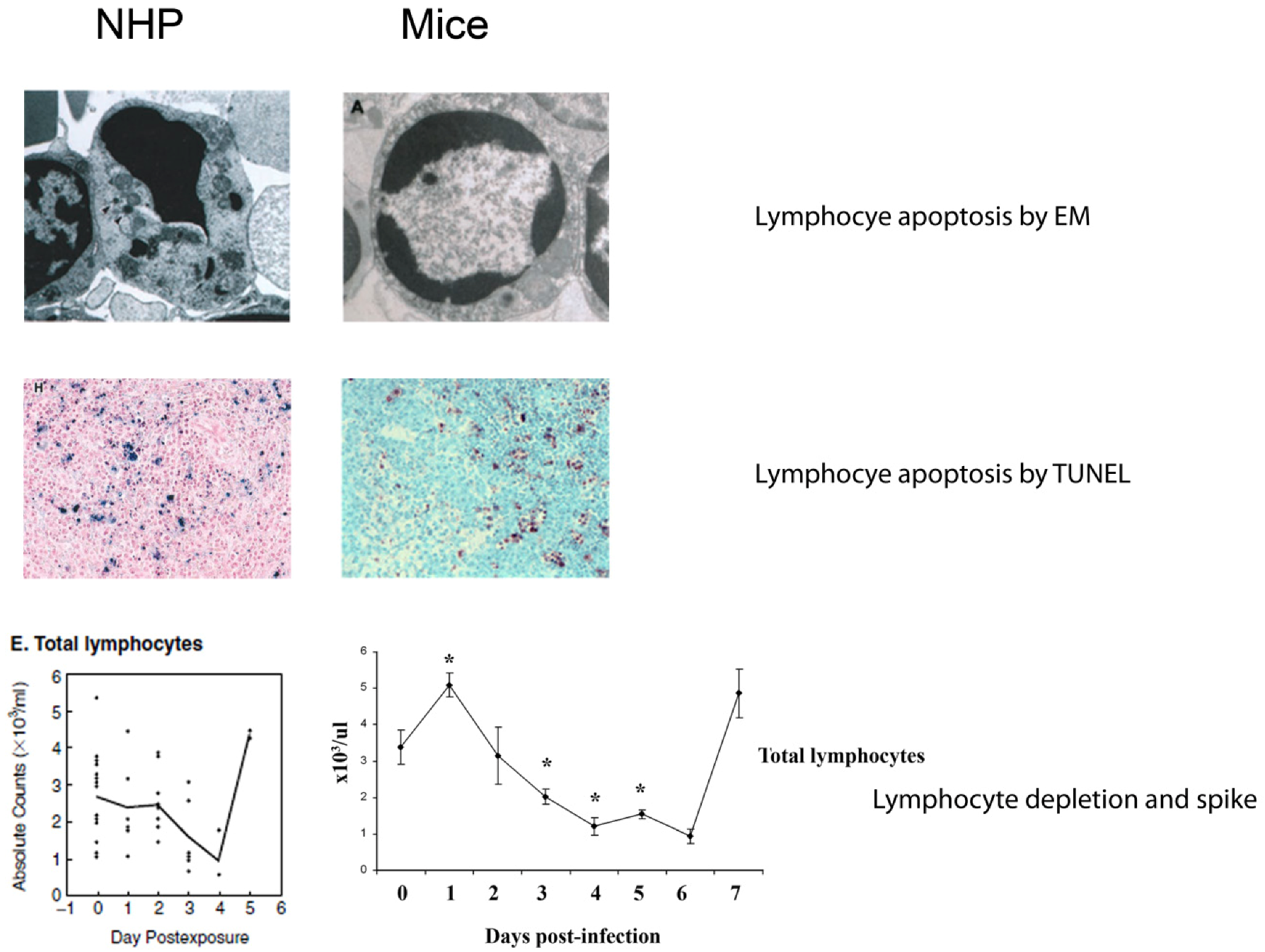
4.4. Lymphocyte Apoptosis
4.6. The Role of CD45 in Resistance to Infection
5. Vaccine Testing in Mouse Models
5.1. Vaccines

{kind=link}
{kind=link}
| Vaccine | Mouse | Guinea Pig | NHP | Human | References |
|---|---|---|---|---|---|
| DNA | Yes | Yes | Partial | Immunogenic | [76,77,78] |
| VRP | Yes | Yes | Yes | NT | [76,79,80,81] |
| Adenovirus | Yes | Yes | Yes | Immunogenic | [74,82,83,84,85,86,87] |
| DNA/Adenovirus prime-boost | NT | Yes | Yes | NT | [77] |
| Virus-like particle | Yes | Yes | Yes | NT | [57,58,69,70,71,73,88,89] |
| Parainfluenza | NT | Yes | Yes | NT | [90,91,92] |
| VSV | Yes | Yes | Yes | NT | [93,94,95,96,97] |
| Vaccinia | NT | Yes | No | NT | [80,98] |
| Purified protein | Yes | Yes | Partial with DNA boost | NT | [75,76,79,99] |
| Inactivated virus | Mixed results (partial to complete protection) | Mixed results (partial to complete protection) | Mixed results (no to partial protection) | NT | [80,100,101] |
5.2. Mechanisms of Vaccine-Induced Immunity
5.3. Predictive Accuracy of Vaccine Testing in Mice
6. Antiviral Drug Testing
| Compound/Drug | Mouse | Guinea Pig | NHP | References |
|---|---|---|---|---|
| S-adenosylhomocysteine hydrolase inhibitors | Yes | NT | No | [116,117] |
| rIFN-αlpha2b | NT | NT | Delay to death | [43] |
| Convalescent blood | NT | NT | No | [118,119] |
| Monoclonal Antibodies | Yes 1 | Yes | Partial 2 | [56,120,121] |
| Polyclonal antibody/sera | Yes 1 | Yes | Yes 3 | [43,122,123,124,125,126,127] |
| PMO Antisense | Yes | Yes | Yes | [60,128,129] |
| siRNA | Yes | Yes | Yes | [130,131] |
| rNAPc2 | NT | NT | 3/9 | [31,34] |
| rhAPC | NT | NT | 2/11 | [35] |
| FGI-103, 104, 106 | Yes | NT | NT | [54,132,133] |
6.1. Passive Transfer of Antibodies or Immune Sera
6.2. Small-Molecule Drugs
6.3. siRNA and Antisense DNA
6.4. Interferon and Interferon Inducers
7. Strengths and Weaknesses of Murine Models
7.1. Compartmentalization
7.2. The Need for Adaptation of Filoviruses for Lethality in Mice
8. Goals for Future Research
8.1. Humanized Mice
8.2. Characterization of Intermediate-Passage Variants
9. Conclusion
Conflict of Interest
References and Notes
- Kuhn, J.H. Filoviruses. A compendium of 40 years of epidemiological, clinical, and laboratory studies. Arch. Virol. Suppl. 2008, 20, 13–360. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Johnson, K.M.; Kawaoka, Y.; Lipkin, W.I.; Negredo, A.I.; Netesov, S.V.; Nichol, S.T.; et al. Proposal for a revised taxonomy of the family Filoviridae: Classification, names of taxa and viruses, and virus abbreviations. Arch. Virol. 2010, 155, 2083–2103. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Becker, S.; Ebihara, H.; Geisbert, T.W.; Jahrling, P.B.; Kawaoka, Y.; Netesov, S.V.; Nichol, S.T.; Peters, C.J.; Volchkov, V.E.; Ksiazek, T.G. Family Filoviridae. In Virus Taxonomy—Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier/Academic Press: San Diego, CA, USA, 2011; pp. 665–671. [Google Scholar]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Dodd, L.E.; Wahl-Jensen, V.; Radoshitzky, S.R.; Bavari, S.; Jahrling, P.B. Evaluation of perceived threat differences posed by filovirus variants. Biosecur. Bioterror. 2011, 9, 361–371. [Google Scholar] [CrossRef]
- Bowen, E.T.; Platt, G.S.; Simpson, D.I.; McArdell, L.B.; Raymond, R.T. Ebola haemorrhagic fever: Experimental infection of monkeys. Trans. R. Soc. Trop. Med. Hyg. 1978, 72, 188–191. [Google Scholar] [CrossRef]
- Hofmann, H.; Kunz, C. A strain of “Marburg virus” (Rhabdovirus simiae) pathogenic to mice. Arch. Gesamte Virusforsch. 1970, 32, 244–248. [Google Scholar] [CrossRef]
- Pattyn, S.; van der Groen, G.; Courteille, G.; Jacob, W.; Piot, P. Isolation of Marburg-like virus from a case of haemorrhagic fever in Zaire. Lancet 1977, 1, 573–574. [Google Scholar]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Kagan, E.; Hensley, L.E. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: Overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003, 188, 1618–1629. [Google Scholar] [CrossRef]
- van der Groen, G.; Jacob, W.; Pattyn, S.R. Ebola virus virulence for newborn mice. J. Med. Virol. 1979, 4, 239–240. [Google Scholar] [CrossRef]
- Bowen, E.T.; Lloyd, G.; Harris, W.J.; Platt, G.S.; Baskerville, A.; Vella, E.E. Viral haemorrhagic fever in southern Sudan and northern Zaire. Preliminary studies on the aetiological agent. Lancet 1977, 1, 571–573. [Google Scholar]
- Moe, J.B.; Lambert, R.D.; Lupton, H.W. Plaque assay for Ebola virus. J. Clin. Microbiol. 1981, 13, 791–793. [Google Scholar]
- Bray, M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001, 82, 1365–1373. [Google Scholar]
- Warfield, K.L.; Bradfute, S.B.; Wells, J.; Lofts, L.; Cooper, M.T.; Alves, D.A.; Reed, D.K.; VanTongeren, S.A.; Mech, C.A.; Bavari, S. Development and characterization of a mouse model for Marburg hemorrhagic fever. J. Virol. 2009, 83, 6404–6415. [Google Scholar] [CrossRef]
- Bray, M.; Davis, K.; Geisbert, T.; Schmaljohn, C.; Huggins, J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 1998, 178, 651–661. [Google Scholar]
- Ebihara, H.; Takada, A.; Kobasa, D.; Jones, S.; Neumann, G.; Theriault, S.; Bray, M.; Feldmann, H.; Kawaoka, Y. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2006, 2, e73. [Google Scholar] [CrossRef]
- Ramanan, P.; Shabman, R.S.; Brown, C.S.; Amarasinghe, G.K.; Basler, C.F.; Leung, D.W. Filoviral immune evasion mechanisms. Viruses 2011, 3, 1634–1649. [Google Scholar] [CrossRef]
- Gibb, T.R.; Bray, M.; Geisbert, T.W.; Steele, K.E.; Kell, W.M.; Davis, K.J.; Jaax, N.K. Pathogenesis of experimental Ebola Zaire virus infection in BALB/c mice. J. Comp. Pathol. 2001, 125, 233–242. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Warfield, K.L.; Bavari, S. Functional CD8+ T cell responses in lethal Ebola virus infection. J. Immunol. 2008, 180, 4058–4066. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Gibb, T.R.; Steele, K.E.; Jaax, N.K.; Jahrling, P.B. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 2000, 80, 171–186. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.; Kagan, E.; Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: Evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003, 163, 2347–2370. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Braun, D.R.; Shamblin, J.D.; Geisbert, J.B.; Paragas, J.; Garrison, A.; Hensley, L.E.; Geisbert, T.W. Lymphocyte death in a mouse model of Ebola virus infection. J. Infect. Dis. 2007, 196, S296–S304. [Google Scholar] [CrossRef]
- Ebola haemorrhagic fever in Zaire, 1976. Bull. World Health Organ. 1978, 56, 271–293.
- Ebola haemorrhagic fever in Sudan, 1976. Report of a WHO/International Study Team. Bull. World Health Organ. 1978, 56, 247–270.
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.C.; Capron, M.; Lansoud-Soukate, J.; Debre, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar]
- Baize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.C.; Capron, M.; Bedjabaga, I.; Lansoud-Soukate, J.; Mavoungou, E. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002, 128, 163–168. [Google Scholar] [CrossRef]
- Villinger, F.; Rollin, P.E.; Brar, S.S.; Chikkala, N.F.; Winter, J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R.; Ansari, A.A.; Peters, C.J. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis. 1999, 179, S188–S191. [Google Scholar]
- Wauquier, N.; Becquart, P.; Padilla, C.; Baize, S.; Leroy, E.M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis. 2010, 4, pii:e837. [Google Scholar]
- Bradfute, S.B.; Bavari, S. Correlates of immunity to filovirus infection. Viruses 2011, 3, 982–1000. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Greiner, D.L.; Shultz, L.D.; Bavari, S. Humanized mice as models for ebolavirus infection. Unpublished work, USAMRIID, Fort Detrick, MD, USA. manuscript in preparation, 2012.
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Young, H.A.; Formenty, P.; Fritz, E.A.; Larsen, T.; Hensley, L.E. Marburg virus Angola infection of rhesus macaques: Pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J. Infect. Dis. 2007, 196, S372–S381. [Google Scholar] [CrossRef]
- Raymond, J.; Bradfute, S.; Bray, M. Filovirus infection of STAT-1 knockout mice. J. Infect. Dis. 2011, 204, S986–S990. [Google Scholar] [CrossRef]
- Warfield, K.L.; Alves, D.A.; Bradfute, S.B.; Reed, D.K.; VanTongeren, S.; Kalina, W.V.; Olinger, G.G.; Bavari, S. Development of a model for marburgvirus based on severe-combined immunodeficiency mice. Virol. J. 2007, 4, 108. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Jahrling, P.B.; Larsen, T.; Geisbert, J.B.; Paragas, J.; Young, H.A.; Fredeking, T.M.; Rote, W.E.; Vlasuk, G.P. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: A study in rhesus monkeys. Lancet 2003, 362, 1953–1958. [Google Scholar]
- Hensley, L.E.; Stevens, E.L.; Yan, S.B.; Geisbert, J.B.; Macias, W.L.; Larsen, T.; Daddario-DiCaprio, K.M.; Cassell, G.H.; Jahrling, P.B.; Geisbert, T.W. Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever. J. Infect. Dis. 2007, 196, S390–S399. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Swanson, P.E.; Smith, M.A.; Watanabe, E.; McDunn, J.E.; Hotchkiss, R.S.; Bavari, S. Mechanisms and consequences of ebolavirus-induced lymphocyte apoptosis. J. Immunol. 2010, 184, 327–335. [Google Scholar] [CrossRef]
- Reed, D.S.; Hensley, L.E.; Geisbert, J.B.; Jahrling, P.B.; Geisbert, T.W. Depletion of peripheral blood T lymphocytes and NK cells during the course of ebola hemorrhagic fever in cynomolgus macaques. Viral Immunol. 2004, 17, 390–400. [Google Scholar] [CrossRef]
- Lofts, L.L.; Wells, J.B.; Bavari, S.; Warfield, K.L. Key genomic changes necessary for an in vivo lethal mouse marburgvirus variant selection process. J. Virol. 2011, 85, 3905–3917. [Google Scholar] [CrossRef]
- Lever, M.S.; Piercy, T.J.; Steward, J.A.; Eastaugh, L.; Smither, S.J.; Taylor, C.; Salguero, F.J.; Phillpotts, R.J. Lethality and pathogenesis of airborne infection with filoviruses in A129 alpha/beta -/- interferon receptor-deficient mice. J. Med. Microbiol. 2012, 61, 8–15. [Google Scholar] [CrossRef]
- de Wit, E.; Munster, V.J.; Metwally, S.A.; Feldmann, H. Assessment of rodents as animal models for Reston ebolavirus. J. Infect. Dis. 2011, 204, S968–S972. [Google Scholar] [CrossRef]
- Bray, M.; Raymond, J.L.; Geisbert, T.; Baker, R.O. 3-deazaneplanocin A induces massively increased interferon-alpha production in Ebola virus-infected mice. Antivir. Res. 2002, 55, 151–159. [Google Scholar]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Canc. Therapeut. 2009, 8, 1579–1588. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Geisbert, T.W.; Geisbert, J.B.; Swearengen, J.R.; Bray, M.; Jaax, N.K.; Huggins, J.W.; LeDuc, J.W.; Peters, C.J. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 1999, 179, S224–S234. [Google Scholar]
- Mahanty, S.; Gupta, M.; Paragas, J.; Bray, M.; Ahmed, R.; Rollin, P.E. Protection from lethal infection is determined by innate immune responses in a mouse model of Ebola virus infection. Virology 2003, 312, 415–424. [Google Scholar] [CrossRef]
- Gupta, M.; Mahanty, S.; Greer, P.; Towner, J.S.; Shieh, W.J.; Zaki, S.R.; Ahmed, R.; Rollin, P.E. Persistent infection with ebola virus under conditions of partial immunity. J. Virol. 2004, 78, 958–967. [Google Scholar] [CrossRef]
- Kalina, W.V.; Warfield, K.L.; Olinger, G.G.; Bavari, S. Discovery of common marburgvirus protective epitopes in a BALB/c mouse model. Virol. J. 2009, 6, 132. [Google Scholar] [CrossRef]
- Sanchez, A.; Wagoner, K.E.; Rollin, P.E. Sequence-based human leukocyte antigen-B typing of patients infected with Ebola virus in Uganda in 2000: Identification of alleles associated with fatal and nonfatal disease outcomes. J. Infect. Dis. 2007, 196, S329–S336. [Google Scholar] [CrossRef]
- Parrino, J.; Hotchkiss, R.; Bray, M. Prevention of immune cell apoptosis as a potential therapeutic strategy for severe infections. Emerg. Infect. Dis. 2007, 13, 191–198. [Google Scholar] [CrossRef]
- Hensley, L.E.; Young, H.A.; Jahrling, P.B.; Geisbert, T.W. Proinflammatory response during Ebola virus infection of primate models: Possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 2002, 80, 169–179. [Google Scholar] [CrossRef]
- Gupta, M.; Spiropoulou, C.; Rollin, P.E. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology 2007, 364, 45–54. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Young, H.A.; Jahrling, P.B.; Davis, K.J.; Larsen, T.; Kagan, E.; Hensley, L.E. Pathogenesis of Ebola hemorrhagic fever in primate models: Evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 2003, 163, 2371–2382. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Mavoungou, E.; Fisher-Hoch, S.P. Apoptosis in fatal Ebola infection. Does the virus toll the bell for immune system? Apoptosis 2000, 5, 5–7. [Google Scholar] [CrossRef]
- Sanchez, A.; Lukwiya, M.; Bausch, D.; Mahanty, S.; Sanchez, A.J.; Wagoner, K.D.; Rollin, P.E. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: Cellular responses, virus load, and nitric oxide levels. J. Virol. 2004, 78, 10370–10377. [Google Scholar] [CrossRef]
- Aman, M.J.; Kinch, M.S.; Warfield, K.; Warren, T.; Yunus, A.; Enterlein, S.; Stavale, E.; Wang, P.; Chang, S.; Tang, Q.; et al. Development of a broad-spectrum antiviral with activity against Ebola virus. Antivir. Res. 2009, 83, 245–251. [Google Scholar]
- Warfield, K.L.; Perkins, J.G.; Swenson, D.L.; Deal, E.M.; Bosio, C.M.; Aman, M.J.; Yokoyama, W.M.; Young, H.A.; Bavari, S. Role of natural killer cells in innate protection against lethal ebola virus infection. J. Exp. Med. 2004, 200, 169–179. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hart, M.K. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef]
- Warfield, K.L.; Olinger, G.; Deal, E.M.; Swenson, D.L.; Bailey, M.; Negley, D.L.; Hart, M.K.; Bavari, S. Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J. Immunol. 2005, 175, 1184–1191. [Google Scholar]
- Warfield, K.L.; Posten, N.A.; Swenson, D.L.; Olinger, G.G.; Esposito, D.; Gillette, W.K.; Hopkins, R.F.; Costantino, J.; Panchal, R.G.; Hartley, J.L.; et al. Filovirus-like particles produced in insect cells: Immunogenicity and protection in rodents. J. Infect. Dis. 2007, 196, S421–S429. [Google Scholar] [CrossRef]
- Warfield, K.L.; Swenson, D.L.; Olinger, G.G.; Kalina, W.V.; Viard, M.; Aitichou, M.; Chi, X.; Ibrahim, S.; Blumenthal, R.; Raviv, Y.; et al. Ebola virus inactivation with preservation of antigenic and structural integrity by a photoinducible alkylating agent. J. Infect. Dis. 2007, 196, S276–S283. [Google Scholar] [CrossRef]
- Warfield, K.L.; Swenson, D.L.; Olinger, G.G.; Nichols, D.K.; Pratt, W.D.; Blouch, R.; Stein, D.A.; Aman, M.J.; Iversen, P.L.; Bavari, S. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2006, 2, e1. [Google Scholar] [CrossRef]
- Gupta, M.; Greer, P.; Mahanty, S.; Shieh, W.J.; Zaki, S.R.; Ahmed, R.; Rollin, P.E. CD8-mediated protection against Ebola virus infection is perforin dependent. J. Immunol. 2005, 174, 4198–4202. [Google Scholar]
- Panchal, R.G.; Ulrich, R.L.; Bradfute, S.B.; Lane, D.; Ruthel, G.; Kenny, T.A.; Iversen, P.L.; Anderson, A.O.; Gussio, R.; Raschke, W.C.; et al. Reduced expression of CD45 protein tyrosine phosphatase provides protection against anthrax pathogenesis. J. Biol. Chem. 2009, 284, 12874–12885. [Google Scholar]
- Panchal, R.G.; Bradfute, S.B.; Peyser, B.D.; Warfield, K.L.; Ruthel, G.; Lane, D.; Kenny, T.A.; Anderson, A.O.; Raschke, W.C.; Bavari, S. Reduced levels of protein tyrosine phosphatase CD45 protect mice from the lethal effects of Ebola virus infection. Cell Host Microbe 2009, 6, 162–173. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Dye, J.M., Jr.; Bavari, S. Filovirus vaccines. Hum. Vaccin. 2011, 7, 701–711. [Google Scholar] [CrossRef]
- Vanderzanden, L.; Bray, M.; Fuller, D.; Roberts, T.; Custer, D.; Spik, K.; Jahrling, P.; Huggins, J.; Schmaljohn, A.; Schmaljohn, C. DNA vaccines expressing either the GP or NP genes of Ebola virus protect mice from lethal challenge. Virology 1998, 246, 134–144. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Vanderzanden, L.; Tammariello, R.F.; Geisbert, J.; Schmaljohn, C.; Smith, J.F.; Jahrling, P.B.; Schmaljohn, A. Marburg virus vaccines, comparing classical and new approaches. Vaccine 2002, 20, 586–593. [Google Scholar]
- Riemenschneider, J.; Garrison, A.; Geisbert, J.; Jahrling, P.; Hevey, M.; Negley, D.; Schmaljohn, A.; Lee, J.; Hart, M.K.; Vanderzanden, L.; et al. Comparison of individual and combination DNA vaccines for B. anthracis, Ebola virus, Marburg virus and Venezuelan equine encephalitis virus. Vaccine 2003, 21, 4071–4080. [Google Scholar] [CrossRef]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid raft microdomains: A gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar] [CrossRef]
- Swenson, D.L.; Warfield, K.L.; Kuehl, K.; Larsen, T.; Hevey, M.C.; Schmaljohn, A.; Bavari, S.; Aman, M.J. Generation of Marburg virus-like particles by co-expression of glycoprotein and matrix protein. FEMS Immunol. Med. Microbiol. 2004, 40, 27–31. [Google Scholar] [CrossRef]
- Warfield, K.L.; Bosio, C.M.; Welcher, B.C.; Deal, E.M.; Mohamadzadeh, M.; Schmaljohn, A.; Aman, M.J.; Bavari, S. Ebola virus-like particles protect from lethal Ebola virus infection. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15889–15894. [Google Scholar]
- Swenson, D.L.; Warfield, K.L.; Negley, D.L.; Schmaljohn, A.; Aman, M.J.; Bavari, S. Virus-like particles exhibit potential as a pan-filovirus vaccine for both Ebola and Marburg viral infections. Vaccine 2005, 23, 3033–3042. [Google Scholar] [CrossRef]
- Warfield, K.L.; Olinger, G.G.; Deal, E.M.; Swenson, D.L.; Bailey, M.; Negley, D.L.; Hart, M.K.; Bavari, S. Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J. Immunol. 2005, 175, 1184–1191. [Google Scholar]
- Warfield, K.L.; Swenson, D.L.; Olinger, G.G.; Kalina, W.V.; Aman, M.J.; Bavari, S. Ebola virus-like particle-based vaccine protects nonhuman primates against lethal Ebola virus challenge. J. Infect. Dis. 2007, 196, S430–S437. [Google Scholar] [CrossRef]
- Swenson, D.L.; Wang, D.; Luo, M.; Warfield, K.L.; Woraratanadharm, J.; Holman, D.H.; Dong, J.Y.; Pratt, W.D. Vaccine to confer to nonhuman primates complete protection against multistrain Ebola and Marburg virus infections. Clin. Vaccine Immunol. 2008, 15, 460–467. [Google Scholar] [CrossRef]
- Konduru, K.; Bradfute, S.B.; Jacques, J.; Manangeeswaran, M.; Nakamura, S.; Morshed, S.; Wood, S.C.; Bavari, S.; Kaplan, G.G. Ebola virus glycoprotein Fc fusion protein confers protection against lethal challenge in vaccinated mice. Vaccine 2011, 29, 2968–2977. [Google Scholar]
- Hevey, M.; Negley, D.; VanderZanden, L.; Tammariello, R.F.; Geisbert, J.; Schmaljohn, C.; Smith, J.F.; Jahrling, P.B.; Schmaljohn, A.L. Marburg virus vaccines: Comparing classical and new approaches. Vaccine 2001, 20, 586–593. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sanchez, A.; Rollin, P.E.; Yang, Z.Y.; Nabel, G.J. Development of a preventive vaccine for Ebola virus infection in primates. Nature 2000, 408, 605–609. [Google Scholar]
- Martin, J.E.; Sullivan, N.J.; Enama, M.E.; Gordon, I.J.; Roederer, M.; Koup, R.A.; Bailer, R.T.; Chakrabarti, B.K.; Bailey, M.A.; Gomez, P.L.; et al. A DNA vaccine for Ebola virus is safe and immunogenic in a phase I clinical trial. Clin. Vaccine Immunol. 2006, 13, 1267–1277. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Pushko, P.; Smith, J.; Schmaljohn, A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 1998, 251, 28–37. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Pushko, P.; Anderson, K.; Smith, J.; Davis, K.J.; Jahrling, P.B. Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg. Infect. Dis. 2002, 8, 503–507. [Google Scholar] [CrossRef]
- Olinger, G.G.; Hart, M.K. Filoviruses: Recent advances and future challenges. In Protective Immunity to Ebola Infection by Venezuelan Equine Encephalitis Virus Replicons Expressing Ebola Virus Proteins; An ICID Global Symposium: Winnipeg, Manitoba, Canada, 2006. [Google Scholar]
- Croyle, M.A.; Patel, A.; Tran, K.N.; Gray, M.; Zhang, Y.; Strong, J.E.; Feldmann, H.; Kobinger, G.P. Nasal delivery of an adenovirus-based vaccine bypasses pre-existing immunity to the vaccine carrier and improves the immune response in mice. PLoS One 2008, 3, e3548. [Google Scholar]
- Kobinger, G.P.; Feldmann, H.; Zhi, Y.; Schumer, G.; Gao, G.; Feldmann, F.; Jones, S.; Wilson, J.M. Chimpanzee adenovirus vaccine protects against Zaire Ebola virus. Virology 2006, 346, 394–401. [Google Scholar] [CrossRef]
- Patel, A.; Zhang, Y.; Croyle, M.; Tran, K.; Gray, M.; Strong, J.; Feldmann, H.; Wilson, J.M.; Kobinger, G.P. Mucosal delivery of adenovirus-based vaccine protects against Ebola virus infection in mice. J. Infect. Dis. 2007, 196, S413–S420. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Geisbert, T.W.; Geisbert, J.B.; Shedlock, D.J.; Xu, L.; Lamoreaux, L.; Custers, J.H.; Popernack, P.M.; Yang, Z.Y.; Pau, M.G.; et al. Immune protection of nonhuman primates against Ebola virus with single low-dose adenovirus vectors encoding modified GPs. PLoS Med. 2006, 3, e177. [Google Scholar] [CrossRef]
- Wang, D.; Hevey, M.; Juompan, L.Y.; Trubey, C.M.; Raja, N.U.; Deitz, S.B.; Woraratanadharm, J.; Luo, M.; Yu, H.; Swain, B.M.; et al. Complex adenovirus-vectored vaccine protects guinea pigs from three strains of Marburg virus challenges. Virology 2006, 353, 324–332. [Google Scholar] [CrossRef]
- Wang, D.; Schmaljohn, A.L.; Raja, N.U.; Trubey, C.M.; Juompan, L.Y.; Luo, M.; Deitz, S.B.; Yu, H.; Woraratanadharm, J.; Holman, D.H.; et al. De novo syntheses of Marburg virus antigens from adenovirus vectors induce potent humoral and cellular immune responses. Vaccine 2006, 24, 2975–2986. [Google Scholar] [CrossRef]
- Warfield, K.L.; Swenson, D.L.; Negley, D.L.; Schmaljohn, A.L.; Aman, M.J.; Bavari, S. Marburg virus-like particles protect guinea pigs from lethal Marburg virus infection. Vaccine 2004, 22, 3495–3502. [Google Scholar] [CrossRef]
- Sun, Y.; Carrion, R., Jr.; Ye, L.; Wen, Z.; Ro, Y.T.; Brasky, K.; Ticer, A.E.; Schwegler, E.E.; Patterson, J.L.; Compans, R.W.; et al. Protection against lethal challenge by Ebola virus-like particles produced in insect cells. Virology 2009, 383, 12–21. [Google Scholar] [CrossRef]
- Bukreyev, A.; Marzi, A.; Feldmann, F.; Zhang, L.; Yang, L.; Ward, J.M.; Dorward, D.W.; Pickles, R.J.; Murphy, B.R.; Feldmann, H.; et al. Chimeric human parainfluenza virus bearing the Ebola virus glycoprotein as the sole surface protein is immunogenic and highly protective against Ebola virus challenge. Virology 2009, 383, 348–361. [Google Scholar] [CrossRef]
- Bukreyev, A.; Rollin, P.E.; Tate, M.K.; Yang, L.; Zaki, S.R.; Shieh, W.J.; Murphy, B.R.; Collins, P.L.; Sanchez, A. Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 2007, 81, 6379–6388. [Google Scholar]
- Bukreyev, A.; Yang, L.; Zaki, S.R.; Shieh, W.J.; Rollin, P.E.; Murphy, B.R.; Collins, P.L.; Sanchez, A. A single intranasal inoculation with a paramyxovirus-vectored vaccine protects guinea pigs against a lethal-dose Ebola virus challenge. J. Virol. 2006, 80, 2267–2279. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-Dicaprio, K.M.; Geisbert, J.B.; Reed, D.S.; Feldmann, F.; Grolla, A.; Stroher, U.; Fritz, E.A.; Hensley, L.E.; Jones, S.M.; et al. Vesicular stomatitis virus-based vaccines protect nonhuman primates against aerosol challenge with Ebola and Marburg viruses. Vaccine 2008, 26, 6894–6900. [Google Scholar] [CrossRef]
- Jones, S.M.; Stroher, U.; Fernando, L.; Qiu, X.; Alimonti, J.; Melito, P.; Bray, M.; Klenk, H.D.; Feldmann, H. Assessment of a vesicular stomatitis virus-based vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J. Infect. Dis. 2007, 196, S404–S412. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Geisbert, J.B.; Stroher, U.; Hensley, L.E.; Grolla, A.; Fritz, E.A.; Feldmann, F.; Feldmann, H.; Jones, S.M. Cross-protection against Marburg virus strains by using a live, attenuated recombinant vaccine. J. Virol. 2006, 80, 9659–9666. [Google Scholar]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Stroher, U.; Geisbert, J.B.; Grolla, A.; Fritz, E.A.; Fernando, L.; Kagan, E.; Jahrling, P.B.; Hensley, L.E.; et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: An efficacy assessment. Lancet 2006, 367, 1399–1404. [Google Scholar]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Williams, K.J.; Geisbert, J.B.; Leung, A.; Feldmann, F.; Hensley, L.E.; Feldmann, H.; Jones, S.M. Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J. Virol. 2008, 82, 5664–5668. [Google Scholar] [CrossRef]
- Hart, M.K. Vaccine research efforts for filoviruses. Int. J. Parasitol. 2003, 33, 583–595. [Google Scholar] [CrossRef]
- Hevey, M.; Negley, D.; Geisbert, J.; Jahrling, P.; Schmaljohn, A. Antigenicity and vaccine potential of Marburg virus glycoprotein expressed by baculovirus recombinants. Virology 1997, 239, 206–216. [Google Scholar] [CrossRef]
- Lupton, H.W.; Lambert, R.D.; Bumgardner, D.L.; Moe, J.B.; Eddy, G.A. Inactivated vaccine for Ebola virus efficacious in guineapig model. Lancet 1980, 2, 1294–1295. [Google Scholar]
- Mikhailov, V.V.; Borisevich, I.V.; Chernikova, N.K.; Potryvaeva, N.V.; Krasnianskii, V.P. The evaluation in hamadryas baboons of the possibility for the specific prevention of Ebola fever. Vopr. Virusol. 1994, 39, 82–84. [Google Scholar]
- Hevey, M.; Negley, D.; Pushko, P.; Smith, J.; Schmaljohn, A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 1998, 251, 28–37. [Google Scholar] [CrossRef]
- Wilson, J.A.; Bray, M.; Bakken, R.; Hart, M.K. Vaccine potential of Ebola virus VP24, VP30, VP35, and VP40 proteins. Virology 2001, 286, 384–390. [Google Scholar] [CrossRef]
- Pushko, P.; Geisbert, J.; Parker, M.; Jahrling, P.; Smith, J. Individual and bivalent vaccines based on alphavirus replicons protect guinea pigs against infection with Lassa and Ebola viruses. J. Virol. 2001, 75, 11677–11685. [Google Scholar] [CrossRef]
- Pushko, P.; Bray, M.; Ludwig, G.V.; Parker, M.; Schmaljohn, A.; Sanchez, A.; Jahrling, P.B.; Smith, J.F. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine 2000, 19, 142–153. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Geisbert, T.W.; Geisbert, J.B.; Xu, L.; Yang, Z.Y.; Roederer, M.; Koup, R.A.; Jahrling, P.B.; Nabel, G.J. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature 2003, 424, 681–684. [Google Scholar] [CrossRef]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.; Jones, S.; Moller, P.; Wagner, R.; Volchkov, V.; Klenk, H.D.; Feldmann, H.; Stroher, U. Properties of replication-competent vesicular stomatitis virus vectors expressing glycoproteins of filoviruses and arenaviruses. J. Virol. 2004, 78, 5458–5465. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hart, M.K. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef]
- Rao, M.; Bray, M.; Alving, C.R.; Jahrling, P.; Matyas, G.R. Induction of immune responses in mice and monkeys to Ebola virus after immunization with liposome-encapsulated irradiated Ebola virus: Protection in mice requires CD4(+) T cells. J. Virol. 2002, 76, 9176–9185. [Google Scholar] [CrossRef]
- Xu, L.; Sanchez, A.; Yang, Z.; Zaki, S.R.; Nabel, E.G.; Nichol, S.T.; Nabel, G.J. Immunization for Ebola virus infection. Nat. Med. 1998, 4, 37–42. [Google Scholar] [CrossRef]
- Vanderzanden, L.; Bray, M.; Fuller, D.; Roberts, T.; Custer, D.; Spik, K.; Jahrling, P.; Huggins, J.; Schmaljohn, A.; Schmaljohn, C. DNA vaccines expressing either the GP or NP genes of Ebola virus protect mice from lethal challenge. Virology 1998, 246, 134–144. [Google Scholar] [CrossRef]
- Simmons, G.; Lee, A.; Rennekamp, A.J.; Fan, X.; Bates, P.; Shen, H. Identification of murine T-cell epitopes in Ebola virus nucleoprotein. Virology 2004, 318, 224–230. [Google Scholar] [CrossRef]
- Warfield, K.L.; Olinger, G.G. Protective role of cytotoxic T lymphocytes in filovirus hemorrhagic fever. J. Biomed. Biotechnol. 2011, 2011, 984241. [Google Scholar]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.C.; Capron, M.; Lansoud-Soukate, J.; Debre, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef]
- Chupurnov, A.A.; Chernukhin, I.V.; Ternovoi, V.A.; Kudoiarova, N.M.; Makhova, N.M.; Azaev, M.; Smolina, M.P. Attempts to develop a vaccine against Ebola fever. Vopr. Virusol. 1995, 40, 257–260. [Google Scholar]
- Huggins, J.; Zhang, Z.X.; Bray, M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J. Infect. Dis. 1999, 179, S240–S247. [Google Scholar] [CrossRef]
- Bray, M.; Driscoll, J.; Huggins, J.W. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antivir. Res. 2000, 45, 135–147. [Google Scholar]
- Jahrling, P.B.; Geisbert, J.B.; Swearengen, J.R.; Larsen, T.; Geisbert, T.W. Ebola hemorrhagic fever: Evaluation of passive immunotherapy in nonhuman primates. J. Infect. Dis. 2007, 196, S400–S403. [Google Scholar] [CrossRef]
- Mupapa, K.; Massamba, M.; Kibadi, K.; Kuvula, K.; Bwaka, A.; Kipasa, M.; Colebunders, R.; Muyembe-Tamfum, J.J. Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. International Scientific and Technical Committee. J. Infect. Dis. 1999, 179, S18–S23. [Google Scholar]
- Shahhosseini, S.; Das, D.; Qiu, X.; Feldmann, H.; Jones, S.M.; Suresh, M.R. Production and characterization of monoclonal antibodies against different epitopes of Ebola virus antigens. J. Virol. Methods 2007, 143, 29–37. [Google Scholar] [CrossRef]
- Takada, A.; Ebihara, H.; Jones, S.; Feldmann, H.; Kawaoka, Y. Protective efficacy of neutralizing antibodies against Ebola virus infection. Vaccine 2007, 25, 993–999. [Google Scholar] [CrossRef]
- Bray, M.; Davis, K.; Geisbert, T.; Schmaljohn, C.; Huggins, J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 1999, 179, S248–S258. [Google Scholar]
- Gupta, M.; Mahanty, S.; Bray, M.; Ahmed, R.; Rollin, P.E. Passive transfer of antibodies protects immunocompetent and imunodeficient mice against lethal Ebola virus infection without complete inhibition of viral replication. J. Virol. 2001, 75, 4649–4654. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Geisbert, J.; Swearengen, J.R.; Jaax, G.P.; Lewis, T.; Huggins, J.W.; Schmidt, J.J.; LeDuc, J.W.; Peters, C.J. Passive immunization of Ebola virus-infected cynomolgus monkeys with immunoglobulin from hyperimmune horses. Arch. Virol. Suppl. 1996, 11, 135–140. [Google Scholar]
- Dye, J.M.; Herbert, A.S.; Kuehne, A.I.; Barth, J.F.; Muhammad, M.A.; Zak, S.E.; Ortiz, R.A.; Prugar, L.I.; Pratt, W.D. Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5034–5039. [Google Scholar]
- Oswald, W.B.; Geisbert, T.W.; Davis, K.J.; Geisbert, J.B.; Sullivan, N.J.; Jahrling, P.B.; Parren, P.W.; Burton, D.R. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. 2007, 3, e9. [Google Scholar] [CrossRef]
- Parren, P.W.; Geisbert, T.W.; Maruyama, T.; Jahrling, P.B.; Burton, D.R. Pre- and postexposure prophylaxis of Ebola virus infection in an animal model by passive transfer of a neutralizing human antibody. J. Virol. 2002, 76, 6408–6412. [Google Scholar] [CrossRef]
- Swenson, D.L.; Warfield, K.L.; Warren, T.K.; Lovejoy, C.; Hassinger, J.N.; Ruthel, G.; Blouch, R.E.; Moulton, H.M.; Weller, D.D.; Iversen, P.L.; et al. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemother. 2009, 53, 2089–2099. [Google Scholar]
- Warfield, K.L.; Panchal, R.G.; Aman, M.J.; Bavari, S. Antisense treatments for biothreat agents. Curr. Opin. Mol. Ther. 2006, 8, 93–103. [Google Scholar]
- Geisbert, T.W.; Hensley, L.E.; Kagan, E.; Yu, E.Z.; Geisbert, J.B.; Daddario-DiCaprio, K.; Fritz, E.A.; Jahrling, P.B.; McClintock, K.; Phelps, J.R.; et al. Postexposure protection of guinea pigs against a lethal ebola virus challenge is conferred by RNA interference. J. Infect. Dis. 2006, 193, 1650–1657. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Lee, A.C.; Robbins, M.; Geisbert, J.B.; Honko, A.N.; Sood, V.; Johnson, J.C.; de Jong, S.; Tavakoli, I.; Judge, A.; et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: A proof-of-concept study. Lancet 2010, 375, 1896–1905. [Google Scholar]
- Kinch, M.S.; Yunus, A.S.; Lear, C.; Mao, H.; Chen, H.; Fesseha, Z.; Luo, G.; Nelson, E.A.; Li, L.; Huang, Z.; et al. FGI-104: A broad-spectrum small molecule inhibitor of viral infection. Am. J. Transl. Res. 2009, 1, 87–98. [Google Scholar]
- Warren, T.K.; Warfield, K.L.; Wells, J.; Enterlein, S.; Smith, M.; Ruthel, G.; Yunus, A.S.; Kinch, M.S.; Goldblatt, M.; Aman, M.J.; et al. Antiviral activity of a small-molecule inhibitor of filovirus infection. Antimicrob. Agents Chemother. 2010, 54, 2152–2159. [Google Scholar]
- Wilson, J.A.; Hevey, M.; Bakken, R.; Guest, S.; Bray, M.; Schmaljohn, A.L.; Hart, M.K. Epitopes involved in antibody-mediated protection from Ebola virus. Science 2000, 287, 1664–1666. [Google Scholar] [CrossRef]
- Qiu, X.; Alimonti, J.B.; Melito, P.L.; Fernando, L.; Stroher, U.; Jones, S.M. Characterization of Zaire ebolavirus glycoprotein-specific monoclonal antibodies. Clin. Immunol. 2011, 141, 218–227. [Google Scholar] [CrossRef]
- Qiu, X.; Fernando, L.; Melito, P.L.; Audet, J.; Feldmann, H.; Kobinger, G.; Alimonti, J.B.; Jones, S.M. Ebola GP-specific monoclonal antibodies protect mice and guinea pigs from lethal Ebola virus infection. PLoS Negl. Trop. Dis. 2012, 6, e1575. [Google Scholar] [CrossRef]
- Kudoyarova-Zubavichene, N.M.; Sergeyev, N.N.; Chepurnov, A.A.; Netesov, S.V. Preparation and use of hyperimmune serum for prophylaxis and therapy of Ebola virus infections. J. Infect. Dis. 1999, 179, S218–S223. [Google Scholar]
- Hevey, M.; Negley, D.; Schmaljohn, A. Characterization of monoclonal antibodies to Marburg virus (strain Musoke) glycoprotein and identification of two protective epitopes. Virology 2003, 314, 350–357. [Google Scholar] [CrossRef]
- Borisevich, I.V.; Mikhailov, V.V.; Krasnianskii, V.P.; Gradoboev, V.N.; Lebedinskaia, E.V.; Potryvaeva, N.V.; Timan'kova, G.D. Development and study of the properties of immunoglobulin against Ebola fever. Vopr. Virusol. 1995, 40, 270–273. [Google Scholar]
- Takada, A.; Kawaoka, Y. Antibody-dependent enhancement of viral infection: Molecular mechanisms and in vivo implications. Rev. Med. Virol. 2003, 13, 387–398. [Google Scholar] [CrossRef]
- Takada, A.; Feldmann, H.; Ksiazek, T.G.; Kawaoka, Y. Antibody-dependent enhancement of Ebola virus infection. J. Virol. 2003, 77, 7539–7544. [Google Scholar] [CrossRef]
- Marzi, A.; Yoshida, R.; Miyamoto, H.; Ishijima, M.; Suzuki, Y.; Higuchi, M.; Matsuyama, Y.; Igarashi, M.; Nakayama, E.; Kuroda, M.; et al. Protective efficacy of neutralizing monoclonal antibodies in a nonhuman primate model of ebola hemorrhagic Fever. PLoS One 2012, 7, e36192. [Google Scholar]
- Qiu, X.; Audet, J.; Wong, G.; Pillet, S.; Bello, A.; Cabral, T.; Strong, J.E.; Plummer, F.; Corbett, C.R.; Alimonti, J.B.; et al. Successful treatment of ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci. Transl. Med. 2012, 4, 138ra181. [Google Scholar]
- Bray, M.; Driscoll, J.; Huggins, J.W. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antivir. Res. 2000, 45, 135–147. [Google Scholar]
- Barrientos, L.G.; O'Keefe, B.R.; Bray, M.; Sanchez, A.; Gronenborn, A.M.; Boyd, M.R. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antivir. Res. 2003, 58, 47–56. [Google Scholar]
- Aman, M.J.; Kinch, M.S.; Warfield, K.; Warren, T.; Yunus, A.; Enterlein, S.; Stavale, E.; Wang, P.; Chang, S.; Tang, Q.; et al. Development of a broad-spectrum antiviral with activity against Ebola virus. Antivir. Res. 2009, 83, 245–251. [Google Scholar] [CrossRef]
- Spurgers, K.B.; Sharkey, C.M.; Warfield, K.L.; Bavari, S. Oligonucleotide antiviral therapeutics: Antisense and RNA interference for highly pathogenic RNA viruses. Antivir. Res. 2008, 78, 26–36. [Google Scholar]
- Warren, T.K.; Warfield, K.L.; Wells, J.; Swenson, D.L.; Donner, K.S.; van Tongeren, S.A.; Garza, N.L.; Dong, L.; Mourich, D.V.; Crumley, S.; et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 2010, 16, 991–994. [Google Scholar]
- Enterlein, S.; Warfield, K.L.; Swenson, D.L.; Stein, D.A.; Smith, J.L.; Gamble, C.S.; Kroeker, A.D.; Iversen, P.L.; Bavari, S.; Muhlberger, E. VP35 knockdown inhibits Ebola virus amplification and protects against lethal infection in mice. Antimicrob. Agents Chemother. 2006, 50, 984–993. [Google Scholar]
- Riabchikova, E.I.; Kolesnikova, L.V.; Rassadkin Iu, N. Microscopic study of species specific features of hemostatic impairment in Ebola virus infected monkeys. Vestn. Ross. Akad. Med. Nauk. 1998, 3, 51–55. [Google Scholar]
- Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999, 179, S199–S202. [Google Scholar]
- Luchko, S.V.; Dadaeva, A.A.; Ustinova, E.N.; Sizikova, L.P.; Riabchikova, E.I.; Sandakhchiev, L.S. Experimental study of Ebola hemorrhagic fever in baboon models. Biull. Eksp. Biol. Med. 1995, 120, 302–304. [Google Scholar]
- P'Iankov O, V.; Sergeev, A.N.; P'Iankova O, G.; Chepurnov, A.A. Experimental Ebola fever in Macaca mulatta. Vopr. Virusol. 1995, 40, 113–115. [Google Scholar]
- Baskerville, A.; Bowen, E.T.; Platt, G.S.; McArdell, L.B.; Simpson, D.I. The pathology of experimental Ebola virus infection in monkeys. J. Pathol. 1978, 125, 131–138. [Google Scholar] [CrossRef]
- Carrion, R., Jr.; Ro, Y.; Hoosien, K.; Ticer, A.; Brasky, K.; de la Garza, M.; Mansfield, K.; Patterson, J.L. A small nonhuman primate model for filovirus-induced disease. Virology 2011, 420, 117–124. [Google Scholar] [CrossRef]
- Fisher-Hoch, S.P.; Brammer, T.L.; Trappier, S.G.; Hutwagner, L.C.; Farrar, B.B.; Ruo, S.L.; Brown, B.G.; Hermann, L.M.; Perez-Oronoz, G.I.; Goldsmith, C.S.; et al. Pathogenic potential of filoviruses: Role of geographic origin of primate host and virus strain. J. Infect. Dis. 1992, 166, 753–763. [Google Scholar] [CrossRef]
- Reed, D.S.; Lackemeyer, M.G.; Garza, N.L.; Sullivan, L.J.; Nichols, D.K. Aerosol exposure to Zaire ebolavirus in three nonhuman primate species: Differences in disease course and clinical pathology. Microbes Infect. 2011, 13, 930–936. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Geisbert, T.W.; Dalgard, D.W.; Johnson, E.D.; Ksiazek, T.G.; Hall, W.C.; Peters, C.J. Preliminary report: Isolation of Ebola virus from monkeys imported to USA. Lancet 1990, 335, 502–505. [Google Scholar]
- Connolly, B.M.; Steele, K.E.; Davis, K.J.; Geisbert, T.W.; Kell, W.M.; Jaax, N.K.; Jahrling, P.B. Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 1999, 179, S203–S217. [Google Scholar]
- Tsuda, Y.; Safronetz, D.; Brown, K.; LaCasse, R.; Marzi, A.; Ebihara, H.; Feldmann, H. Protective efficacy of a bivalent recombinant vesicular stomatitis virus vaccine in the Syrian hamster model of lethal Ebola virus infection. J. Infect. Dis. 2011, 204, S1090–S1097. [Google Scholar] [CrossRef]
- Olinger, G.G.; Bailey, M.A.; Dye, J.M.; Bakken, R.; Kuehne, A.; Kondig, J.; Wilson, J.; Hogan, R.J.; Hart, M.K. Protective cytotoxic T-cell responses induced by venezuelan equine encephalitis virus replicons expressing Ebola virus proteins. J. Virol. 2005, 79, 14189–14196. [Google Scholar] [CrossRef]
- Reid, S.P.; Valmas, C.; Martinez, O.; Sanchez, F.M.; Basler, C.F. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 2007, 81, 13469–13477. [Google Scholar] [CrossRef]
- Valmas, C.; Basler, C.F. Marburg virus VP40 antagonizes interferon signaling in a species-specific manner. J. Virol. 2011, 85, 4309–4317. [Google Scholar] [CrossRef]
- Martinez, O.; Leung, L.W.; Basler, C.F. The role of antigen-presenting cells in filoviral hemorrhagic fever: Gaps in current knowledge. Antivir. Res. 2012, 93, 416–428. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Humanized SCID mouse models for biomedical research. Curr. Top. Microbiol. Immunol. 2008, 324, 25–51. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bradfute, S.B.; Warfield, K.L.; Bray, M. Mouse Models for Filovirus Infections. Viruses 2012, 4, 1477-1508. https://doi.org/10.3390/v4091477
Bradfute SB, Warfield KL, Bray M. Mouse Models for Filovirus Infections. Viruses. 2012; 4(9):1477-1508. https://doi.org/10.3390/v4091477
Chicago/Turabian StyleBradfute, Steven B., Kelly L. Warfield, and Mike Bray. 2012. "Mouse Models for Filovirus Infections" Viruses 4, no. 9: 1477-1508. https://doi.org/10.3390/v4091477
APA StyleBradfute, S. B., Warfield, K. L., & Bray, M. (2012). Mouse Models for Filovirus Infections. Viruses, 4(9), 1477-1508. https://doi.org/10.3390/v4091477