Influenza Virus-Mediated Membrane Fusion: Determinants of Hemagglutinin Fusogenic Activity and Experimental Approaches for Assessing Virus Fusion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
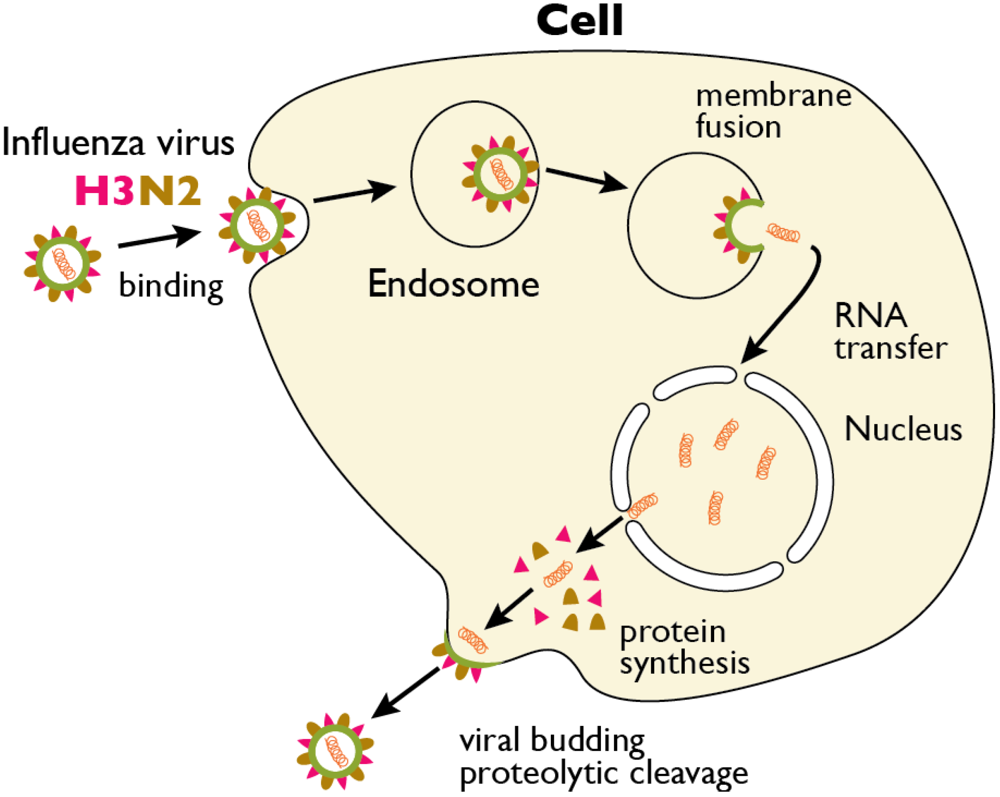
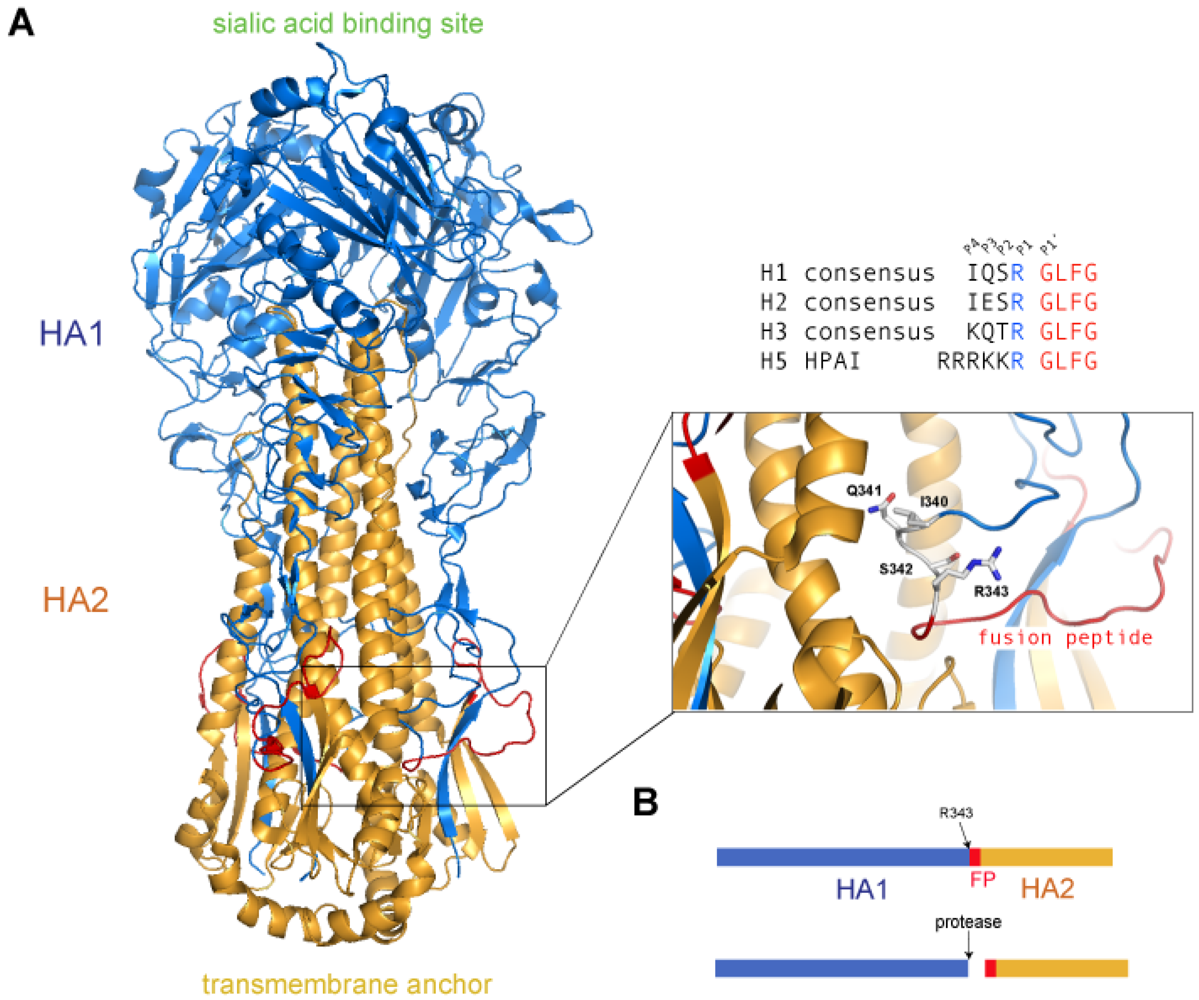
2. Proteolytic Cleavage of HA
2.1. Overview of HA Cleavage by Host Proteases
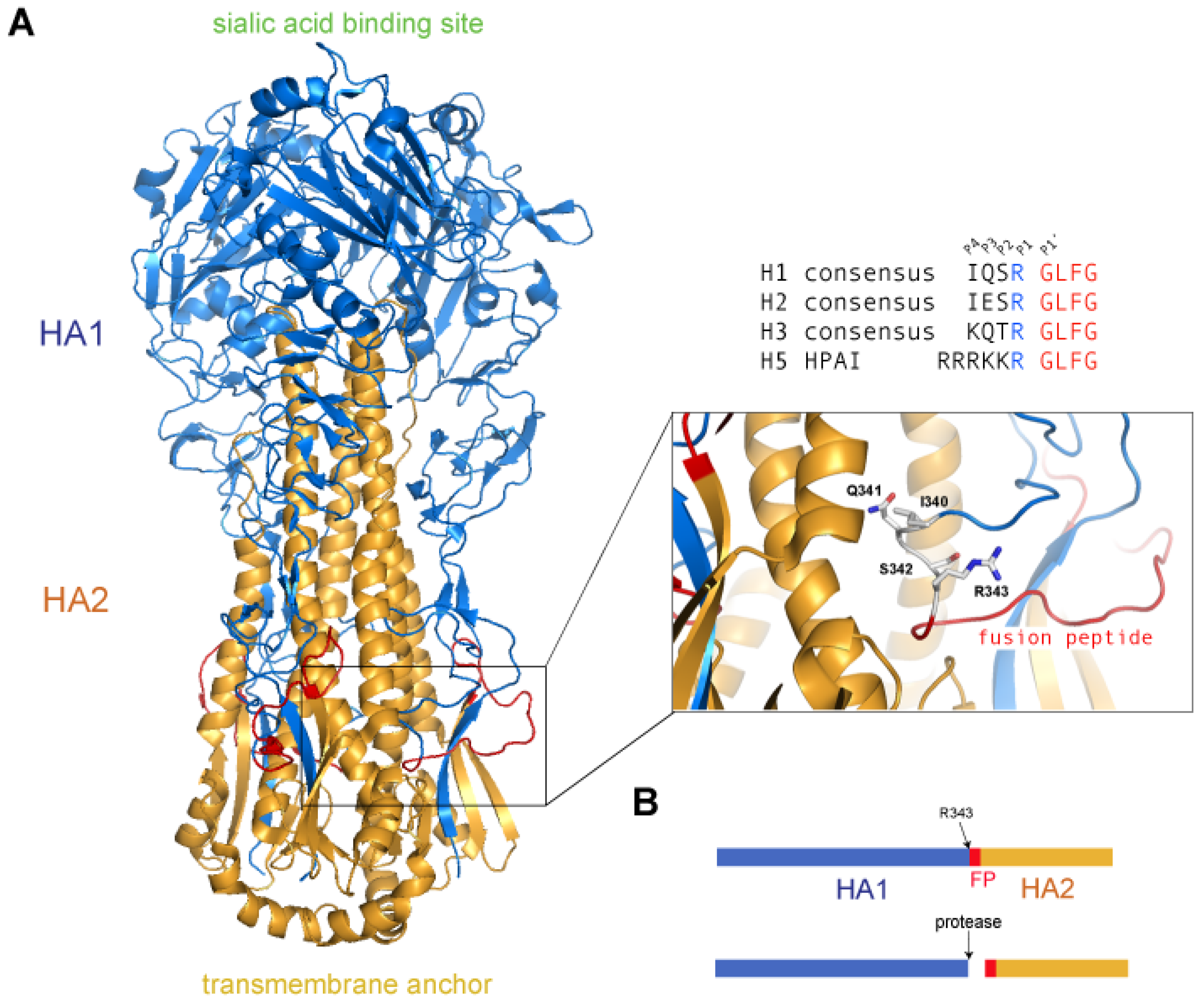
2.1.1. HA Cleavage by Transmembrane Serine Proteases (TTSP’s)
2.1.2. HA Cleavage by Secreted Serine Proteases (TTSP’s)
2.2. Other Factors Contributing to HA Activation and Function
2.3. Molecular Determinants of HA Cleavage
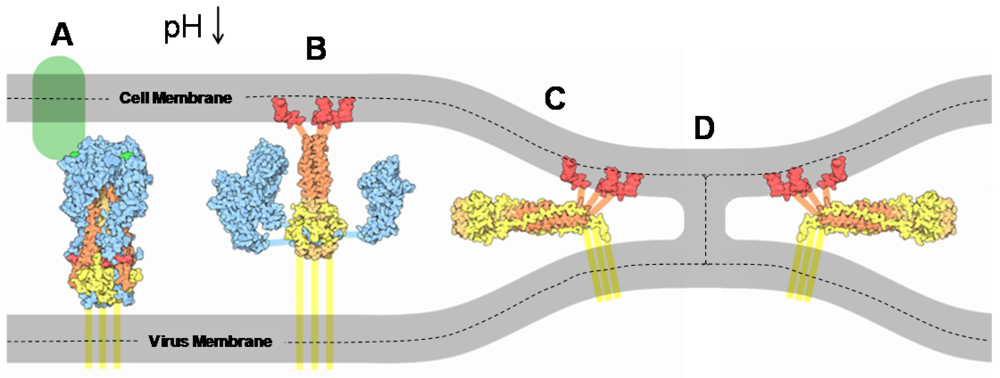
3. Influenza Entry Processes
3.1. Overview of the Roles of HA in Viral Entry
3.2. HA Mediated Fusion Mechanism
3.3. Open Questions in Fusion
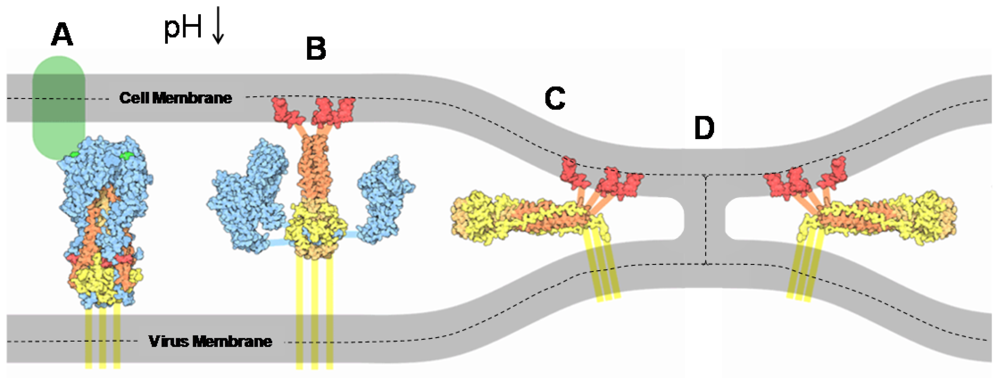
4. Studying HA Fusion Kinetics
4.1. Overview
4.2. In Vitro Ensemble Fusion Assays
4.3. Early Individual Virion Imaging of Virus Fusion to Cell Membrane Mimics
4.4. Stochastic Fusion Assays
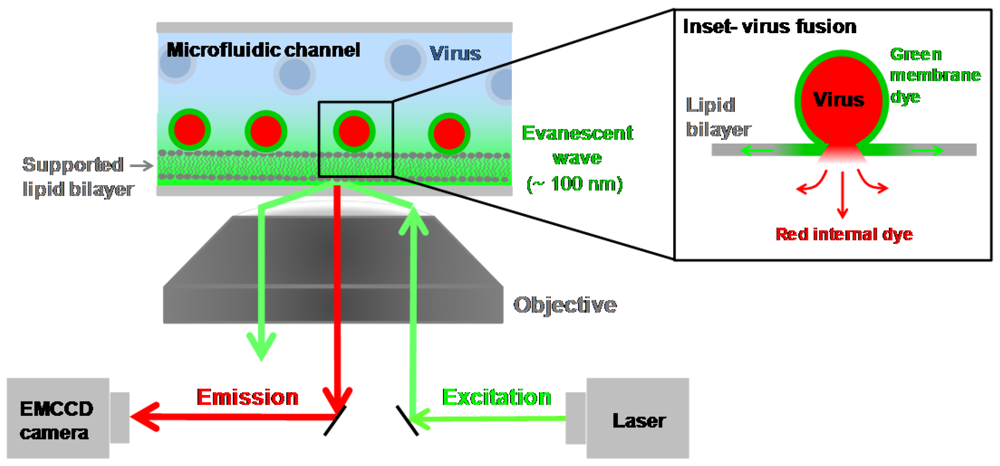
4.5. Single Virion Fusion Using Total Internal Reflection Fluorescence Microscopy

4.6. Data Treatment and Stochastic Analysis
4.7. Limitations of Current IVI Assays and Considerations for Future Improvements
5. Looking Ahead
Acknowledgments
Conflict of Interest
References
- Horimoto, T.; Kawaoka, Y. Influenza: Lessons from past pandemics, warnings from current incidents. Nat. Rev. Microbiol. 2005, 3, 591–600. [Google Scholar]
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The Viruses and Their Replication. In Fields Virology; Knipe, D.M., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Fouchier, R.A.M.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A.D.M.E. Characterization of a novel influenza a virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar]
- Nelson, M.I.; Holmes, E.C. The evolution of epidemic influenza. Nat. Rev. Genet. 2007, 8, 196–205. [Google Scholar]
- Medina, R.A.; Garcia-Sastre, A. Influenza a viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar]
- Fauci, A.S. Emerging and re-emerging infectious diseases: Influenza as a prototype of the host-pathogen balancing act. Cell 2006, 124, 665–670. [Google Scholar]
- Palese, P. Influenza: Old and new threats. Nat. Med. 2004, 10, S82–S87. [Google Scholar]
- Garcia-Sastre, A. Influenza virus receptor specificity: Disease and transmission. Am. J. Pathol. 2010, 176, 1584–1585. [Google Scholar]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammal. J. Virol. 2000, 74, 8502–8512. [Google Scholar]
- Stevens, J.; Blixt, O.; Tumpey, T.M.; Taubenberger, J.K.; Paulson, J.C.; Wilson, I.A. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science 2006, 312, 404–410. [Google Scholar]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Endocytosis of influenza viruses. Microbes Infect. 2004, 6, 929–936. [Google Scholar]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 2006, 124, 997–1009. [Google Scholar]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar]
- Chen, C.; Zhuang, X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc. Natl. Acad. Sci. USA 2008, 105, 11790–11795. [Google Scholar]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza a virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar]
- De Conto, F.; Covan, S.; Arcangeletti, M.C.; Orlandini, G.; Gatti, R.; Dettori, G.; Chezzi, C. Differential infectious entry of human influenza A/WSN/33 virus (H1N1) in mammalian kidney cells. Virus Res. 2011, 155, 221–230. [Google Scholar]
- Sieczkarski, S.B.; Brown, H.A.; Whittaker, G.R. The role of protein kinase C bii in influenza virus entry via late endosomes. J. Virol. 2003, 77, 460–469. [Google Scholar]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza a viruses (IAV) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar]
- Ehrhardt, C.; Marjuki, H.; Wolff, T.; Nurnberg, B.; Planz, O.; Pleschka, S.; Ludwig, S. Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol. 2006, 8, 1336–1348. [Google Scholar]
- Chu, V.C.; Whittaker, G.R. Influenza virus entry and infection require host cell n-linked glycoprotein. Proc. Natl. Acad. Sci. USA 2004, 101, 18153–18158. [Google Scholar]
- Stray, S.; Cummings, R.D.; Air, G.M. Influenza virus infection of desialylated cells. Glycobiology 2000, 10, 649–658. [Google Scholar]
- Sun, X.; Whittaker, G.R. Role of the actin cytoskeleton during influenza virus internalization into polarized epithelial cells. Cell Microbiol. 2007, 9, 1672–1682. [Google Scholar]
- Arcangeletti, M.C.; de Conto, F.; Ferraglia, F.; Pinardi, F.; Gatti, R.; Orlandini, G.; Covan, S.; Motta, F.; Rodighiero, I.; Dettori, G.; et al. Host-cell-dependent role of actin cytoskeleton during the replication of a human strain of influenza a virus. Arch. Virol. 2008, 153, 1209–1221. [Google Scholar]
- Sieczkarski, S.B.; Whittaker, G.R. Differential requirements of Rab5 and Rab7 for endocytosis of influenza and other enveloped viruses. Traffic 2003, 4, 333–343. [Google Scholar]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar]
- Liu, S.L.; Zhang, Z.L.; Tian, Z.Q.; Zhao, H.S.; Liu, H.; Sun, E.Z.; Xiao, G.F.; Zhang, W.; Wang, H.Z.; Pang, D.W. Effectively and efficiently dissecting the infection of influenza virus by quantum-dot-based single-particle tracking. ACS Nano 2012, 6, 141–150. [Google Scholar]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Ann. Rev. Biochem. 1987, 56, 365–394. [Google Scholar]
- Bui, M.; Whittaker, G.; Helenius, A. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 1996, 70, 8391–8401. [Google Scholar]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar]
- Kemler, I.; Whittaker, G.; Helenius, A. Nuclear import of microinjected influenza virus ribonucleoproteins. Virology 1994, 202, 1028–1033. [Google Scholar]
- Babcock, H.P.; Chen, C.; Zhuang, X. Using single-particle tracking to study nuclear trafficking of viral genes. Biophys. J. 2004, 87, 2749–2758. [Google Scholar]
- Boulay, F.; Doms, R.W.; Wilson, I.; Helenius, A. The influenza hemagglutinin precursor as an acid-sensitive probe of the biosynthetic pathway. EMBO J. 1987, 6, 2643–2650. [Google Scholar]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar]
- Garten, W.; Klenk, H.-D. Cleavage Activation of the Influenza Virus Hemagglutinin and Its Role in Pathogenesis. In Avian Influenza; Klenk, H.-D., Matrosovich, M.N., Stech, J., Eds.; Karger: Basel, Switzerland, 2008. [Google Scholar]
- Stevens, J.; Corper, A.L.; Basler, C.F.; Taubenberger, J.K.; Palese, P.; Wilson, I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 2004, 303, 1866–1870. [Google Scholar]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.-D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar]
- Bottcher-Friebertshauser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.-D.; Garten, W. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar]
- Bertram, S.; Glowacka, I.; Blazejewska, P.; Soilleux, E.; Allen, P.; Danisch, S.; Steffen, I.; Choi, S.-Y.; Park, Y.; Schneider, H.; et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J. Virol. 2010, 84, 10016–10025. [Google Scholar]
- Bahgat, M.; Blazejewska, P.; Schughart, K. Inhibition of lung serine proteases in mice: A potentially new approach to control influenza infection. J. Virol. 2011, 8. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Solomon Tsegaye, T.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 2009, 83, 3200–3211. [Google Scholar]
- Okumura, Y.; Takahashi, E.; Yano, M.; Ohuchi, M.; Daidoji, T.; Nakaya, T.; Bottcher, E.; Garten, W.; Klenk, H.-D.; Kido, H. Novel type II transmembrane serine proteases, MSPl and TMPRSS13, proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J. Virol. 2010, 84, 5089–5096. [Google Scholar] [CrossRef]
- Kido, H.; Okumura, Y.; Takahashi, E.; Pan, H.-Y.; Wang, S.; Yao, D.; Yao, M.; Chida, J.; Yano, M. Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failure. Biochim. Biophys. Acta Proteins Prot. 2012, 1824, 186–194. [Google Scholar]
- Kido, H.; Yokogoshi, Y.; Sakai, K.; Tashiro, M.; Kishino, Y.; Fukutomi, A.; Katunuma, N. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial clara cells. A possible activator of the viral fusion glycoprotein. J. Biol. Chem. 1992, 267, 13573–13579. [Google Scholar]
- Scheiblauer, H.; Reinacher, M.; Tashiro, M.; Rott, R. Interactions between bacteria and influenza a virus in the development of influenza pneumonia. J. Infect. Dis. 1992, 166, 783–791. [Google Scholar]
- Tashiro, M.; Ciborowski, P.; Klenk, H.D.; Pulverer, G.; Rott, R. Role of staphylococcus protease in the development of influenza pneumonia. Nature 1987, 325, 536–537. [Google Scholar]
- Tashiro, M.; Ciborowski, P.; Reinacher, M.; Pulverer, G.; Klenk, H.D.; Rott, R. Synergistic role of staphylococcal proteases in the induction of influenza virus pathogenicity. Virology 1987, 157, 421–430. [Google Scholar]
- Su, B.; Wurtzer, S.; Rameix-Welti, M.-A.; Dwyer, D.; van der Werf, S.; Naffakh, N.; Clavel, F.; Labrosse, B. Enhancement of the influenza a hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS One 2009, 4, e8495. [Google Scholar]
- Reed, M.L.; Bridges, O.A.; Seiler, P.; Kim, J.-K.; Yen, H.-L.; Salomon, R.; Govorkova, E.A.; Webster, R.G.; Russell, C.J. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity and transmissibility in ducks. J. Virol. 2010, 84, 1527–1535. [Google Scholar]
- Goto, H.; Kawaoka, Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. USA 1998, 95, 10224–10228. [Google Scholar]
- Goto, H.; Wells, K.; Takada, A.; Kawaoka, Y. Plasminogen-binding activity of neuraminidase determines the pathogenicity of influenza a virus. J. Virol. 2001, 75, 9297–9301. [Google Scholar]
- Li, S.; Schulman, J.; Itamura, S.; Palese, P. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J. Virol. 1993, 67, 6667–6673. [Google Scholar]
- Tumpey, T.M.; Basler, C.F.; Aguilar, P.V.; Zeng, H.; Solórzano, A.; Swayne, D.E.; Cox, N.J.; Katz, J.M.; Taubenberger, J.K.; Palese, P.; et al. Characterization of the reconstructed 1918 spanish influenza pandemic virus. Science 2005, 310, 77–80. [Google Scholar]
- Garten, W.; Bosch, F.X.; Linder, D.; Rott, R.; Klenk, H.-D. Proteolytic activation of the influenza virus hemagglutinin: The structure of the cleavage site and the enzymes involved in cleavage. Virology 1981, 115, 361–374. [Google Scholar]
- Garten, W.; Klenk, H.-D. Characterization of the carboxypeptidase involved in the proteolytic cleavage of the influenza haemagglutinin. J. Gen. Virol. 1983, 64, 2127–2137. [Google Scholar]
- Sun, X.; Tse, L.V.; Ferguson, A.D.; Whittaker, G.R. Modifications to the hemagglutinin cleavage site control the virulence of a neurotropic H1N1 influenza virus. J. Virol. 2010, 84, 8683–8690. [Google Scholar]
- Fleury, D.; Barrere, B.; Bizebard, T.; Daniels, R.S.; Skehel, J.J.; Knossow, M. A complex of influenza hemagglutinin with a neutralizing antibody that binds outside the virus receptor binding site. Nat. Struct. Mol. Biol. 1999, 6, 530–534. [Google Scholar]
- Bizebard, T.; Gigant, B.; Rigolet, P.; Rasmussen, B.; Diat, O.; Baseckei, P.; Wharton, S.A.; Skehel, J.J.; Knossow, M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995, 376, 92–94. [Google Scholar]
- Daniels, R.S.; Jeffries, S.; Yates, P.; Schild, G.C.; Rogers, G.N.; Paulson, J.C.; Wharton, S.A.; Douglas, A.R.; Skehel, J.J.; Wiley, D.C. The receptor-binding and membrane-fusion properties of influenza virus variants selected using anti-haemagglutinin monoclonal antibodies. EMBO J. 1987, 6, 1459–1465. [Google Scholar]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar]
- Ekiert, D.C.; Friesen, R.H.E.; Bhabha, G.; Kwaks, T.; Jongeneelen, M.; Yu, W.; Ophorst, C.; Cox, F.; Korse, H.J.M.M.; Brandenburg, B.; et al. Hhighly conserved neutralizing eptitope on group 2 influenza A viruses. Science 2011, 333, 843–850. [Google Scholar]
- Barbey-Martin, C.; Gigant, B.; Bizebard, T.; Calder, L.J.; Wharton, S.A.; Skehel, J.J.; Knossow, M. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology 2002, 294, 70–74. [Google Scholar]
- Sui, J.; Hwang, W.C.; Perez, S.; Wei, G.; Aird, D.; Chen, L.-m.; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza a viruses. Nat. Struct. Mol. Biol. 2009, 16, 265–273. [Google Scholar] [CrossRef]
- Ekiert, D.C.; Bhabha, G.; Elsliger, M.-A.; Friesen, R.H.E.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science 2009, 324, 246–251. [Google Scholar]
- Modis, Y. How influenza virus is locked out the cell. Proc. Natl. Acad. Sci. USA 2008, 105, 18647–18648. [Google Scholar]
- Bodian, D.L.; Yamasaki, R.B.; Buswell, R.L.; Streans, J.F.; White, J.M.; Kuntz, I.D. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 1993, 32, 2967–2978. [Google Scholar]
- Shangguan, T.; Alford, D.; Bentz, J. Influenza virus-liposome lipid mixing is leaky and largely insensitive to the material properties of the target membrane. Biochemistry 1996, 35, 4956–4965. [Google Scholar]
- Hoffman, L.R.; Kuntz, I.D.; White, J.M. Structure-based identification of an inducer of the low-pH conformational change in the influenza virus hemagglutinin: Irreversible inhibition of infectivity. J. Virol. 1997, 71, 8808–8820. [Google Scholar]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gambin, S.J.; Skehel, J.J. Structure of influenza hemaglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar]
- Vanderlinden, E.; Goktas, F.; Cesur, Z.; Froeyen, M.; Reed, M.L.; Russell, R.J.; Cesur, N.; Naesens, L. Novel inhibitors of influenza fusion: Structure-activity relationships and interaction with the viral hemagglutinin. J. Virol. 2010, 84, 4277–4288. [Google Scholar]
- Zhu, L.; Li, Y.; Li, S.; Li, H.; Qiu, Z.; Lee, C.; Lu, H.; Lin, X.; Zhao, R.; Chen, L.; et al. Inhibition of influenza a virus (H1N1) fusion by benzenesulfonamide derivatives targeting viral hemagglutinin. PLoS One 2011, 6, e29120. [Google Scholar]
- Lee, K.K.; Pessi, A.; Gui, L.; Santoprete, A.; Talekar, A.; Moscana, A.; Porotto, M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011, 286, 42141–42149. [Google Scholar]
- Harrison, S.C. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res. 2005, 64, 231–261. [Google Scholar]
- Ramalho-Santos, J.; Pedroso de Lima, M.C. The influenza virus hemaglutinin: A model protein in the study of membrane fusion. Biochim. Biophys. Acta 1998, 1376, 147–154. [Google Scholar]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar]
- Tamm, L.K.; Crane, J.; Kiessling, V. Membrane fusion: A structural perspective on the interplay of lipids and proteins. Curr. Opin. Struct. Biol. 2003, 13, 453–466. [Google Scholar]
- Xu, R.; Wilson, I.A. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 2011, 85, 5172–5182. [Google Scholar]
- White, J.M.; Wilson, I.A. Anti-peptide antibodies detect steps in a protein conformational change: Low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 1987, 105, 2887–2896. [Google Scholar]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar]
- Han, X.; Bushwell, J.H.; Cafisco, D.S.; Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001, 8, 715–720. [Google Scholar]
- Stegmann, T.; White, J.M.; Helenius, A. Intermediates in influenza induced membrane fusion. EMBO J. 1990, 9, 4231–4241. [Google Scholar]
- Markovic, I.; Leikina, E.; Zhukovsky, M.; Zimmerberg, J.; Chernomordik, L.V. Synchronized activation and refolding of influenza hemagglutinin in multimeric fusion machines. J. Cell Biol. 2001, 155, 833–843. [Google Scholar]
- Danieli, T.; Pelletier, S.; Henis, Y.I.; White, J.M. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 1996, 133, 559–569. [Google Scholar]
- Blumenthal, R.; Sarkar, D.; Durell, S.; Howard, D.E.; Morris, S.J. Dilation of the influenza hemagglutinin fusion pore revealed by kinetics of individual cell-cell fusion events. J. Cell Biol. 1996, 135, 63–71. [Google Scholar]
- Doms, R.W.; Helenius, A. Quaternary structure of influenza virus hemagglutinin after acid treatment. J. Virol. 1986, 60, 833–839. [Google Scholar]
- Imai, M.; Mizuno, T.; Kawasaki, K. Membrane fusion by single influenza hemagglutinin trimers: Kinetic evidence from image analysis of hemagglutinin-reconstituted vesicles. J. Biol. Chem. 2006, 281, 12729–12735. [Google Scholar]
- Dobay, M.P.; Dobay, A.; Bantang, J.; Mendoza, E. How many trimers? Modeling influenza virus fusion yields a minimum aggregate size of six trimers, three of which are fusogenic. Mol. BioSyst. 2011, 7, 2741–2749. [Google Scholar]
- Chernomordik, L.V.; Kozlov, M.M. Membrane hemifusion: Crossing a chasm in two leaps. Cell 2005, 123, 375–382. [Google Scholar]
- Goodsell, D.S. Molecule of the month: Hemagglutinin. RCSB PDB Molecule of the Month. 2006. Available online: http://www.rcsb.org/pdb/101/motm.do?momID=76.
- Baker, D.; Agard, D.A. Influenza hemagglutinin: Kinetic control of protein function. Structure 1994, 2, 907–910. [Google Scholar]
- Li, Y.; Han, X.; Tamm, L.K. Thermodynamics of fusion peptide-membrane interactions. Biochemistry 2003, 42, 7245–7251. [Google Scholar]
- Niles, W.D.; Cohen, F.S. Single event recording shows that docking onto receptor alters the kinetics of membrane fusion mediate by influenza hemaglutinin. Biophys. J. 1993, 65, 171–176. [Google Scholar]
- Niles, W.D.; Cohen, F.S. The role of n-acetylneuraminic (sialic) acid in the pH dependence of influenza virion fusion with planar phospholipid membranes. J. Gen. Physiol. 1991, 97, 1121–1140. [Google Scholar]
- Kim, C.S.; Epand, R.F.; Leikina, E.; Epand, R.M.; Chernomordik, L.V. The final conformation of the complete ectodomain of the HA2 subunit of influenza hemagglutinin can by itself drive low pH-dependent fusion. J. Biol. Chem. 2011, 286, 13226–13234. [Google Scholar]
- Nussbaum, O.; Rott, R.; Loyter, A. Fusion of influenza virus particles with liposomes: Requirement for cholesterol and virus receptors to allow fusion with and lysis of neutral but not of negatively charged liposomes. J. Gen. Virol. 1992, 73, 2831–2837. [Google Scholar]
- Chernomordik, L.V.; Leikina, E.; Frolov, V.; Bronk, P.; Zimmerberg, J. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 1997, 136, 81–93. [Google Scholar]
- Bailey, A.; Zhukovsky, M.; Gliozzi, A.; Chernomordik, L.V. Liposome composition effects on lipid mixing between cells expressing influenza virus hemagglutinin and bound liposomes. Arch. Biochem. Biophys. 2005, 439, 211–221. [Google Scholar]
- Razinkov, V.I.; Cohen, F.S. Sterols and sphingolipids strongly affect the growth of fusion pores induced by the hemagglutinin of influenza virus. Biochemistry 2000, 39, 13462–13468. [Google Scholar]
- Chernomordik, L.V.; Frolov, V.; Leikina, E.; Bronk, P.; Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: Restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998, 140, 1369–1382. [Google Scholar] [CrossRef]
- Bonnafous, P.; Stegmann, T. Membrane perturbation and fusion pore formation in influenza hemagglutinin-mediated membrane fusion. J. Biol. Chem. 2000, 275, 6160–6166. [Google Scholar]
- Takeda, M.; Leser, G.P.; Russell, C.L.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar]
- Mittal, A.; Leikina, E.; Chernomordik, L.V.; Bentz, J. Kinetically differentiating influenza hemagglutinin fusion and hemifusion machines. Biophys. J. 2003, 85, 1713–1724. [Google Scholar]
- Mittal, A.; Leikina, E.; Bentz, J.; Chernomordik, L.V. Kinetics of influenza hemagglutinin-mediated membrane fusion as a function of technique. Anal. Biochem. 2002, 303, 145–152. [Google Scholar]
- Hoekstra, D.; de Boer, T.; Klappe, K.; Wilschut, J. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry 1984, 23, 5675–5681. [Google Scholar]
- Loyter, A.; Citovsky, V.; Blumenthal, R. The Use of Fluorscence Dequenching Measurements to Follow Viral Membrane Fusion Events. In Methods of Biochemical Analysis; Glick, D., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988. [Google Scholar]
- Hoekstra, D.; Klappe, K. Fluorescence assays to monitor fusion of enveloped viruses. Methods Enzymol. 1993, 220, 261–276. [Google Scholar]
- Polozov, I.V.; Berrukov, L.; Gawrisch, K.; Zimmerberg, J. Progressive ordering with decreasing temperature of the phospholipids of influenza virus. Nat. Chem. Biol. 2008, 4, 248–255. [Google Scholar]
- Ramalho-Santos, J.; Nir, S.; Duzgunes, N.; Pato de Carvalho, A.; da Conceicao Pedroso de Lima, M. A common mechanism for influenza virus fusion activity and inactivation. Biochemistry 1993, 32, 2771–2779. [Google Scholar]
- Clague, M.J.; Schoch, C.; Blumenthal, R. Delay time for influenza virus hemagglutinin-induced membrane fusion depends on hemagglutinin surface density. J. Virol. 1991, 65, 2402–2407. [Google Scholar]
- Stegmann, T.; Hoekstra, D.; Scherphof, G.; Wilschut, J. Kinetics of pH-dependent fusion between influenza virus and liposomes. Biochemistry 1985, 24, 3107–3113. [Google Scholar]
- Georgiou, G.N.; Morrison, I.E.G.; Cherry, R.J. Digital fluorescence imaging of fusion of influenza virus with erythrocytes. FEBS Lett. 1989, 250, 487–492. [Google Scholar]
- Sarkar, D.P.; Morris, S.J.; Eidelman, O.; Zimmerberg, J.; Blumenthal, R. Initial stages of influenza hemagglutinin-induced cell fusion monitored simultaneously by two fluorescent events: Cytoplasmic continuity and lipid mixing. J. Cell Biol. 1989, 109, 113–122. [Google Scholar]
- Kaplan, D.; Zimmerberg, J.; Puri, A.; Sarkar, D.P.; Blumenthal, R. Single cell fusion events induced by influenza hemagglutinin: Studies with rapid-flow, quantitative fluorescence microscopy. Exp. Cell Res. 1991, 195, 137–144. [Google Scholar]
- Niles, W.D.; Cohen, F.S. Fusion of influenza virions with a planar lipid membrane detected by video fluorescence microscopy. J. Gen. Physiol. 1991, 97, 1101–1119. [Google Scholar]
- Montal, M.; Mueller, P. Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc. Natl. Acad. Sci. USA 1972, 69, 3561–3566. [Google Scholar]
- Wunderli-Allenspach, H.; Ott, S. Kinetics of fusion and lipid transfer between virus receptor-containing liposomes and influenza viruses as measured with the octadecylrhodamine B chloride assay. Biochemistry 1990, 29, 1990–1997. [Google Scholar]
- Wharton, S.A.; Skehel, J.J.; Wiley, D.C. Studies of influenza haemagglutinin-mediated membrane fusion. Virology 1986, 149, 27–35. [Google Scholar]
- Ramalho-Santos, J.; Pedroso de Lima, M.C. The role of target membrane sialic acid residues in the fusion activity of the influenza virus: The effect of two types of ganglioside on the kinetics of membrane merging. Ceullar Mol. Biol. Lett. 2004, 9, 337–351. [Google Scholar]
- Melikyan, G.B.; Niles, W.D.; Cohen, F.S. Influenza virus hemagglutinin-induced cell-planar bilayer fusion: Quantitative dissection of fusion pore kinetics into stages. J. Gen. Physiol. 1993, 102, 1151–1170. [Google Scholar]
- Zimmerberg, J.; Blumenthal, R.; Sarkar, D.; Curran, M.; Morris, S.J. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J. Cell Biol. 1994, 127, 1885–1894. [Google Scholar]
- Jha, N.K.; Latinovic, O.; Martin, E.; Novitskiy, G.; Marin, M.; Miyauchi, K.; Naughton, J.; Young, J.A.T.; Melikyan, G.B. Imaging single retrovirus entry through alternative receptor isoforms and intermediates of virus-endosome fusion. PLoS Pathog. 2011, 7, e1001260. [Google Scholar]
- Padilla-Parra, S.; Marin, M.; Kondo, N.; Melikyan, G.B. Synchronized retrovirus fusion in cells expressing alternative receptor isoforms releases the viral core into distinct sub-cellular compartments. PLoS Pathog. 2012, 8, e1002694. [Google Scholar]
- Floyd, D.L.; Ragain, J.R.; Skehel, J.J.; Harrison, S.C.; van Oijen, A.M. Single-particle kinetics of influenza virus membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 15382–15387. [Google Scholar]
- Ivanovic, T.; Rozendaal, R.; Floyd, D.L.; Popvic, M.; van Oijen, A.M.; Harrison, S.C. Kinetics of proton transport into influenza virions by the viral M2 channel. PLoS One 2012, 7, e31566. [Google Scholar]
- Axelrod, D.; Burghardt, T.P.; Thompson, N.L. Total internal reflection fluorescence. Ann. Rev. Biophys. Bioeng. 1984, 13, 247–268. [Google Scholar]
- Brian, A.A.; McConnell, H.M. Allogeneic stimulation of cytotoxic T cells by supported planar membranes. Proc. Natl. Acad. Sci. USA 1984, 81, 6159–6163. [Google Scholar]
- Castellana, E.T.; Cremer, P.S. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf. Sci. Rep. 2006, 61, 429–444. [Google Scholar]
- Dietrich, C.; Volovyk, Z.N.; Levi, M.; Thompson, N.L.; Jacobson, K. Partitioning of THY-1, Gm1, and cross-linked phospholipid analogs into lipid rafts reconstituted in supported model membrane monolayer. Proc. Natl. Acad. Sci. USA 2001, 98, 10642–10647. [Google Scholar]
- Diaz, A.J.; Albertorio, F.; Daniel, S.; Cremer, P.S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir 2008, 24, 6820–6826. [Google Scholar]
- Elender, G.; Kuhner, M.; Sackmann, E. Functionalisation of Si/SiO2 and glass surfaces with ultrathin dextran films and deposition of lipid bilayers. Biosens. Bioelectron. 1996, 11, 565–577. [Google Scholar]
- Wagner, M.L.; Tamm, L.K. Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: Silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys. J. 2000, 79, 1400–1414. [Google Scholar]
- Johnson, J.M.; Ha, T.; Chu, S.; Boxer, S.G. Early steps of supported bilayer formation probed by single vesicle fluorescence assays. Biophys. J. 2002, 83, 3371–3379. [Google Scholar]
- Murcia, M.J.; Garg, S.; Naumann, C.A. Single-molecule fluorescence microscopy to determine phospholipid lateral diffusion. Methods Mol. Biol. 2007, 400, 277–294. [Google Scholar]
- Saxton, M.J.; Jacobson, K. Single-particle tracking: Applications to membrane dynamics. Ann. Rev. Biophys. Biomol. Struct. 1997, 26, 373–399. [Google Scholar]
- Groves, J.T.; Parthasarathy, R.; Forstner, M.B. Fluorescence imaging of membrane dynamics. Ann. Rev. Biomed. Eng. 2008, 10, 311–338. [Google Scholar]
- Ewers, H.; Smith, A.E.; Sbalzarini, I.F.; Lilie, H.; Koumoutsakos, P.; Helenius, A. Single-particle tracking of murine polyoma virus-like particles on live cells and artificial membranes. Proc. Natl. Acad. Sci. USA 2005, 102, 15110–15115. [Google Scholar]
- Fix, M.; Melia, T.J.; Jaiswal, J.K.; Rappoport, J.Z.; You, D.; Sollner, T.H.; Rothman, J.E.; Simon, S.M. Imaging single membrane fusion events mediated by snare proteins. Proc. Natl. Acad. Sci. USA 2006, 101, 7311–7316. [Google Scholar]
- Domanska, M.K.; Kiessling, V.; Stein, A.; Fasshauer, D.; Tamm, L.K. Single vesicle millisecond fusion kinetics reveals number of snare complexes optimal for fast snare-mediated membrane fusion. J. Biol. Chem. 2009, 284, 32158–32166. [Google Scholar]
- Wang, T.; Smith, E.A.; Chapman, E.R.; Weisshaar, J.C. Lipid mixing and content release in single-vesicle, snare-driven fusion assay with 1-5 ms resolution. Biophys. J. 2009, 96, 4122–4131. [Google Scholar]
- Rawle, R.J.; van Lengerich, B.; Chung, M.; Bendix, P.M.; Boxer, S.G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys. J. 2011, 101, L37–L39. [Google Scholar]
- Wessels, L.; Elting, M.W.; Scimeca, D.; Weninger, K. Rapid membrane fusion of individual virus particles with supported lipid bilayers. Biophys. J. 2007, 93, 526–538. [Google Scholar]
- Krumbiegel, M.; Herrman, A.; Blumenthal, R. Kinetics of the low pH-induced conformational changes and fusogenic activity of influenza hemagglutinin. Biophys. J. 1994, 67, 2355–2360. [Google Scholar]
- Brandenburg, B.; Lee, L.Y.; Lakadamyali, M.; Rust, M.J.; Zhuang, X.; Hogle, J.M. Imaging poliovirus entry in live cells. PLoS Biol. 2007, 5, e183. [Google Scholar]
- Mangenot, S.; Hochrein, M.; Radler, J.; Letellier, L. Real-time imaging of DNA ejection from single phage particles. Curr. Biol. 2005, 15, 430–435. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hamilton, B.S.; Whittaker, G.R.; Daniel, S. Influenza Virus-Mediated Membrane Fusion: Determinants of Hemagglutinin Fusogenic Activity and Experimental Approaches for Assessing Virus Fusion. Viruses 2012, 4, 1144-1168. https://doi.org/10.3390/v4071144
Hamilton BS, Whittaker GR, Daniel S. Influenza Virus-Mediated Membrane Fusion: Determinants of Hemagglutinin Fusogenic Activity and Experimental Approaches for Assessing Virus Fusion. Viruses. 2012; 4(7):1144-1168. https://doi.org/10.3390/v4071144
Chicago/Turabian StyleHamilton, Brian S., Gary R. Whittaker, and Susan Daniel. 2012. "Influenza Virus-Mediated Membrane Fusion: Determinants of Hemagglutinin Fusogenic Activity and Experimental Approaches for Assessing Virus Fusion" Viruses 4, no. 7: 1144-1168. https://doi.org/10.3390/v4071144
APA StyleHamilton, B. S., Whittaker, G. R., & Daniel, S. (2012). Influenza Virus-Mediated Membrane Fusion: Determinants of Hemagglutinin Fusogenic Activity and Experimental Approaches for Assessing Virus Fusion. Viruses, 4(7), 1144-1168. https://doi.org/10.3390/v4071144