Epstein-Barr Virus (EBV)-associated Gastric Carcinoma



Abstract
:1. Introduction
2. Definition, Epidemiology, and Clinical Features
2.1. Definition
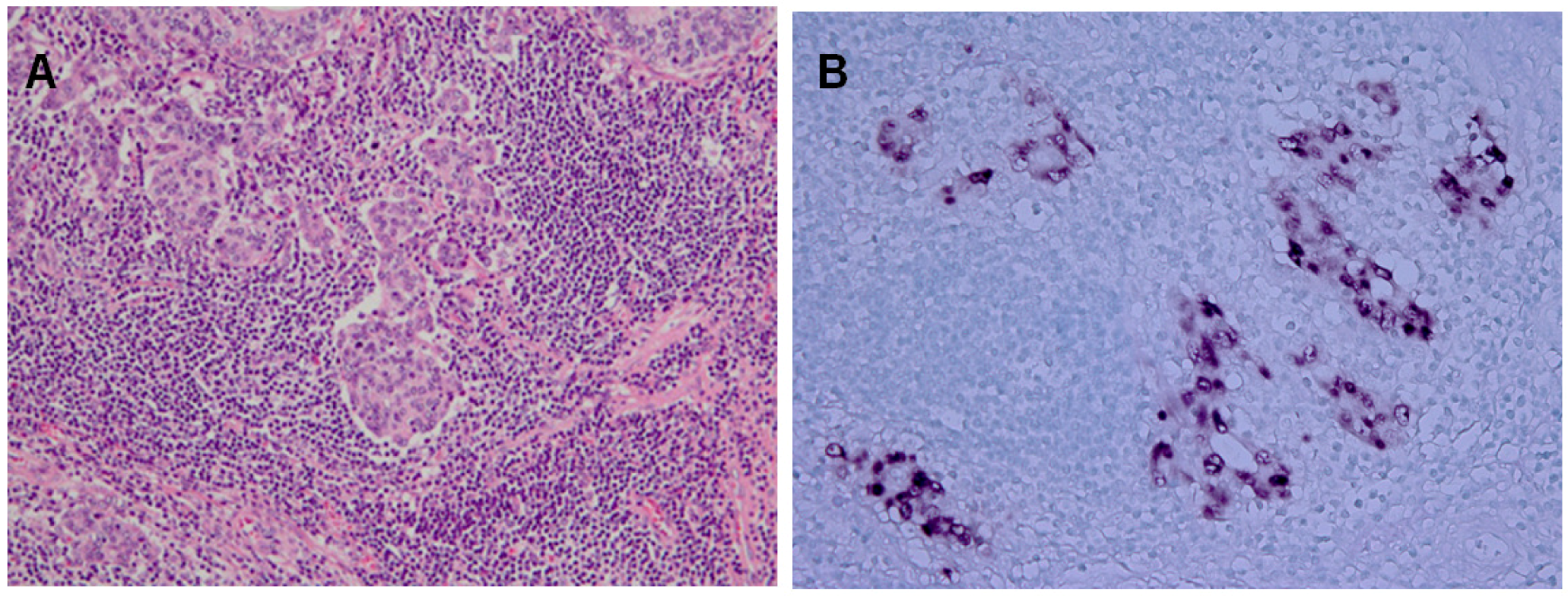
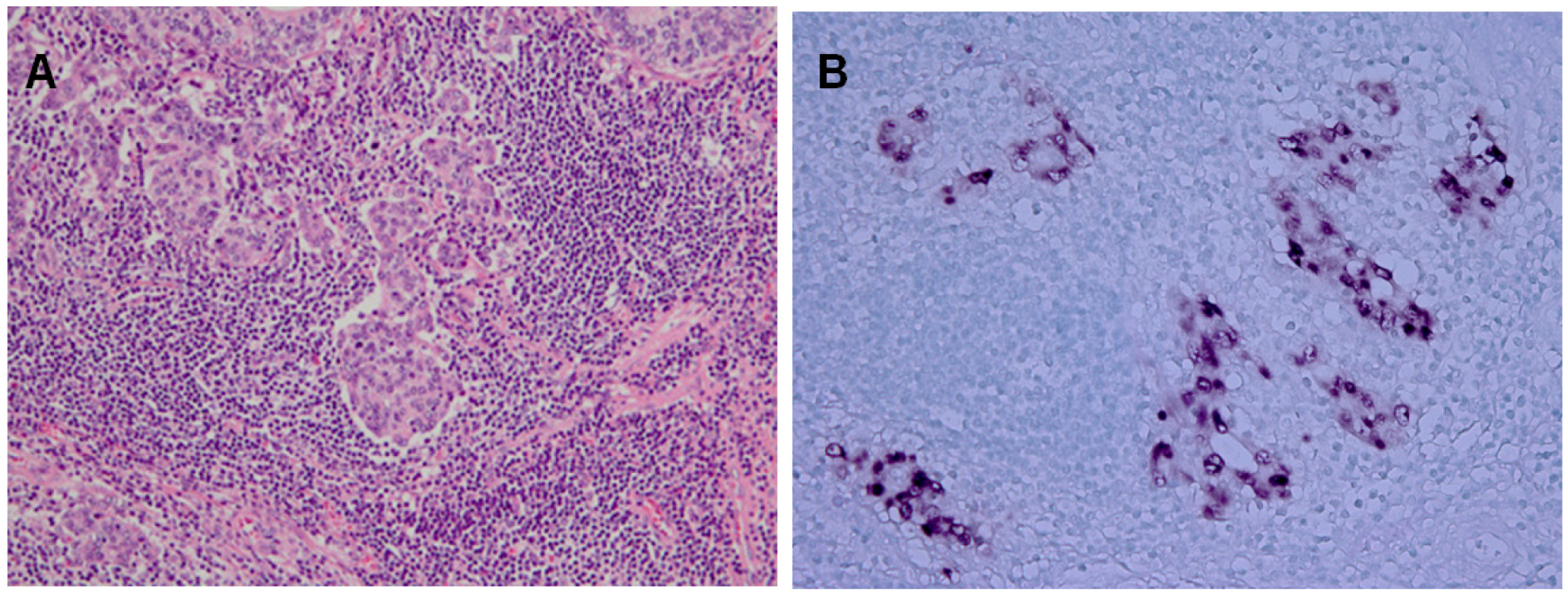
2.2. Epidemiology
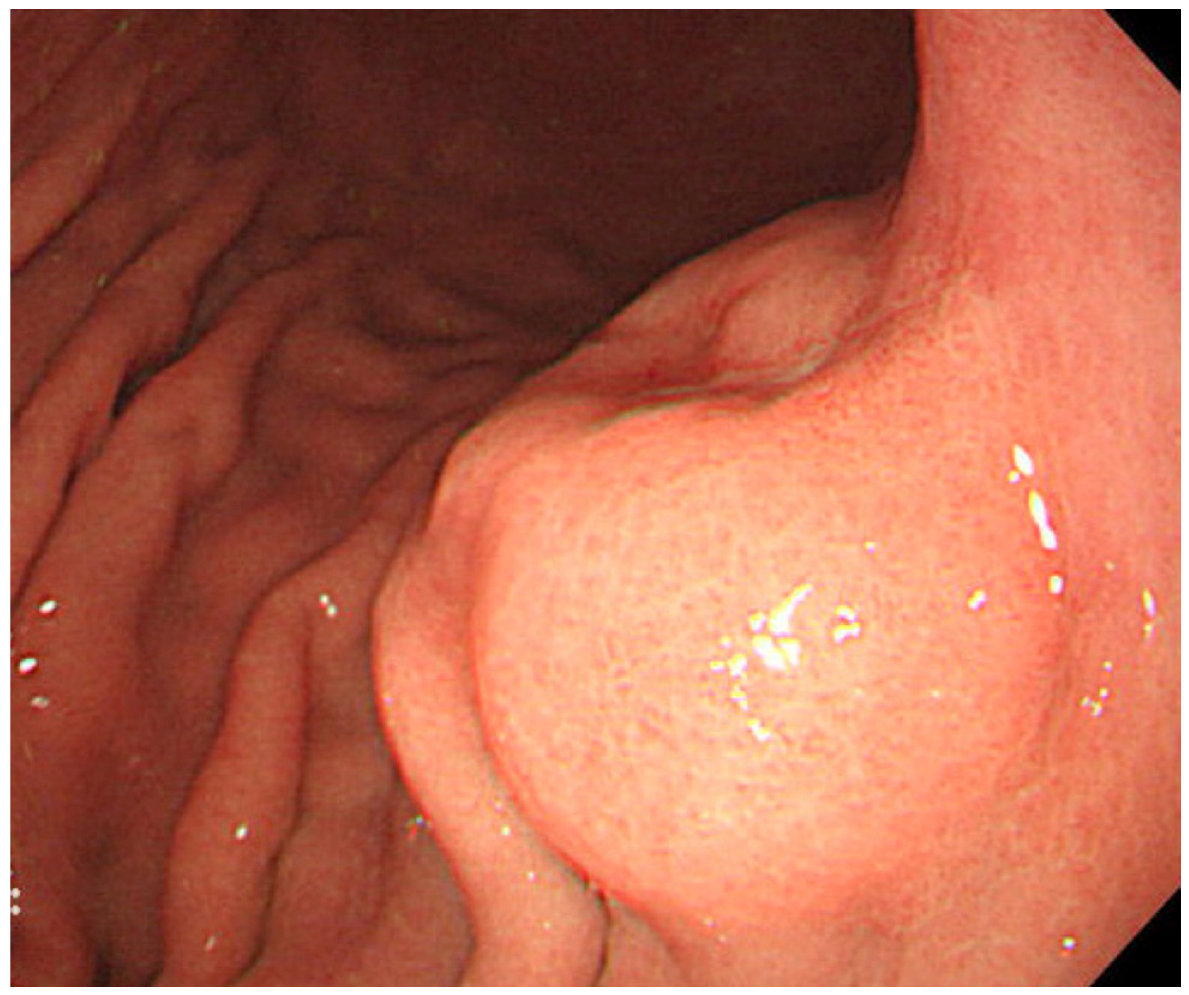
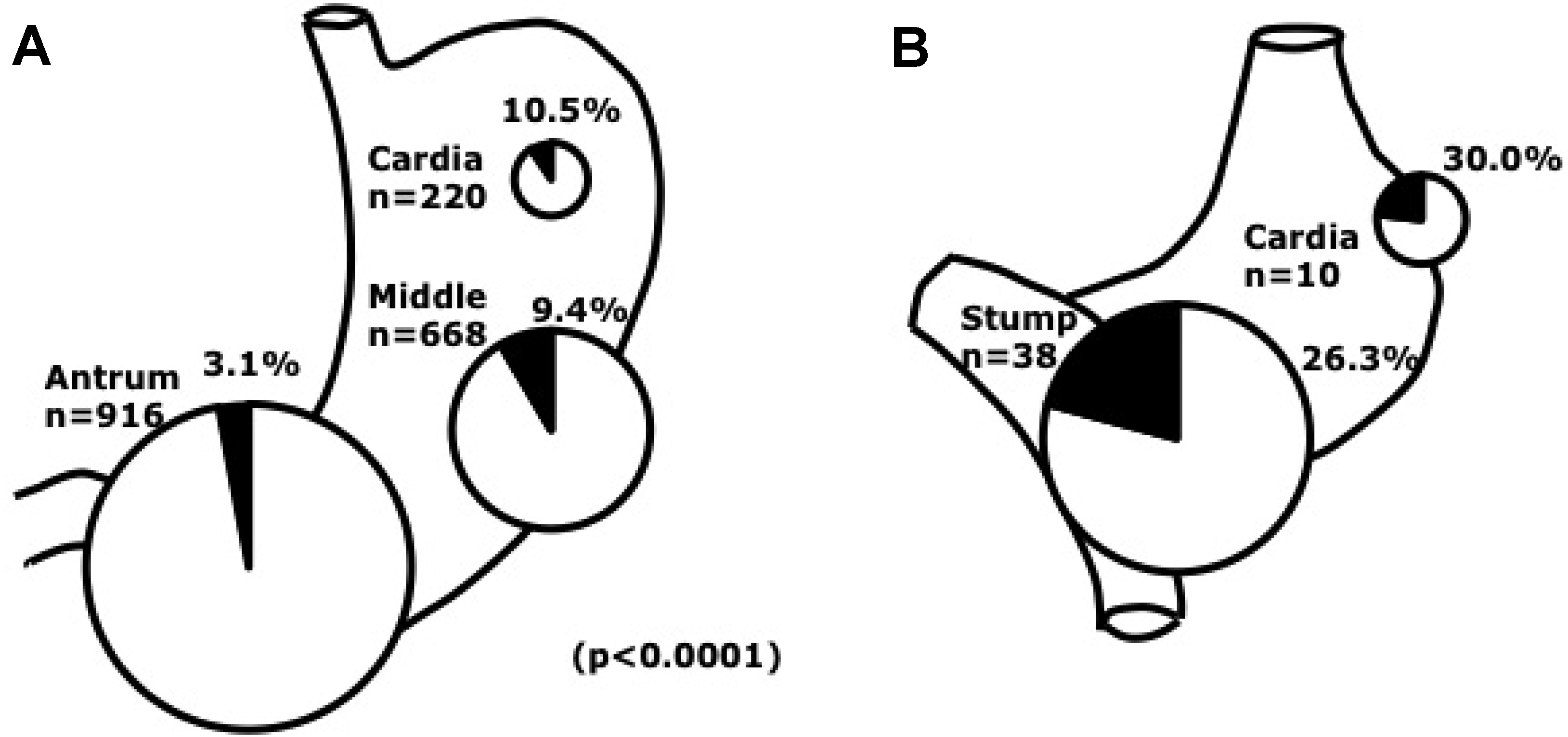
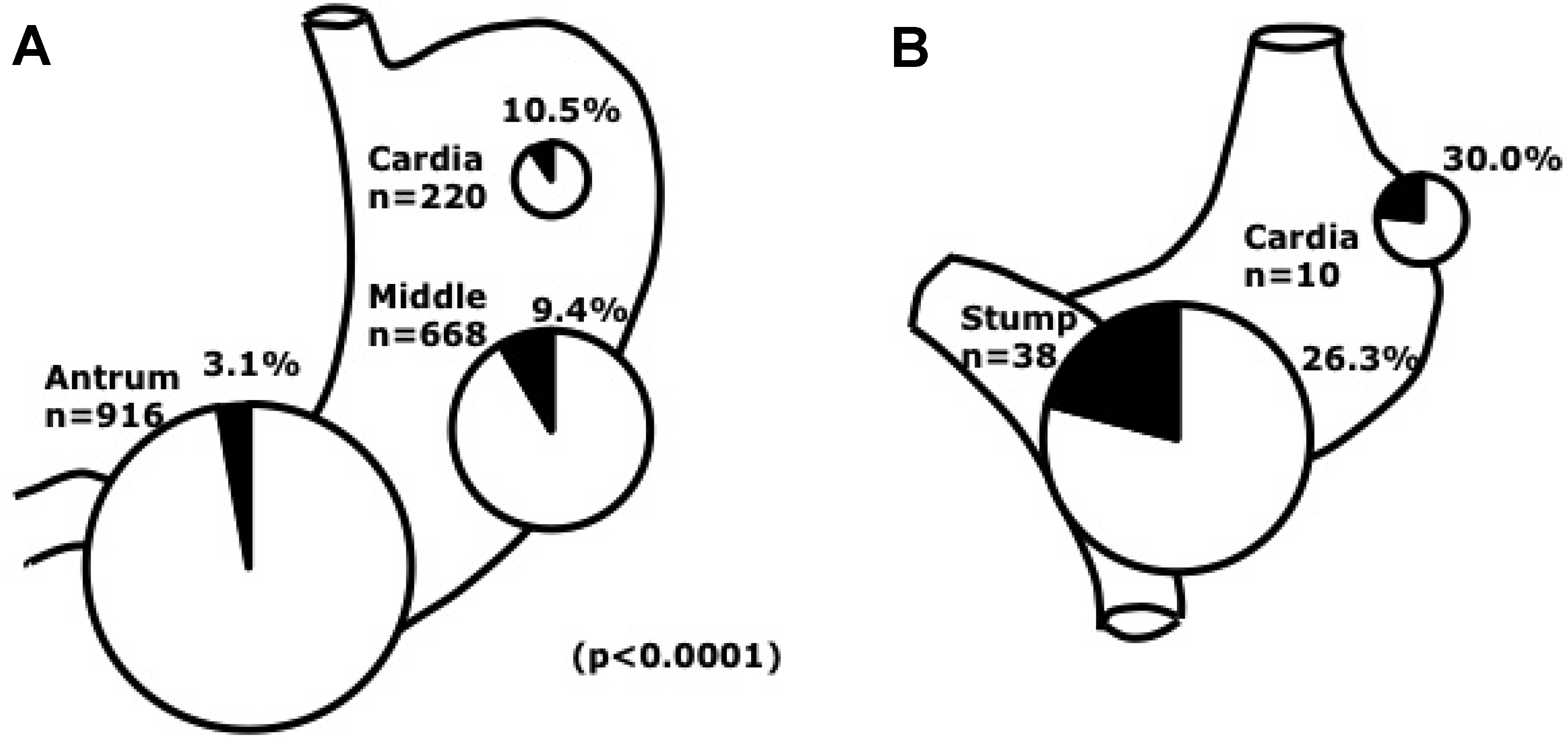
2.3. Clinical Features
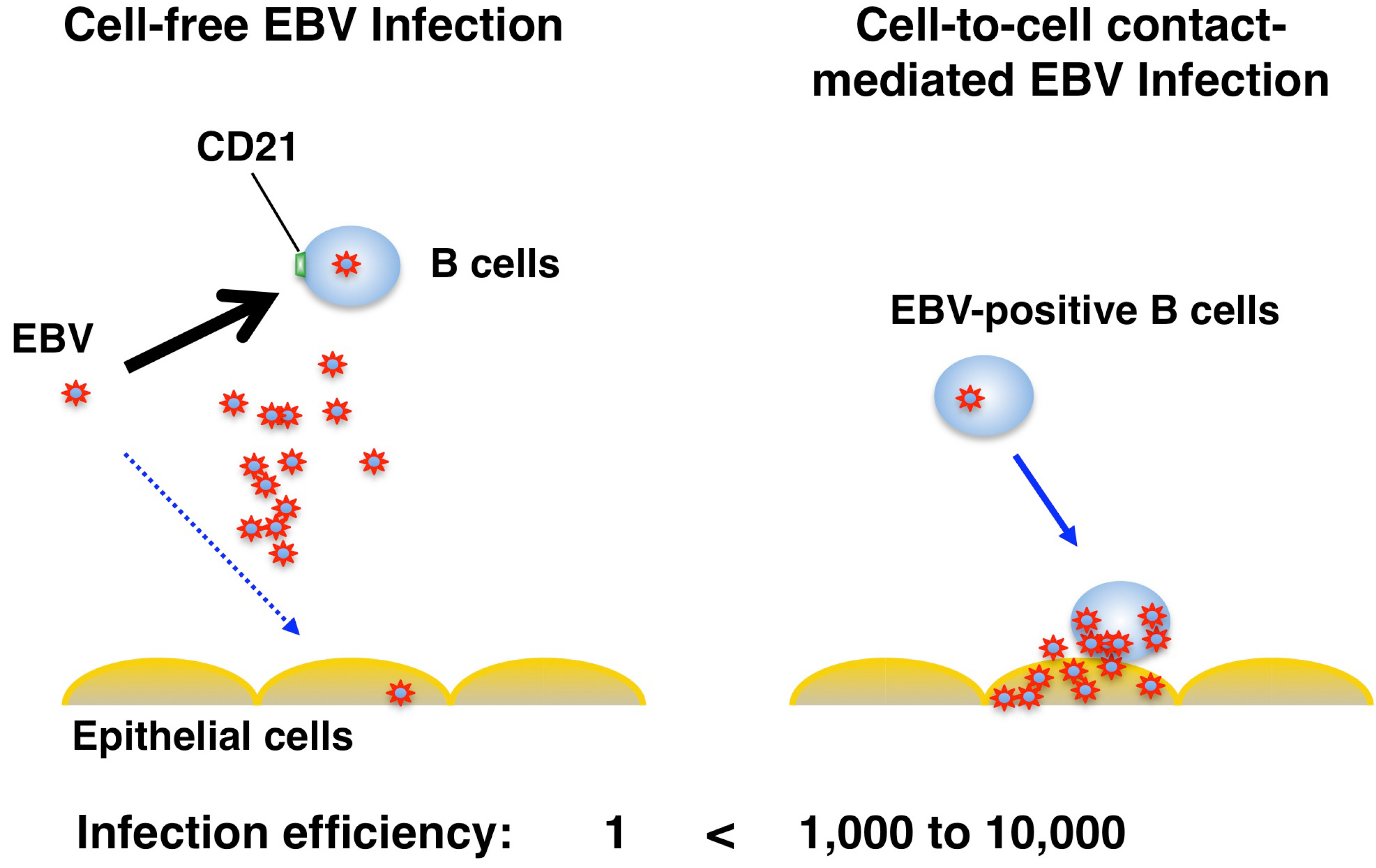
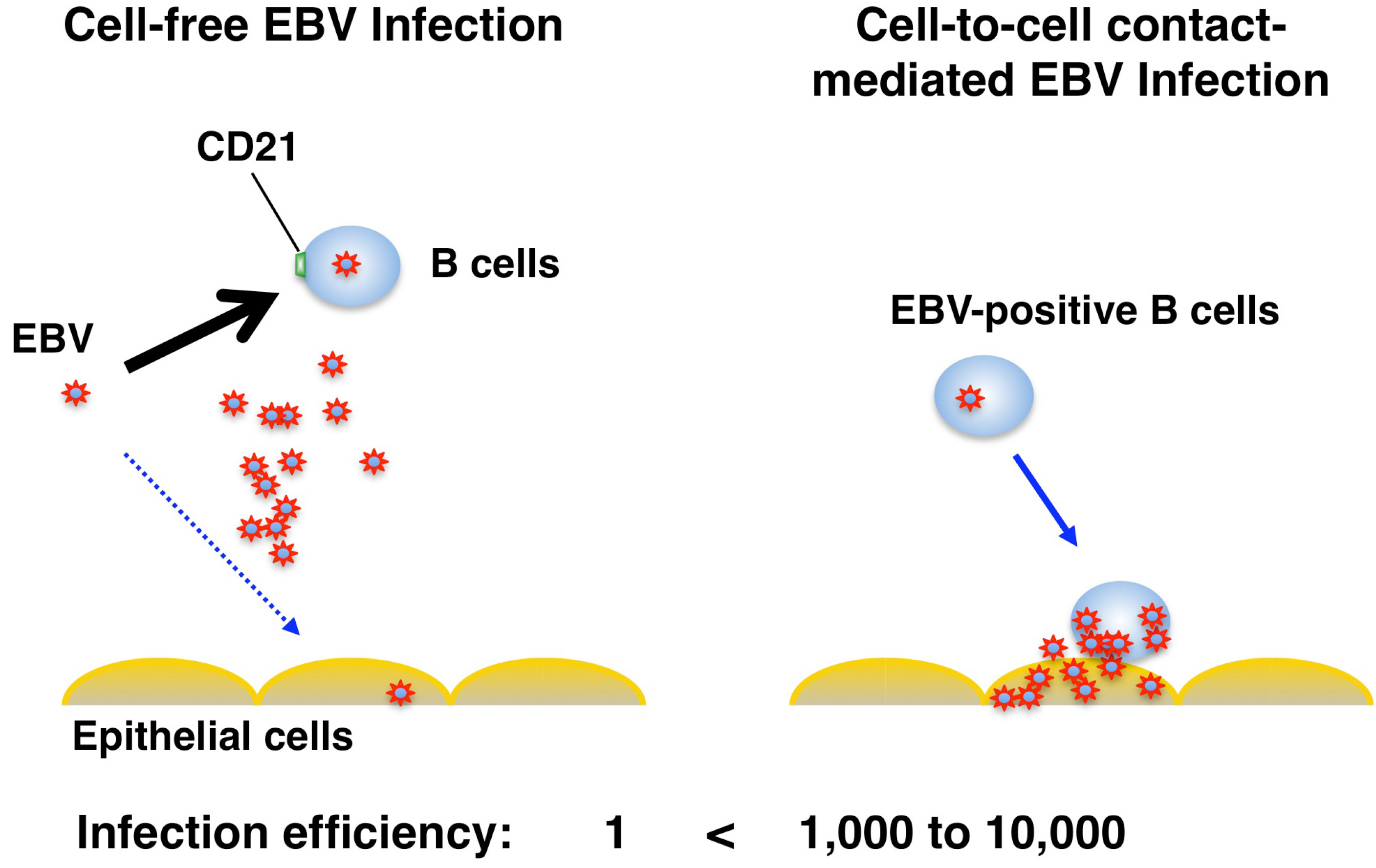
3. Route of Epithelial Infection
4. Viral Genes and Carcinogenesis
4.1. Models of EBV infection of gastric epithelial cells
4.2. Growth promoting effects of EBV
5. Virus and Host Interactions at Molecular Level
5.1. DNA Hypermethylation in EBV and Host Genomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
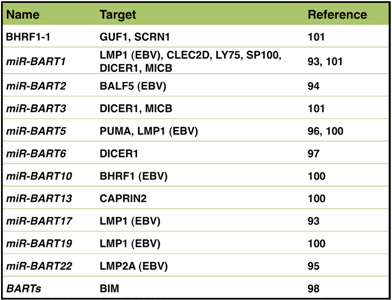
5.2. miRNA and Carcinogenesis
 |
6. Diagnosis and Treatment of EBV-associated GC
6.1. Procedure
6.2. Prognosis
7. Conclusion
Acknowledgments
Conflict of Interests
References
- zur Hausen, H.; Schulte–Holthausen, H.; Klein, G.; Henle, W.; Henle, G.; Clifford, P.; Santesson, L. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 1970, 228, 1056–1058. [Google Scholar] [CrossRef]
- Young, L.S.; Murray, P.G. Epstein–Barr virus and oncogenesis: from latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef]
- Burke, A.P.; Yen, T.S.; Shekitka, K.M.; Sobin, L.H. Lymphoepithelial carcinoma of the stomach with Epstein–Barr virus demonstrated by polymerase chain reaction. Modern Pathol. 1990, 3, 377–380. [Google Scholar]
- Shibata, D.; Weiss, L.M. Epstein–Barr virus–associated gastric adenocarcinoma. Am. J. Pathol. 1992, 140, 769–774. [Google Scholar]
- Tokunaga, M.; Land, C.E.; Uemura, Y.; Tokudome, T.; Tanaka, S.; Sato, E. Epstein–Barr virus in gastric carcinoma. Am. J. Pathol. 1993, 143, 1250–1254. [Google Scholar]
- Takada, K. Epstein–Barr virus and gastric carcinoma. Mol. Pathol. 2000, 53, 255–261. [Google Scholar]
- Young, L.S.; Rickinson, A.B. Epstein–Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Fukayama, M.; Hayashi, Y.; Iwasaki, Y.; Chong, J.; Ooba, T.; Takizawa, T.; Koike, M.; Mizutani, S.; Miyaki, M.; Hirai, K. Epstein–Barr virus–associated gastric carcinoma and Epstein–Barr virus infection of the stomach. Lab. Invest. 1994, 71, 73–81. [Google Scholar]
- Imai, S.; Koizumi, S.; Sugiura, M.; Tokunaga, M.; Uemura, Y.; Yamamoto, N.; Tanaka, S.; Sato, E.; Osato, T. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein–Barr virus latent infection protein. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 9131–9135. [Google Scholar]
- Rickinson, A.B.; Kieff, E. Epstein–Barr virus, 5thFields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott–Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2655–2700. [Google Scholar]
- Robertson, K.D.; Manns, A.; Swinnen, L.J.; Zong, J.C.; Gulley, M.L.; Ambinder, R.F. CpG methylation of the major Epstein–Barr virus latency promoter in Burkitt's lymphoma and Hodgkin's disease. Blood 1996, 88, 3129–3136. [Google Scholar]
- Fukayama, M. Epstein–Barr virus and gastric carcinoma. Pathol. Int. 2010, 60, 337–350. [Google Scholar] [CrossRef]
- Chong, J.M.; Sakuma, K.; Sudo, M.; Ushiku, T.; Uozaki, H.; Shibahara, J.; Nagai, H.; Funata, N.; Taniguchi, H.; Aburatani, H.; Fukayama, M. Global and non–random CpG–island methylation in gastric carcinoma associated with Epstein–Barr virus. Cancer Sci. 2003, 94, 76–80. [Google Scholar] [CrossRef]
- Kang, G.H.; Lee, S.; Kim, W.H.; Lee, H.W.; Kim, J.C.; Rhyu, M.G.; Ro, J.Y. Epstein–Barr virus–positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype–positive gastric carcinoma. Am. J. Pathol. 2002, 160, 787–794. [Google Scholar] [CrossRef]
- Vo, Q.N.; Geradts, J.; Gulley, M.L.; Boudreau, D.A.; Bravo, J.C.; Schneider, B.G. Epstein–Barr virus in gastric adenocarcinomas: association with ethnicity and CDKN2A promoter methylation. J. Clin. Pathol. 2002, 55, 669–675. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Ramalingam, D.; Kieffer–Kwon, P.; Ziegelbauer, J.M. Emerging themes from EBV and KSHV microRNA targets. Viruses 2012, 4, 1687–1710. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.H.; Han, S.H.; An, J.S.; Lee, E.S.; Kim, Y.S. Clinicopathological and molecular characteristics of Epstein–Barr virus–associated gastric carcinoma: a meta–analysis. J. Gastroenterol. Hepatol. 2009, 24, 354–365. [Google Scholar]
- Akiba, S.; Koriyama, C.; Herrera–Goepfert, R.; Eizuru, Y. Epstein–Barr virus associated gastric carcinoma: epidemiological and clinicopathological features. Cancer Sci. 2008, 99, 195–201. [Google Scholar]
- Koriyama, C.; Akiba, S.; Minakami, Y.; Eizuru, Y. Environmental factors related to Epstein–Barr virus–associated gastric cancer in Japan. J. Exp. Clin. Cancer Res. 2005, 24, 547–553. [Google Scholar]
- Camargo, M.C.; Murphy, G.; Koriyama, C.; Pfeiffer, R.M.; Kim, W.H.; Herrera–Goepfert, R.; Corvalan, A.H.; Carrascal, E.; Abdirad, A.; Anwar, M.; Hao, Z.; Kattoor, J.; Yoshiwara–Wakabayashi, E.; Eizuru, Y.; Rabkin, C.S.; Akiba, S. Determinants of Epstein–Barr virus–positive gastric cancer: An international pooled analysis. Br. J. Cancer 2011, 105, 38–43. [Google Scholar] [CrossRef]
- Tashiro, Y.; Arikawa, J.; Itho, T.; Tokunaga, M. Clinico–pathological findings of Epstein–Barr virus–related gastric cancer. In Epstein–Barr Virus and Human Cancer (Gann Monograph on Cancer Research, No 45); Osato, T., Takada, K., Tokunaga, M., Eds.; S. Karger Ag: Barsel, Switzerland, 1998; pp. 87–97. [Google Scholar]
- Tokunaga, M.; Uemura, Y.; Tokudome, T.; Ishidate, T.; Masuda, H.; Okazaki, E.; Kaneko, K.; Naoe, S.; Ito, M.; Okamura, A.; Shimada, A.; Sato, E.; Land, C.E. Epstein–Barr virus related gastric cancer in Japan: A molecular patho–epidemiological study. Acta. Pathol. Jpn. 1993, 43, 574–581. [Google Scholar]
- Yanai, H.; Murakami, T.; Yoshiyama, H.; Takeuchi, H.; Nishikawa, J.; Nakamura, H.; Okita, K.; Miura, O.; Shimizu, N.; Takada, K. Epstein–Barr virus–associated gastric carcinoma and atrophic gastritis. J. Clin. Gastroenterol. 1999, 29, 39–43. [Google Scholar] [CrossRef]
- Hirano, A.; Yanai, H.; Shimizu, N.; Okamoto, T.; Matsubara, Y.; Yamamoto, K.; Okita, K. Evaluation of Epstein–Barr virus DNA load in gastric mucosa with chronic atrophic gastritis using a real–time quantitative PCR assay. Int. J. Gastrointest. Cancer. 2003, 34, 87–94. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Weis, J.J.; Tedder, T.F.; Strominger, J.L.; Biro, P.A.; Fearon, D.T. Epstein–Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc. Natl. Acad. Sci. U. S. A. 1984, 81, 4510–4514. [Google Scholar] [CrossRef]
- Nemerow, G.R.; Mold, C.; Schwend, V.K.; Tollefson, V.; Cooper, N.R. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein–Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J. Virol. 1987, 61, 1416–1420. [Google Scholar]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein–Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Tanner, J.; Whang, Y.; Sample, J.; Sears, A.; Kieff, E. Soluble gp350/220 and deletion mutant glycoproteins block Epstein–Barr virus adsorption to lymphocytes. J. Virol. 1988, 62, 4452–4464. [Google Scholar]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt–Fletcher, L.M. Epstein–Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt–Fletcher, L.M. Epstein–Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef]
- Oda, T.; Imai, S.; Chiba, S.; Takada, K. Epstein–Barr virus lacking glycoprotein gp85 cannot infect B cells and epithelial cells. Virology 2000, 276, 52–58. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Diamond, M.E.; Sage, D.R.; Hayman, J. CD21–Dependent infection of an epithelial cell line, 293, by Epstein–Barr virus. J. Virol. 1999, 73, 2115–2125. [Google Scholar]
- Imai, S.; Nishikawa, J.; Takada, K. Cell–to–cell contact as an efficient mode of Epstein–Barr virus infection of diverse human epithelial cells. J. Virol. 1998, 72, 4371–4378. [Google Scholar]
- Sixbey, J.W.; Yao, Q.Y. Immunoglobulin A–induced shift of Epstein–Barr virus tissue tropism. Science 1992, 255, 1578–1580. [Google Scholar]
- Gan, Y.J.; Chodosh, J.; Morgan, A.; Sixbey, J.W. Epithelial cell polarization is a determinant in the infectious outcome of immunoglobulin A–mediated entry by Epstein–Barr virus. J. Virol. 1997, 71, 519–526. [Google Scholar]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt–Fletcher, L.M. Fusion of epithelial cells by epstein–barr virus proteins is triggered by binding of viral glycoproteins ghgl to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 20464–20469. [Google Scholar]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein–Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Berline, J.; Tugizov, S.M. The Epstein–Barr virus BMRF–2 protein facilitates virus attachment to oral epithelial cells. Virology 2008, 370, 430–442. [Google Scholar] [CrossRef]
- Xiao, J.; Palefsky, J.M.; Herrera, R.; Tugizov, S.M. Characterization of the Epstein–Barr virus glycoprotein BMRF–2. Virology 2007, 359, 382–396. [Google Scholar] [CrossRef]
- Haan, K.M.; Lee, S.K.; Longnecker, R. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein–Barr virus–induced membrane fusion. Virology 2001, 290, 106–114. [Google Scholar] [CrossRef]
- McShane, M.P.; Longnecker, R. Cell–surface expression of a mutated Epstein–Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 17474–17479. [Google Scholar] [CrossRef]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir–McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein–Barr virus. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16286–16291. [Google Scholar]
- Borza, C.M.; Hutt–Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein–Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef]
- Borza, C.M.; Morgan, A.J.; Turk, S.M.; Hutt–Fletcher, L.M. Use of gHgL for attachment of Epstein–Barr virus to epithelial cells compromises infection. J. Virol. 2004, 78, 5007–5014. [Google Scholar]
- Omerovic, J.; Lev, L.; Longnecker, R. The amino terminus of Epstein–Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J. Virol. 2005, 79, 12408–12415. [Google Scholar] [CrossRef]
- Wang, X.; Kenyon, W.J.; Li, Q.; Mullberg, J.; Hutt–Fletcher, L.M. Epstein–Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 1998, 72, 5552–5558. [Google Scholar]
- Wu, L.; Borza, C.M.; Hutt–Fletcher, L.M. Mutations of Epstein–Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J. Virol. 2005, 79, 10923–10930. [Google Scholar]
- Wu, L.; Hutt–Fletcher, L.M. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology 2007, 363, 148–155. [Google Scholar] [CrossRef]
- Yoshiyama, H.; Imai, S.; Shimizu, N.; Takada, K. Epstein–Barr virus infection to human gastric carcinoma cells : Implication of the existence of a new virus receptor different from CD21. J. Virol. 1997, 71, 5688–5691. [Google Scholar]
- Chang, Y.; Tung, C.H.; Huang, Y.T.; Lu, J.; Chen, J.Y.; Tsai, C.H. Requirement for cell–to–cell contact in Epstein–Barr virus infection of nasopharyngeal carcinoma cells and keratinocytes. J. Virol. 1999, 73, 8857–8866. [Google Scholar]
- Nanbo, A.; Terada, H.; Kachi, K.; Takada, K.; Matsuda, T. Roles of cell signaling pathways in cell–to–cell contact–mediated Epstein–Barr virus transmission. J. Virol. 2012, 86, 9285–9296. [Google Scholar] [CrossRef]
- Shannon–Lowe, C.; Rowe, M. Epstein–Barr virus infection of polarized epithelial cells via the basolateral surface by memory B cell–mediated transfer infection. PLoS Pathog. 2011, 7, e1001338. [Google Scholar] [CrossRef]
- Shannon–Lowe, C.D.; Neuhierl, B.; Baldwin, G.; Rickinson, A.B.; Delecluse, H.J. Resting B cells as a transfer vehicle for Epstein–Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7065–7070. [Google Scholar]
- Speck, P.; Longnecker, R. Infection of breast epithelial cells with Epstein–Barr virus via cell–to–cell contact. J. Natl. Cancer Inst. 2000, 92, 1849–1851. [Google Scholar] [CrossRef]
- Jolly, C.; Sattentau, Q.J. Retroviral spread by induction of virological synapses. Traffic 2004, 5, 643–650. [Google Scholar] [CrossRef]
- Li, Q.X.; Young, L.S.; Niedobitek, G.; Dawson, C.W.; Birkenbach, M.; Wang, F.; Rickinson, A.B. Epstein–Barr virus infection and replication in a human epithelial cell system. Nature 1992, 356, 347–350. [Google Scholar]
- Yoshiyama, H.; Shimizu, N.; Takada, K. Persistent Epstein–Barr virus infection in a human T–cell line : Unique program of latent virus expression. EMBO. J. 1995, 14, 3706–3711. [Google Scholar]
- Shimizu, N.; Yoshiyama, H.; Takada, K. Clonal propagation of Epstein–Barr virus (EBV) recombinants in EBV–negative Akata cells. J. Virol. 1996, 70, 7260–7263. [Google Scholar]
- Oh, S.T.; Seo, J.S.; Moon, U.Y.; Kang, K.H.; Shin, D.J.; Yoon, S.K.; Kim, W.H.; Park, J.G.; Lee, S.K. A naturally derived gastric cancer cell line shows latency I Epstein–Barr virus infection closely resembling EBV–associated gastric cancer. Virology 2004, 320, 330–336. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Chong, J.M.; Hayashi, Y.; Ikeno, R.; Arai, K.; Kitamura, M.; Koike, M.; Hirai, K.; Fukayama, M. Establishment and characterization of a human Epstein–Barr virus–associated gastric carcinoma in SCID mice. J. Virol. 1998, 72, 8321–8326. [Google Scholar]
- Chong, J.M.; Sakuma, K.; Sudo, M.; Osawa, T.; Ohara, E.; Uozaki, H.; Shibahara, J.; Kuroiwa, K.; Tominaga, S.; Hippo, Y.; Aburatani, H.; Funata, N.; Fukayama, M. Interleukin–1beta expression in human gastric carcinoma with Epstein–Barr virus infection. J. Virol. 2002, 76, 6825–6831. [Google Scholar]
- Nishikawa, J.; Imai, S.; Oda, T.; Kojima, T.; Okita, K.; Takada, K. Epstein–Barr virus promotes epithelial cell growth in the absence of EBNA2 and LMP1 expression. J. Virol. 1999, 73, 1286–1292. [Google Scholar]
- Iwakiri, D.; Eizuru, Y.; Tokunaga, M.; Takada, K. Autocrine growth of Epstein–Barr virus–positive gastric carcinoma cells mediated by an Epstein–Barr virus–encoded small RNA. Cancer Res. 2003, 63, 7062–7067. [Google Scholar]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of Epstein–Barr virus–encoded RNAs in Burkitt's lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar]
- Nanbo, A.; Yoshiyama, H.; Takada, K. Epstein–Barr virus–encoded poly(A)– RNA confers resistance to apoptosis mediated through Fas by blocking the PKR pathway in human epithelial intestine 407 cells. J. Virol. 2005, 79, 12280–12285. [Google Scholar] [CrossRef]
- Iwakiri, D.; Sheen, T.S.; Chen, J.Y.; Huang, D.P.; Takada, K. Epstein–Barr virus–encoded small RNA induces insulin–like growth factor 1 and supports growth of nasopharyngeal carcinoma–derived cell lines. Oncogene 2005, 24, 1767–1773. [Google Scholar] [CrossRef]
- Seto, E.; Yang, L.; Middeldorp, J.; Sheen, T.S.; Chen, J.Y.; Fukayama, M.; Eizuru, Y.; Ooka, T.; Takada, K. Epstein–Barr virus (EBV)–encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV–associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 2005, 76, 82–88. [Google Scholar] [CrossRef]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; Fukayama, M. Aivation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef]
- Shinozaki, A.; Sakatani, T.; Ushiku, T.; Hino, R.; Isogai, M.; Ishikawa, S.; Uozaki, H.; Takada, K.; Fukayama, M. Downregulation of microRNA–200 in EBV–associated gastric carcinoma. Cancer Res. 2010, 70, 4719–4727. [Google Scholar] [CrossRef]
- Herman, J.G. Hypermethylation of tumor suppressor genes in cancer. Semin. Can. Biol. 1999, 9, 359–367. [Google Scholar] [CrossRef]
- Li, Q.L.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; Kim, H.M.; Kim, W.J.; Yamamoto, H.; Yamashita, N.; Yano, T.; Ikeda, T.; Itohara, S.; Inazawa, J.; Abe, T.; Hagiwara, A.; Yamagishi, H.; Ooe, A.; Kaneda, A.; Sugimura, T.; Ushijima, T.; Bae, S.C.; Ito, Y. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar]
- Kaneda, A.; Kaminishi, M.; Yanagihara, K.; Sugimura, T.; Ushijima, T. Identification of silencing of nine genes in human gastric cancers. Cancer Res. 2002, 62, 6645–6650. [Google Scholar]
- Kusano, M.; Toyota, M.; Suzuki, H.; Akino, K.; Aoki, F.; Fujita, M.; Hosokawa, M.; Shinomura, Y.; Imai, K.; Tokino, T. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein–Barr virus. Cancer 2006, 106, 1467–1479. [Google Scholar] [CrossRef]
- Chang, M.S.; Uozaki, H.; Chong, J.M.; Ushiku, T.; Sakuma, K.; Ishikawa, S.; Hino, R.; Barua, R.R.; Iwasaki, Y.; Arai, K.; Fujii, H.; Nagai, H.; Fukayama, M. CpG island methylation status in gastric carcinoma with and without infection of Epstein–Barr virus. Clin. Cancer Res. 2006, 12, 2995–3002. [Google Scholar]
- Kaneda, A.; Matsusaka, K.; Aburatani, H.; Fukayama, M. Epstein–Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012, 72, 3445–50. [Google Scholar] [CrossRef]
- Ushiku, T.; Chong, J.M.; Uozaki, H.; Hino, R.; Chang, M.S.; Sudo, M.; Rani, B.R.; Sakuma, K.; Nagai, H.; Fukayama, M. p73 gene promoter methylation in Epstein–Barr virus–associated gastric carcinoma. Int. J. Cancer 2007, 120, 60–66. [Google Scholar]
- Kang, G.H.; Lee, S.; Cho, N.Y.; Gandamihardja, T.; Long, T.I.; Weisenberger, D.J.; Campan, M.; Laird, P.W. DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab. Invest. 2008, 88, 161–170. [Google Scholar] [CrossRef]
- Matsusaka, K.; Kaneda, A.; Nagae, G.; Ushiku, T.; Kikuchi, Y.; Hino, R.; Uozaki, H.; Seto, Y.; Takada, K.; Aburatani, H.; Fukayama, M. Classification of Epstein–Barr virus–positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011, 71, 7187–7197. [Google Scholar] [CrossRef]
- Kojima, T.; Shimazui, T.; Hinotsu, S.; Joraku, A.; Oikawa, T.; Kawai, K.; Horie, R.; Suzuki, H.; Nagashima, R.; Yoshikawa, K.; Michiue, T.; Asashima, M.; Akaza, H.; Uchida, K. Decreased expression of CXXC4 promotes a malignant phenotype in renal cell carcinoma by activating Wnt signaling. Oncogene 2009, 28, 297–305. [Google Scholar]
- Kawamata, H.; Kawai, K.; Kameyama, S.; Johnson, M.D.; Stetler–Stevenson, W.G.; Oyasu, R. Over–expression of tissue inhibitor of matrix metalloproteinases (TIMP1 and TIMP2) suppresses extravasation of pulmonary metastasis of a rat bladder carcinoma. Int. J. Cancer 1995, 63, 680–687. [Google Scholar]
- Kanai, Y.; Hui, A.M.; Sun, L.; Ushijima, S.; Sakamoto, M.; Tsuda, H.; Hirohashi, S. DNA hypermethylation at the D17S5 locus and reduced HIC–1 mRNA expression are associated with hepatocarcinogenesis. Hepatology 1999, 29, 703–709. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanai, Y.; Sakamoto, M.; Mizokami, M.; Ueda, R.; Hirohashi, S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis––A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 2000, 32, 970–979. [Google Scholar] [CrossRef]
- Luo, B.; Wang, Y.; Wang, X.F.; Liang, H.; Yan, L.P.; Huang, B.H.; Zhao, P. Expression of Epstein–Barr virus genes in EBV–associated gastric carcinomas. World J. Gastroenterol. 2005, 11, 629–633. [Google Scholar]
- Lennette, E.T.; Winberg, G.; Yadav, M.; Enblad, G.; Klein, G. Antibodies to LMP2A/2B in EBV–carrying malignancies. Eur. J. Cancer 1995, 31A, 1875–1878. [Google Scholar]
- Konishi, K.; Maruo, S.; Kato, H.; Takada, K. Role of Epstein–Barr virus–encoded latent membrane protein 2A on virus–induced immortalization and virus activation. J. Gen. Virol. 2001, 82, 1451–1456. [Google Scholar]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Nana–Sinkam, S.P.; Croce, C.M. Non–coding RNAs in cancer initiation and progression and as novel biomarkers. Mol. Oncol. 2011, 5, 483–491. [Google Scholar] [CrossRef]
- Raab–Traub, N.; Flynn, K. The structure of the termini of the Epstein–Barr virus as a marker of clonal cellular proliferation. Cell 1986, 47, 883–889. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; Tuschl, T. Identification of virus–encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar]
- Cai, X.; Schäfer, A.; Lu, S.; Bilello, J.P.; Desrosiers, R.C.; Edwards, R.; Raab–Traub, N.; Cullen, B.R. Epstein–Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006, 2, e23. [Google Scholar] [CrossRef]
- Robertson, E.S.; Tomkinson, B.; Kieff, E. An Epstein–Barr virus with a 58–kilobase–pair deletion that includes BARF0 transforms B lymphocytes in vitro. J. Virol. 1994, 68, 1449–1458. [Google Scholar]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulation of LMP1 protein expression by EBV–encoded microRNAs. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16164–16169. [Google Scholar]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jaker, C.; Hock, J.; Meister, G.; Grasser, F.A. Epstein–Barr virus–encoded microRNA miR–BART2 down–regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar]
- Lung, R.W.; Tong, J.H.; Sung, Y.M.; Leung, P.S.; Ng, D.C.; Chau, S.L.; Chan, A.W.; Ng, E.K.; Lo, K.W.; To, K.F. Modulation of LMP2A expression by a newly identified Epstein–Barr virus–encoded microRNA miR–BART22. Neoplasia. 2009, 11, 1174–1184. [Google Scholar]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein–Barr virus–encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; Lieberman, P.; Nishikura, K. Editing of Epstein–Barr virus–encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J. Biol. Chem. 2010, 285, 33358–33370. [Google Scholar]
- Marquitz, A.R.; Mathur, A.; Nam, C.S.; Raab–Traub, N. The Epstein–Barr Virus BART microRNAs target the pro–apoptotic protein Bim. Virology 2011, 412, 392–400. [Google Scholar] [CrossRef]
- Gottwein, E.; Corcoran, D.L.; Mukherjee, N.; Skalsky, R.L.; Hafner, M.; Nusbaum, J.D.; Shamulailatpam, P.; Love, C.L.; Dave, S.S.; Tuschl, T.; Ohler, U.; Cullen, B.R. Viral microRNA targetome of KSHV–infected primary effusion lymphoma cell lines. Cell Host. Microbe. 2011, 10, 515–526. [Google Scholar] [CrossRef]
- Riley, K.J.; Rabinowitz, G.S.; Yario, T.A.; Luna, J.M.; Darnell, R.B.; Steitz, J.A. EBV and human microRNAs co–target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012, 31, 2207–2221. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; Tuschl, T.; Ohler, U.; Cullen, B.R. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar]
- Seto, E.; Moosmann, A.; Gromminger, S.; Walz, N.; Grundhoff, A.; Hammerschmidt, W. Micro RNAs of Epstein–Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010, 6, e1001063. [Google Scholar] [CrossRef]
- Cameron, J.E.; Yin, Q.; Fewell, C.; Lacey, M.; McBride, J.; Wang, X.; Lin, Z.; Schaefer, B.C.; Flemington, E.K. Epstein–Barr virus latent membrane protein 1 induces cellular MicroRNA miR–146a, a modulator of lymphocyte signaling pathways. J. Virol. 2008, 82, 1946–1958. [Google Scholar] [CrossRef]
- Motsch, N.; Pfuhl, T.; Mrazek, J.; Barth, S.; Grasser, F.A. Epstein–Barr virus–encoded latent membrane protein 1 (LMP1) induces the expression of the cellular microRNA miR–146a. RNA Biol. 2007, 4, 131–137. [Google Scholar] [CrossRef]
- Gatto, G.; Rossi, A.; Rossi, D.; Kroening, S.; Bonatti, S.; Mallardo, M. Epstein–Barr virus latent membrane protein 1 trans–activates miR–155 transcription through the NF–kappaB pathway. Nucleic Acids Res. 2008, 36, 6608–6619. [Google Scholar]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, C.M. Pre–B cell proliferation and lymphoblastic leukemia/high–grade lymphoma in E(mu)–miR155 transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7024–7029. [Google Scholar]
- Godshalk, S.E.; Bhaduri–McIntosh, S.; Slack, F.J. Epstein–Barr virus–mediated dysregulation of human microRNA expression. Cell Cycle 2008, 7, 3595–3600. [Google Scholar] [CrossRef]
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; Deavers, M.T.; Mourad–Zeidan, A.; Wang, H.; Mueller, P.; Lenburg, M.E.; Gray, J.W.; Mok, S.; Birrer, M.J.; Lopez–Berestein, G.; Coleman, R.L.; Bar–Eli, M.; Sood, A.K. Dicer, Drosha, and outcomes in patients with ovarian cancer. New Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar]
- Yanai, H.; Nishikawa, J.; Mizugaki, Y.; Shimizu, N.; Takada, K.; Matsusaki, K.; Toda, T.; Matsumoto, Y.; Tada, M.; Okita, K. Endoscopic and pathologic features of Epstein–Barr virus–associated gastric carcinoma. Gastrointest. Endosc. 1997, 45, 236–242. [Google Scholar] [CrossRef]
- Nishikawa, J.; Yanai, H.; Mizugaki, Y.; Takada, K.; Tada, M.; Okita, K. Hypoechoic submucosal nodules: a sign of Epstein–Barr virus–associated early gastric cancer. J. Gastroenterol. Hepatol. 1998, 13, 585–590. [Google Scholar] [CrossRef]
- Lagergren, J.; Lindam, A.; Mason, R.M. Gastric stump cancer after distal gastrectomy for benign gastric ulcer in a population–based study. Int. J. Cancer 2012, 131, E1048–1052. [Google Scholar]
- Yamamoto, N.; Tokunaga, M.; Uemura, Y.; Tanaka, S.; Shirahama, H.; Nakamura, T.; Land, C.E.; Sato, E. Epstein–Barr virus and gastric remnant cancer. Cancer 1994, 74, 805–809. [Google Scholar] [CrossRef]
- Nishikawa, J.; Yanai, H.; Hirano, A.; Okamoto, T.; Nakamura, H.; Matsusaki, K.; Kawano, T.; Miura, O.; Okita, K. High prevalence of Epstein–Barr virus in gastric remnant carcinoma after Billroth–II reconstruction. Scand. J. Gastroenterol. 2002, 37, 825–829. [Google Scholar]
- Feng, W.H.; Hong, G.; Delecluse, H.J.; Kenney, S.C. Lytic induction therapy for Epstein–Barr virus–positive B–cell lymphomas. J. Virol. 2004, 78, 1893–1902. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Gore, S.D. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 2008, 45, 23–30. [Google Scholar] [CrossRef]
- Nishikawa, J.; Saito, M.; Kiyotoki, S.; Hamabe, K.; Okamoto, T.; Yanai, H.; Sakaida, I. Epstein–Barr virus associated gastric carcinoma. (in Japanese). Biotherapy 2010, 24, 429–434. [Google Scholar]
- van Beek, J.; zur Hausen, A.; Klein Kranenbarg, E.; van de Velde, C.J.; Middeldorp, J.M.; van den Brule, A.J.; Meijer, C.J.; Bloemena, E. EBV–positive gastric adenocarcinomas: a distinct clinicopathologic entity with a low frequency of lymph node involvement. J. Clin. Oncol. 2004, 22, 664–670. [Google Scholar] [CrossRef]
- Abe, H.; Maeda, D.; Hino, R.; Otake, Y.; Isogai, M.; Ushiku, A.S.; Matsusaka, K.; Kunita, A.; Ushiku, T.; Uozaki, H.; Tateishi, Y.; Hishima, T.; Iwasaki, Y.; Ishikawa, S.; Fukayama, M. ARID1A expression loss in gastric cancer: Pathway–dependent roles with and without Epstein–Barr virus infection and microsatellite instability. Virchows Arch. 2012, 461, 367–377. [Google Scholar]
- Wu, M.S.; Huang, S.P.; Chang, Y.T.; Shun, C.T.; Chang, M.C.; Lin, M.T.; Wang, H.P.; Lin, J.T. Tumor necrosis factor–alpha and interleukin–10 promoter polymorphisms in Epstein–Barr virus–associated gastric carcinoma. J. Infect. Dis. 2002, 185, 106–109. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated Gastric Carcinoma. Viruses 2012, 4, 3420-3439. https://doi.org/10.3390/v4123420
Iizasa H, Nanbo A, Nishikawa J, Jinushi M, Yoshiyama H. Epstein-Barr Virus (EBV)-associated Gastric Carcinoma. Viruses. 2012; 4(12):3420-3439. https://doi.org/10.3390/v4123420
Chicago/Turabian StyleIizasa, Hisashi, Asuka Nanbo, Jun Nishikawa, Masahisa Jinushi, and Hironori Yoshiyama. 2012. "Epstein-Barr Virus (EBV)-associated Gastric Carcinoma" Viruses 4, no. 12: 3420-3439. https://doi.org/10.3390/v4123420
APA StyleIizasa, H., Nanbo, A., Nishikawa, J., Jinushi, M., & Yoshiyama, H. (2012). Epstein-Barr Virus (EBV)-associated Gastric Carcinoma. Viruses, 4(12), 3420-3439. https://doi.org/10.3390/v4123420