MicroRNAs, Hepatitis C Virus, and HCV/HIV-1 Co-Infection: New Insights in Pathogenesis and Therapy


Abstract
:1. Introduction
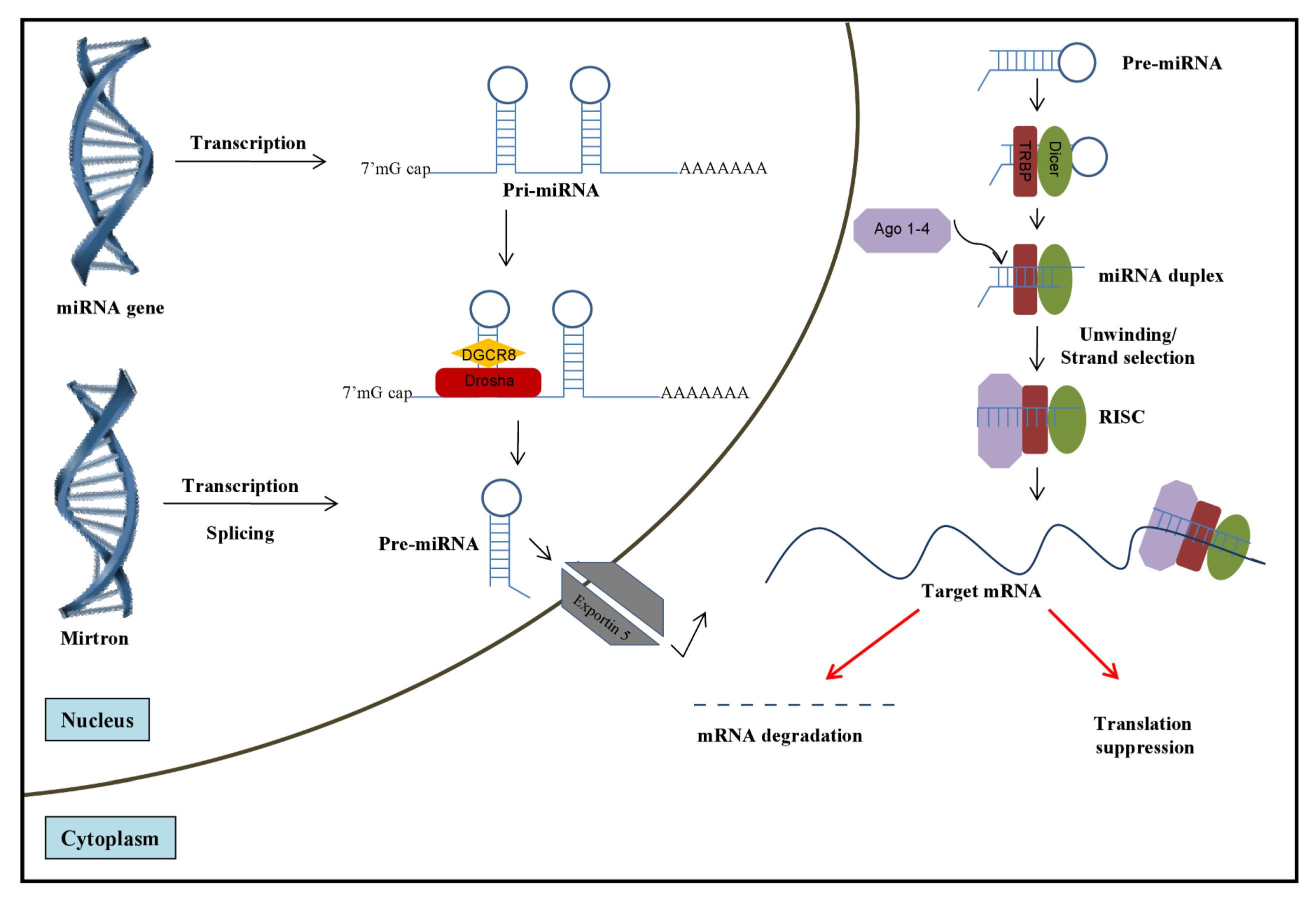
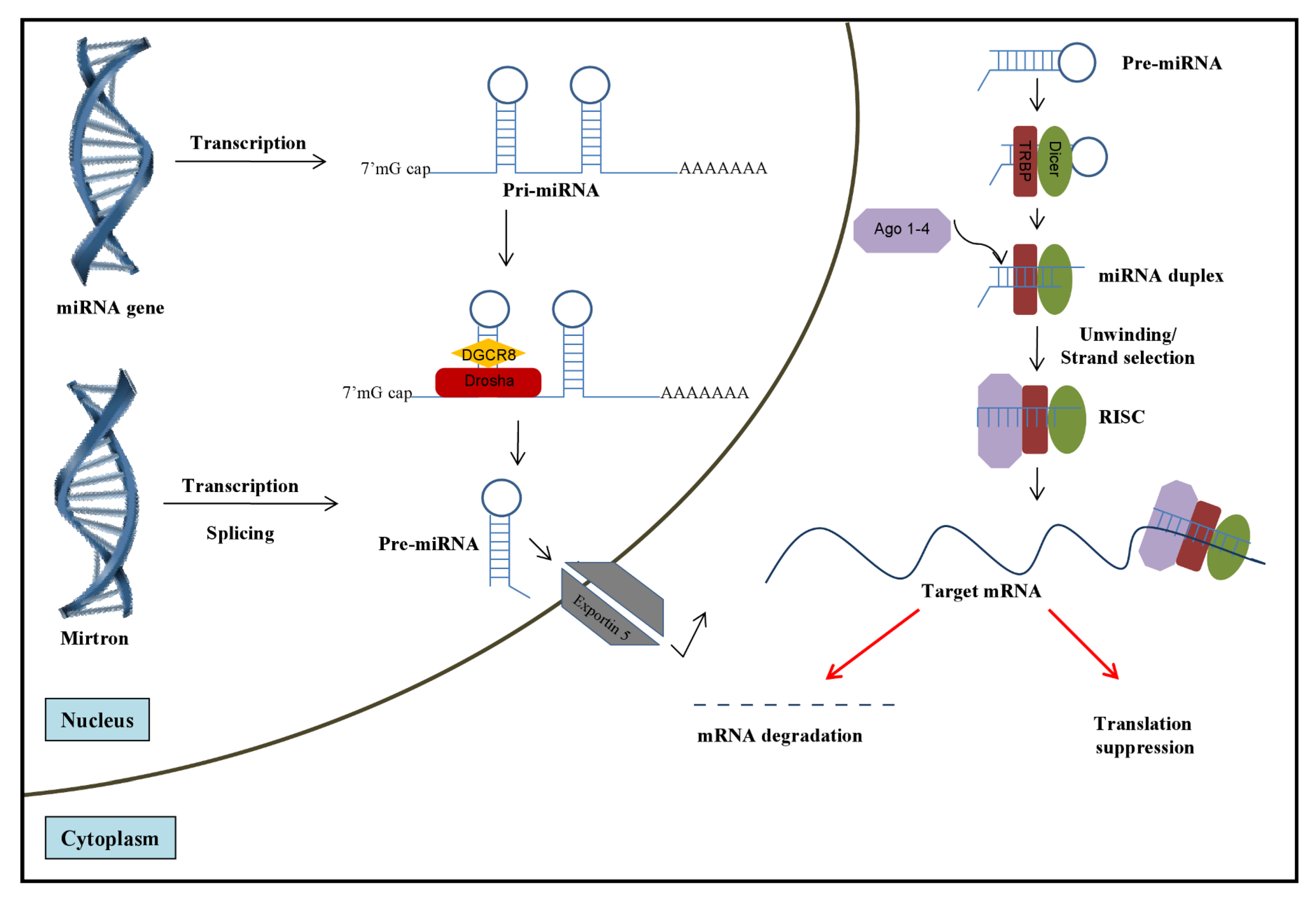
2. Role of miRNAs in HCV Infection
2.1. Role of Cellular miRNAs in Regulating HCV Infection

{kind=link}
{kind=link}
| microRNA | Mechanism | Target on HCV | Target on HCV-infected cells | Other effects in host and targets |
|---|---|---|---|---|
| miR-122 | Binding to HCV; Enhanced translation [29,59,60,61] | 5’UTR | Independent | Liver homeostasis; Tumor suppressor; Lipid metabolism; Anti-inflammatory [62,63,64] |
| miR-491 | Inhibition of the PI3K/Akt pathway [27] | NR | PI3K/Akt pathway | Apoptosis; Bcl-X(L) [65] |
| miR-141 | Inhibition of tumor-suppressor DCL-1[66] | NR | DCL-1[66] | Cancer; oxidative stress response [67] |
| Anti-HBV through peroxisome proliferator-activated receptor alpha (PPARA) [68] |
| microRNA | Relationship with IFN | Mechanism | Target on HCV | Target on HCV-infected cells | Antiviral effect on other viruses |
|---|---|---|---|---|---|
| miR-199a-3p | Independent of IFN [54] | Binding to HCV, [54] | 5’UTR IRES [54] | NR | HBV [24]; Herpesvirus and SFV [25] |
| miR-196 | Induced by IFN [53] | Binding to HCV and host protein [69] | NS5A | Batch1[69] | NR |
| miR-29 | Induced by IFN [53] | NR [70] | NR | Extracellular matrix proteins [70] | Influenza [26]; HIV-1 [71] |
| miR-let-7b | Regulates IFNβ expression [72] | Binding to HCV, no effects on translation [55] | 5’UTR, NS5B [55] | NR | NR |
| miRs-296,-351, -431, and -448 | Induced by IFN [53] | NR[53] | NR | NR | NR |
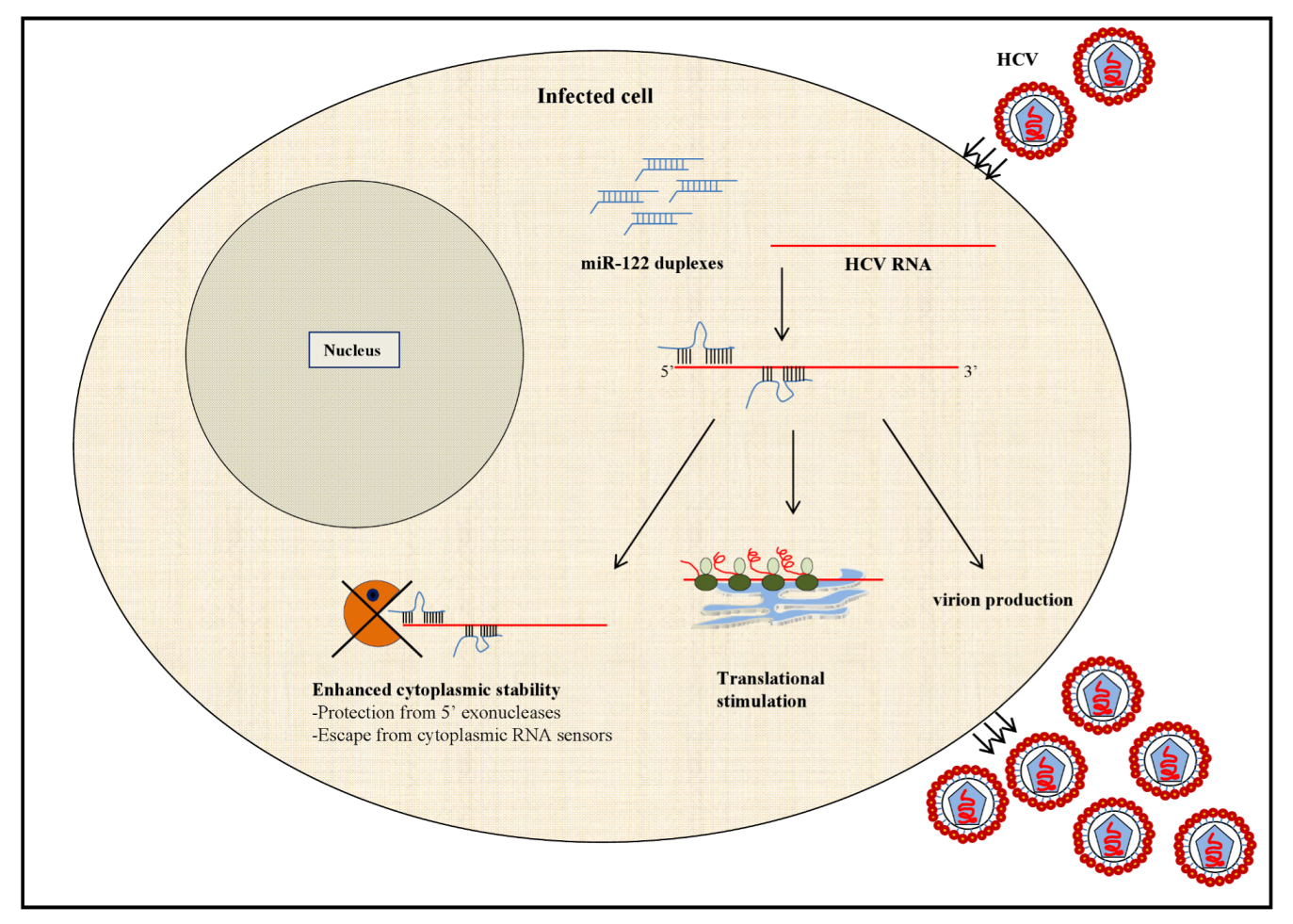
2.1.1. MicroRNA-122: A Master Regulator of HCV Infection?
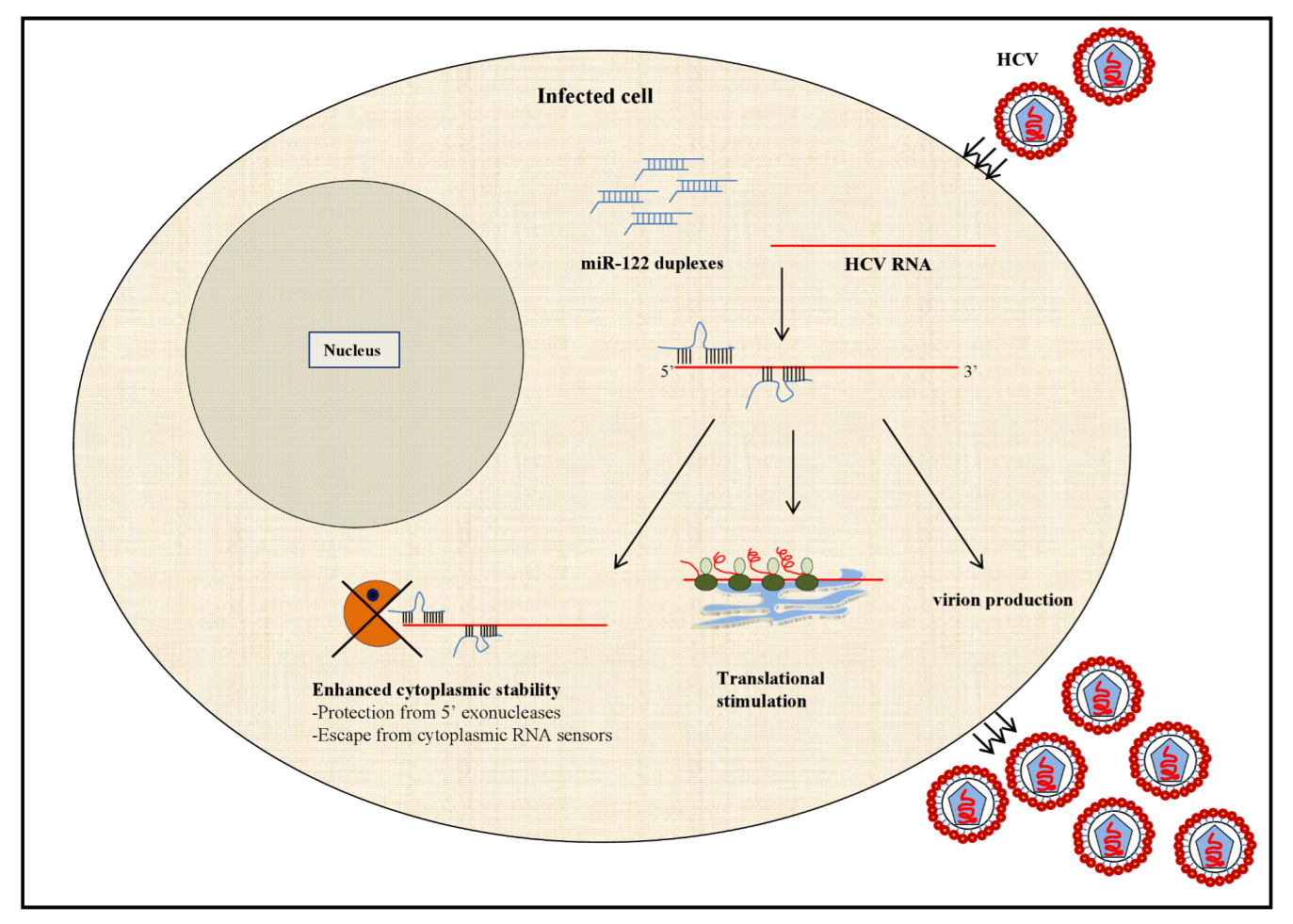
2.2. HCV Modulation of the Cellular miRNAome
3. Potential Role of miRNAs in HCV/HIV-1 Co-Infection
- MicroRNA-122. As mentioned above, miRNA-122 plays a critical role in HCV life cycle [29,30,60,140]. Interestingly, HIV-1 infection of T cell lines, significantly up-regulated the expression of miR-122 [141]. Of note, miR-122 is undetectable in uninfected, quiescent T cells [142], and although miR-122 is detected in human primary macrophages, its expression levels are significantly lower when compared to a human hepatoma cell line (Huh7.5) (Swaminathan, G. et al., unpublished data). Would an increase in miRNA-122 expression upon HIV-1 infection of CD4+ T cells, monocytes and macrophages explain the ability of HCV to infect cells beyond hepatocytes in co-infected patients?
- MicroRNA-29. As previously mentioned, miR-29 (miR 29a, b and c) has been shown to be down-regulated in hepatocytes upon HCV infection, and miR-29 over-expression significantly reduced HCV replication [70]. Interestingly, miR-29a was reported to bind HIV-1 3’UTR by base-pair complementarities and target the viral RNA to P-bodies for degradation [71]. Another independent report characterized that ectopic expression of miR-29a inhibits HIV-1 protein Nef, in addition to reducing viral infectivity [143]. Thus, it would be very interesting to explore the modulation of miR-29 upon HIV-1/HCV co-infection and to determine if over-expression of miR-29 could have dual-inhibitory activity in HIV-1/HCV co-infected cells.
- MicroRNA-149. miR-149 was shown to be up-regulated by about 10-fold upon HCV infection [88]. Intriguingly, miR-149 was predicted to bind to HIV-1 3’-UTR and to the HIV-1 protein Vpr [144]. In addition, HIV-1’s own miRNA denoted as hiv1-miR-H1 has been shown to down regulate the expression of cellular miR-149 through direct interaction [145].
- MicroRNA-199a. The role of miR-199a-3p in liver diseases remains controversial. MiR-199a-3p has been associated with progression to liver fibrosis [146] and other liver injuries [147]. However, other studies have found down-regulated expression of miR-199a/b in the majority of HCCs from their cohorts [148]. Interestingly, while over-expression of miR-199a was shown to inhibit HCV replication by binding to the viral 5’UTR [54], it was also reported that HIV-1 infection resulted in greater than two-fold increase in miR199a levels [149]. Based on these data, it would be interesting to explore the potential role of miR-199a in HCV/HIV-1 co-infection.
- MicroRNA-223. This “anti-HIV-1” microRNA was validated to inhibit HIV-1 replication by binding to HIV-1 3’UTR by several groups [135,136]. It is worth noting that miR-223 was found to be down-regulated by about 4.8-fold in HCV-induced hepatocellular carcinoma from HCV infected patients [95]. However, it has not been investigated whether miR-223 expression is down-regulated in the liver of HCV/HIV-1 co-infected patients with HCC, and if so, whether it would correlate with higher HIV-1 viral load.
- Let7 microRNAs. Let 7b and Let 7g microRNAs were shown to be significantly decreased in PBMCs and CD4+ T cells of HIV-1 infected patients as compared to healthy controls or patients who can naturally control HIV-1 infection (Long Term Non-Progressors, LTNP; and elite suppressors) [150]. Very recently, Let7b has been shown to bind to HCV protein NS5B and to the 5’UTR, and to significantly suppress HCV infection [55]. In a scenario where HIV-1 replication can suppress Let7b levels, it is plausible that a decrease in Let7b miRNA in the context of co-infection could potentially augment HCV replication.
4. Conclusions and Perspectives
Acknowledgments
Conflict of Interest
References
- Selbach, M.; Schwanhausser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008, 455, 58–63. [Google Scholar]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Baltimore, D. MicroRNAs and immunity: tiny players in a big field. Immunity 2007, 26, 133–137. [Google Scholar] [CrossRef]
- Salta, E.; De Strooper, B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet Neurol 2012, 11, 189–200. [Google Scholar]
- Schoof, C.R.; Botelho, E.L.; Izzotti, A.; Vasques Ldos, R. MicroRNAs in cancer treatment and prognosis. Am J Cancer Res 2012, 2, 414–433. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009, 19, 92–105. [Google Scholar]
- Gottwein, E.; Cullen, B.R. Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 2008, 3, 375–387. [Google Scholar] [CrossRef]
- Bazzini, A.A.; Lee, M.T.; Giraldez, A.J. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011, 12, 99–110. [Google Scholar]
- Cullen, B.R. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev 2011, 25, 1881–1894. [Google Scholar] [CrossRef]
- Houzet, L.; Jeang, K.T. MicroRNAs and human retroviruses. Biochim Biophys Acta 2011, 1809, 686–693. [Google Scholar]
- Carnero, E.; Sutherland, J.D.; Fortes, P. Adenovirus and miRNAs. Biochim Biophys Acta 2011, 1809, 660–667. [Google Scholar]
- Israsena, N.; Mahavihakanont, A.; Hemachudha, T. Rabies virus infection and microRNAs. Adv Virus Res 2011, 79, 329–344. [Google Scholar]
- Liu, W.H.; Yeh, S.H.; Chen, P.J. Role of microRNAs in hepatitis B virus replication and pathogenesis. Biochim Biophys Acta 2011, 1809, 678–685. [Google Scholar]
- Tuddenham, L.; Pfeffer, S. Roles and regulation of microRNAs in cytomegalovirus infection. Biochim Biophys Acta 2011, 1089, 613–622. [Google Scholar]
- Haasnoot, J.; Berkhout, B. RNAi and cellular miRNAs in infections by mammalian viruses. Methods Mol Biol 2011, 721, 23–41. [Google Scholar]
- Grundhoff, A.; Sullivan, C.S. Virus-encoded microRNAs. Virology 2011, 411, 325–343. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; Tuschl, T. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Rouha, H.; Thurner, C.; Mandl, C.W. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res 2010, 38, 8328–8337. [Google Scholar] [CrossRef]
- Shapiro, J. S.; Varble, A.; Pham, A.M.; Tenoever, B.R. Noncanonical cytoplasmic processing of viral microRNAs. RNA 2010, 16, 2068–2074. [Google Scholar] [CrossRef]
- Varble, A.; Chua, M.A.; Perez, J.T.; Manicassamy, B.; Garcia-Sastre, A.; tenOever, B. R. Engineered RNA viral synthesis of microRNAs. Proc Natl Acad Sci U S A 2010, 107, 11519–11524. [Google Scholar]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3' untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res 2012, 40, 2210–2223. [Google Scholar] [CrossRef]
- Bhanja Chowdhury, J.; Shrivastava, S.; Steele, R.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Hepatitis C Virus Infection Modulates Expression of Interferon Stimulatory Gene IFITM1 by Upregulating miR-130A. J Virol 2012, 86, 10221–10225. [Google Scholar] [CrossRef]
- Zhang, G.L.; Li, Y.X.; Zheng, S.Q.; Liu, M.; Li, X.; Tang, H. Suppression of hepatitis B virus replication by microRNA-199a-3p and microRNA-210. Antiviral Res 2010, 88, 169–175. [Google Scholar] [CrossRef]
- Santhakumar, D.; Forster, T.; Laqtom, N.N.; Fragkoudis, R.; Dickinson, P.; Abreu-Goodger, C.; Manakov, S.A.; Choudhury, N.R.; Griffiths, S.J.; Vermeulen, A.; Enright, A.J.; Dutia, B.; Kohl, A.; Ghazal, P.; Buck, A.H. Combined agonist-antagonist genome-wide functional screening identifies broadly active antiviral microRNAs. Proc Natl Acad Sci U S A 2010, 107, 13830–13835. [Google Scholar]
- Fang, J.; Hao, Q.; Liu, L.; Li, Y.; Wu, J.; Huo, X.; Zhu, Y. Epigenetic changes mediated by microRNA miR29 activate cyclooxygenase 2 and lambda-1 interferon production during viral infection. J Virol 2012, 86, 1010–1020. [Google Scholar]
- Ishida, H.; Tatsumi, T.; Hosui, A.; Nawa, T.; Kodama, T.; Shimizu, S.; Hikita, H.; Hiramatsu, N.; Kanto, T.; Hayashi, N.; Takehara, T. Alterations in microRNA expression profile in HCV-infected hepatoma cells: involvement of miR-491 in regulation of HCV replication via the PI3 kinase/Akt pathway. Biochem Biophys Res Commun 2011, 412, 92–97. [Google Scholar]
- Hemida, M.G.; Ye, X.; Zhang, H.M.; Hanson, P.J.; Liu, Z.; McManus, B.M.; Yang, D. MicroRNA-203 enhances Coxsackievirus B3 replication through targeting zinc finger protein-148. Cell Mol Life Sci 2012. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Jangra, R.K.; Yi, M.; Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol 2010, 84, 6615–6625. [Google Scholar] [CrossRef]
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005, 5, 558–567. [Google Scholar] [CrossRef]
- Micallef, J.M.; Kaldor, J.M.; Dore, G.J. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat 2006, 13, 34–41. [Google Scholar] [CrossRef]
- Alter, M.J. Epidemiology of hepatitis C in the West. Semin Liver Dis 1995, 15, 5–14. [Google Scholar]
- Munir, S.; Saleem, S.; Idrees, M.; Tariq, A.; Butt, S.; Rauff, B.; Hussain, A.; Badar, S.; Naudhani, M.; Fatima, Z.; Ali, M.; Ali, L.; Akram, M.; Aftab, M.; Khubaib, B.; Awan, Z. Hepatitis C treatment: current and future perspectives. Virol J 2010, 7, 296. [Google Scholar] [CrossRef]
- Asselah, T. Realize the advance in HCV treatment, but remain cautious. J Hepatol 2011, 55, 1457–1460. [Google Scholar] [CrossRef]
- Foote, B.S.; Spooner, L.M.; Belliveau, P.P. Boceprevir: a protease inhibitor for the treatment of chronic hepatitis C. Ann Pharmacother 2011, 45, 1085–1093. [Google Scholar] [CrossRef]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef]
- Reesink, H.W.; Janssen, H.L.A.; Zeuzem, E.; Lawitz, S.; Rodriguez-Torres, M.; Patel, K.; Chen, A.; Davis, C.; King, B.; Levin, A.; Hodges, M.R. Final Results: Randomized, Double-Blind, Placebo-Controlled Safety, Anti-Viral Proof-of-Concept Study of Miravirsen, an Oligonucleotide Targeting miR-122, in Treatment-Naive Patients with Genotype 1 Chronic HCV Infection. 47th International Liver Congress (EASL). Barcelona, Spain, April 18-22 2012. J. Hepatol. 2012, 56: pS1-S614. Suplement 2, A58. [Google Scholar]
- Quinn, S.R.; O'Neill, L.A. A trio of microRNAs that control Toll-like receptor signalling. Int Immunol 2011, 23, 421–425. [Google Scholar] [CrossRef]
- O'Neill, L.A.; Sheedy, F.J.; McCoy, C.E. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol 2011, 11, 163–175. [Google Scholar] [CrossRef]
- Swaminathan, G.; Rossi, F.; Sierra, L.J.; Gupta, A.; Navas-Martin, S.; Martin-Garcia, J. A Role for microRNA-155 Modulation in the Anti-HIV-1 Effects of Toll-Like Receptor 3 Stimulation in Macrophages. PLoS Pathog 2012, 8, e1002937. [Google Scholar] [CrossRef]
- Sulkowski, M.S.; Thomas, D.L. Hepatitis C in the HIV-infected patient. Clin Liver Dis 2003, 7, 179–194. [Google Scholar] [CrossRef]
- Low, E.; Vogel, M.; Rockstroh, J.; Nelson, M. Acute hepatitis C in HIV-positive individuals. AIDS Rev 2008, 10, 245–253. [Google Scholar]
- Koziel, M.J.; Peters, M.G. Viral hepatitis in HIV infection. N Engl J Med 2007, 356, 1445–1454. [Google Scholar] [CrossRef]
- Sulkowski, M.S.; Thomas, D.L. Hepatitis C in the HIV-Infected Person. Ann Intern Med 2003, 138, 197–207. [Google Scholar]
- Kim, A.Y.; Chung, R.T. Coinfection with HIV-1 and HCV--a one-two punch. Gastroenterology 2009, 137, 795–814. [Google Scholar] [CrossRef]
- Sulkowski, M.S.; Moore, R.D.; Mehta, S.H.; Chaisson, R.E.; Thomas, D.L. Hepatitis C and progression of HIV disease. JAMA 2002, 288, 199–206. [Google Scholar] [CrossRef]
- Dorrucci, M.; Valdarchi, C.; Suligoi, B.; Zaccarelli, M.; Sinicco, A.; Giuliani, M.; Vlahov, D.; Pezzotti, P.; Rezza, G. The effect of hepatitis C on progression to AIDS before and after highly active antiretroviral therapy. AIDS 2004, 18, 2313–2318. [Google Scholar]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat Rev Microbiol 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Farci, P. New insights into the HCV quasispecies and compartmentalization. Semin Liver Dis 2011, 31, 356–374. [Google Scholar]
- Argentini, C.; Genovese, D.; Dettori, S.; Rapicetta, M. HCV genetic variability: from quasispecies evolution to genotype classification. Future Microbiol. 2009, 4, 359–373. [Google Scholar]
- Wang, B.X.; Fish, E.N. The yin and yang of viruses and interferons. Trends Immunol 2012, 33, 190–197. [Google Scholar] [CrossRef]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef]
- Murakami, Y.; Aly, H.H.; Tajima, A.; Inoue, I.; Shimotohno, K. Regulation of the hepatitis C virus genome replication by miR-199a. J Hepatol 2009, 50, 453–460. [Google Scholar]
- Cheng, J.C.; Yeh, Y.J.; Tseng, C.P.; Hsu, S.D.; Chang, Y.L.; Sakamoto, N.; Huang, H.D. Let-7b is a novel regulator of hepatitis C virus replication. Cell Mol Life Sci 2012, 69, 2621–2633. [Google Scholar] [CrossRef]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; Taketo, M.M.; Yamamoto, M.; Igarashi, K. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J 2002, 21, 5216–5224. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Britigan, B.E.; Wen, F.; Icardi, M.; McCormick, M.L.; LaBrecque, D.R.; Voigt, M.; Brown, K.E.; Schmidt, W.N. Down-regulation of heme oxygenase-1 by hepatitis C virus infection in vivo and by the in vitro expression of hepatitis C core protein. J Infect Dis 2004, 190, 1109–1118. [Google Scholar] [CrossRef]
- Zhu, Z.; Wilson, A.T.; Mathahs, M.M.; Wen, F.; Brown, K.E.; Luxon, B.A.; Schmidt, W.N. Heme oxygenase-1 suppresses hepatitis C virus replication and increases resistance of hepatocytes to oxidant injury. Hepatology 2008, 48, 1430–1439. [Google Scholar] [CrossRef]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol 2012, 9, 137–142. [Google Scholar]
- Fehr, C.; Conrad, K.D.; Niepmann, M. Differential stimulation of hepatitis C virus RNA translation by microRNA-122 in different cell cycle phases. Cell Cycle 2012, 11, 277–285. [Google Scholar] [CrossRef]
- Villanueva, R.A.; Jangra, R.K.; Yi, M.; Pyles, R.; Bourne, N.; Lemon, S.M. miR-122 does not modulate the elongation phase of hepatitis C virus RNA synthesis in isolated replicase complexes. Antiviral Res 2010, 88, 119–123. [Google Scholar] [CrossRef]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; Hsu, M.T.; Wu, J.C.; Huang, H.D.; Shiao, M.S.; Hsiao, M.; Tsou, A.P. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest 2012, 122, 2884–2897. [Google Scholar]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; Mao, H.; Wei, M.; Clark, K.R.; Mendell, J.R.; Caligiuri, M.A.; Jacob, S.T.; Mendell, J.T.; Ghoshal, K. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest 2012, 122, 2871–2883. [Google Scholar] [CrossRef]
- Wen, J.; Friedman, J.R. miR-122 regulates hepatic lipid metabolism and tumor suppression. J Clin Invest 2012, 122, 2773–2776. [Google Scholar] [CrossRef]
- Nakano, H.; Miyazawa, T.; Kinoshita, K.; Yamada, Y.; Yoshida, T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-X(L) in colorectal cancer cells. Int J Cancer 2010, 127, 1072–1080. [Google Scholar]
- Banaudha, K.; Kaliszewski, M.; Korolnek, T.; Florea, L.; Yeung, M.L.; Jeang, K.T.; Kumar, A. MicroRNA silencing of tumor suppressor DLC-1 promotes efficient hepatitis C virus replication in primary human hepatocytes. Hepatology 2011, 53, 53–61. [Google Scholar] [CrossRef]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J. P.; Cottu, P.; Sastre-Garau, X.; Mechta-Grigoriou, F. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med 2011, 17, 1627–1635. [Google Scholar]
- Hu, W.; Wang, X.; Ding, X.; Li, Y.; Zhang, X.; Xie, P.; Yang, J.; Wang, S. MicroRNA-141 represses HBV replication by targeting PPARA. PLoS One 2012, 7, e34165. [Google Scholar]
- Hou, W.; Tian, Q.; Zheng, J.; Bonkovsky, H.L. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 2010, 51, 1494–1504. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y.; McCaffrey, A.P. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J Infect Dis 2011, 203, 1753–1762. [Google Scholar] [CrossRef]
- Nathans, R.; Chu, C.Y.; Serquina, A.K.; Lu, C.C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol Cell 2009, 34, 696–709. [Google Scholar] [CrossRef]
- Witwer, K.W.; Sisk, J.M.; Gama, L.; Clements, J.E. MicroRNA regulation of IFN-beta protein expression: rapid and sensitive modulation of the innate immune response. J Immunol 2010, 184, 2369–2376. [Google Scholar] [CrossRef]
- Wang, G.K.; Zhu, J.Q.; Zhang, J.T.; Li, Q.; Li, Y.; He, J.; Qin, Y.W.; Jing, Q. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J 2010, 31, 659–666. [Google Scholar] [CrossRef]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; Subramaniam, A.; Propp, S.; Lollo, B.A.; Freier, S.; Bennett, C.F.; Bhanot, S.; Monia, B.P. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 2006, 3, 87–98. [Google Scholar] [CrossRef]
- Elmen, J.; Lindow, M.; Schutz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjarn, M.; Hansen, H.F.; Berger, U.; Gullans, S.; Kearney, P.; Sarnow, P.; Straarup, E.M.; Kauppinen, S. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar]
- Henke, J.I.; Goergen, D.; Zheng, J.; Song, Y.; Schuttler, C.G.; Fehr, C.; Junemann, C.; Niepmann, M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 2008, 27, 3300–3310. [Google Scholar] [CrossRef]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res 2011, 39, 7716–7729. [Google Scholar] [CrossRef]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5' terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc Natl Acad Sci U S A 2011, 108, 3193–3198. [Google Scholar] [CrossRef]
- Chang, J.; Guo, J.T.; Jiang, D.; Guo, H.; Taylor, J.M.; Block, T.M. Liver-specific microRNA miR-122 enhances the replication of hepatitis C virus in nonhepatic cells. J Virol 2008, 82, 8215–8223. [Google Scholar] [CrossRef]
- Narbus, C.M.; Israelow, B.; Sourisseau, M.; Michta, M.L.; Hopcraft, S.E.; Zeiner, G.M.; Evans, M.J. HepG2 cells expressing miR-122 support the entire hepatitis C virus life cycle. J Virol 2011.
- Wilson, J.A.; Zhang, C.; Huys, A.; Richardson, C.D. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol 2011, 85, 2342–2350. [Google Scholar] [CrossRef]
- Kulkarni, M.; Ozgur, S.; Stoecklin, G. On track with P-bodies. Biochem Soc Trans 2010, 38, 242–251. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Kushima, Y.; Osugi, K.; Hijikata, M.; Maki, M.; Ikeda, M.; Kato, N. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol 2011, 85, 6882–6892. [Google Scholar]
- Jangra, R.K.; Yi, M.; Lemon, S.M. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 2010, 84, 6810–6824. [Google Scholar] [CrossRef]
- Hoffmann, T.W.; Duverlie, G.; Bengrine, A. MicroRNAs and hepatitis C virus: Toward the end of miR-122 supremacy. Virol J 2012, 9, 109. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu Rev Microbiol 2010, 64, 123–141. [Google Scholar] [CrossRef]
- Cullen, B.R. Five questions about viruses and microRNAs. PLoS Pathog 2010, 6, e1000787. [Google Scholar]
- Liu, X.; Wang, T.; Wakita, T.; Yang, W. Systematic identification of microRNA and messenger RNA profiles in hepatitis C virus-infected human hepatoma cells. Virology 2010, 398, 57–67. [Google Scholar]
- Peveling-Oberhag, J.; Crisman, G.; Schmidt, A.; Doring, C.; Lucioni, M.; Arcaini, L.; Rattotti, S.; Hartmann, S.; Piiper, A.; Hofmann, W.P.; Paulli, M.; Kuppers, R.; Zeuzem, S.; Hansmann, M.L. Dysregulation of global microRNA expression in splenic marginal zone lymphoma and influence of chronic hepatitis C virus infection. Leukemia 2012, 26, 1654–1662. [Google Scholar] [CrossRef]
- Bihrer, V.; Waidmann, O.; Friedrich-Rust, M.; Forestier, N.; Susser, S.; Haupenthal, J.; Welker, M.; Shi, Y.; Peveling-Oberhag, J.; Polta, A.; von Wagner, M.; Radeke, H.H.; Sarrazin, C.; Trojan, J.; Zeuzem, S.; Kronenberger, B.; Piiper, A. Serum microRNA-21 as marker for necroinflammation in hepatitis C patients with and without hepatocellular carcinoma. PLoS One 2011, 6, e26971. [Google Scholar]
- Bihrer, V.; Friedrich-Rust, M.; Kronenberger, B.; Forestier, N.; Haupenthal, J.; Shi, Y.; Peveling-Oberhag, J.; Radeke, H.H.; Sarrazin, C.; Herrmann, E.; Zeuzem, S.; Waidmann, O.; Piiper, A. Serum miR-122 as a Biomarker of Necroinflammation in Patients With Chronic Hepatitis C Virus Infection. Am J Gastroenterol 2011, 106, 1663–1669. [Google Scholar] [CrossRef]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating MicroRNAs in Patients with Chronic Hepatitis C and Non-Alcoholic Fatty Liver Disease. PLoS One 2011, 6, e23937. [Google Scholar]
- Grek, M.; Piekarska, A.; Bartkowiak, J.; Fendler, W.; Kuydowicz, J.; Wroblewski, P.; Paradowski, M.; Sidorkiewicz, M. Coordinated increase of miRNA-155 and miRNA-196b expression correlates with the detection of the antigenomic strand of hepatitis C virus in peripheral blood mononuclear cells. Int J Mol Med 2011, 28, 875–880. [Google Scholar]
- Bala, S.; Tilahun, Y.; Taha, O.; Alao, H.; Kodys, K.; Catalano, D.; Szabo, G. Increased microRNA-155 expression in the serum and peripheral monocytes in chronic HCV infection. J Transl Med 2012, 10, 151. [Google Scholar]
- Wong, Q.W.; Lung, R.W.; Law, P.T.; Lai, P.B.; Chan, K.Y.; To, K.F.; Wong, N. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology 2008, 135, 257–269. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, W.; Cheng, N.; Wang, K.; Li, B.; Jiang, X.; Sun, S. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012. [Google Scholar]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A. H. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem 2008, 283, 1026–1033. [Google Scholar]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Sarasin-Filipowicz, M.; Krol, J.; Markiewicz, I.; Heim, M.H.; Filipowicz, W. Decreased levels of microRNA miR-122 in individuals with hepatitis C responding poorly to interferon therapy. Nat Med 2009, 15, 31–33. [Google Scholar] [CrossRef]
- Marquez, R.T.; Bandyopadhyay, S.; Wendlandt, E.B.; Keck, K.; Hoffer, B.A.; Icardi, M.S.; Christensen, R.N.; Schmidt, W.N.; McCaffrey, A.P. Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab Invest 2010, 90, 1727–1736. [Google Scholar] [CrossRef]
- Sarasin-Filipowicz, M.; Oakeley, E.J.; Duong, F.H.; Christen, V.; Terracciano, L.; Filipowicz, W.; Heim, M.H. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A 2008, 105, 7034–7039. [Google Scholar]
- Varnholt, H.; Drebber, U.; Schulze, F.; Wedemeyer, I.; Schirmacher, P.; Dienes, H.P.; Odenthal, M. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology 2008, 47, 1223–1232. [Google Scholar]
- Augello, C.; Vaira, V.; Caruso, L.; Destro, A.; Maggioni, M.; Park, Y.N.; Montorsi, M.; Santambrogio, R.; Roncalli, M.; Bosari, S. MicroRNA profiling of hepatocarcinogenesis identifies C19MC cluster as a novel prognostic biomarker in hepatocellular carcinoma. Liver Int 2012, 32, 772–782. [Google Scholar] [CrossRef]
- Zhang, Q.; Pu, R.; Du, Y.; Han, Y.; Su, T.; Wang, H.; Cao, G. Non-coding RNAs in hepatitis B or C-associated hepatocellular carcinoma: potential diagnostic and prognostic markers and therapeutic targets. Cancer Lett 2012, 321, 1–12. [Google Scholar]
- Bouchard, M.J.; Navas-Martin, S. Hepatitis B and C virus hepatocarcinogenesis: lessons learned and future challenges. Cancer Lett 2011, 305, 123–143. [Google Scholar] [CrossRef]
- Ura, S.; Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Sunakozaka, H.; Sakai, Y.; Horimoto, K.; Kaneko, S. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology 2009, 49, 1098–1112. [Google Scholar]
- Bain, C.; Fatmi, A.; Zoulim, F.; Zarski, J. P.; Trepo, C.; Inchauspe, G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology 2001, 120, 512–524. [Google Scholar]
- Simone, O.; Tortorella, C.; Zaccaro, B.; Napoli, N.; Antonaci, S. Impairment of TLR7-dependent signaling in dendritic cells from chronic hepatitis C virus (HCV)-infected non-responders to interferon/ribavirin therapy. J Clin Immunol 2010, 30, 556–565. [Google Scholar]
- Villacres, M. C.; Literat, O.; DeGiacomo, M.; Du, W.; Frederick, T.; Kovacs, A. Defective response to Toll-like receptor 3 and 4 ligands by activated monocytes in chronic hepatitis C virus infection. J Viral Hepat 2008, 15, 137–144. [Google Scholar]
- Scagnolari, C.; Zingariello, P.; Vecchiet, J.; Selvaggi, C.; Racciatti, D.; Taliani, G.; Riva, E.; Pizzigallo, E.; Antonelli, G. Differential expression of interferon-induced microRNAs in patients with chronic hepatitis C virus infection treated with pegylated interferon alpha. Virol J 2010, 7, 311. [Google Scholar]
- Wang, Y.; Kato, N.; Jazag, A.; Dharel, N.; Otsuka, M.; Taniguchi, H.; Kawabe, T.; Omata, M. Hepatitis C virus core protein is a potent inhibitor of RNA silencing-based antiviral response. Gastroenterology 2006, 130, 883–892. [Google Scholar]
- Chen, W.; Zhang, Z.; Chen, J.; Zhang, J.; Wu, Y.; Huang, Y.; Cai, X.; Huang, A. HCV core protein interacts with Dicer to antagonize RNA silencing. Virus Res 2008, 133, 250–258. [Google Scholar]
- Clement, S.; Peyrou, M.; Sanchez-Pareja, A.; Bourgoin, L.; Ramadori, P.; Suter, D.; Vinciguerra, M.; Guilloux, K.; Pascarella, S.; Rubbia-Brandt, L.; Negro, F.; Foti, M. Down-regulation of phosphatase and tensin homolog by hepatitis C virus core 3a in hepatocytes triggers the formation of large lipid droplets. Hepatology 2011, 54, 38–49. [Google Scholar]
- Gummuluru, S.; Emerman, M. Advances in HIV molecular biology. AIDS 2002, 16 Suppl 4, S17–23. [Google Scholar]
- Sanchez-Quijano, A.; Andreu, J.; Gavilan, F.; Luque, F.; Abad, M.A.; Soto, B.; Munoz, J.; Aznar, J.M.; Leal, M.; Lissen, E. Influence of human immunodeficiency virus type 1 infection on the natural course of chronic parenterally acquired hepatitis C. Eur J Clin Microbiol Infect Dis 1995, 14, 949–953. [Google Scholar] [CrossRef]
- Sherman, K.E.; Rouster, S.D.; Chung, R.T.; Rajicic, N. Hepatitis C Virus prevalence among patients infected with Human Immunodeficiency Virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis 2002, 34, 831–837. [Google Scholar] [CrossRef]
- Darby, S.C.; Ewart, D.W.; Giangrande, P.L.; Spooner, R.J.; Rizza, C.R.; Dusheiko, G.M.; Lee, C.A.; Ludlam, C.A.; Preston, F.E. Mortality from liver cancer and liver disease in haemophilic men and boys in UK given blood products contaminated with hepatitis C. UK Haemophilia Centre Directors' Organisation. Lancet 1997, 350, 1425–1431. [Google Scholar]
- Bica, I.; McGovern, B.; Dhar, R.; Stone, D.; McGowan, K.; Scheib, R.; Snydman, D. R. Increasing mortality due to end-stage liver disease in patients with human immunodeficiency virus infection. Clin Infect Dis 2001, 32, 492–497. [Google Scholar]
- Gadalla, S.M.; Preiss, L.R.; Eyster, M.E.; Goedert, J.J. Correlates of high hepatitis C virus RNA load in a cohort of HIV-negative and HIV-positive individuals with haemophilia. J Viral Hepat 2011, 18, 161–169. [Google Scholar] [CrossRef]
- Cribier, B.; Schmitt, C.; Rey, D.; Uhl, G.; Lang, J.M.; Vetter, D.; Kirn, A.; Stoll-Keller, F. HIV increases hepatitis C viraemia irrespective of the hepatitis C virus genotype. Res Virol 1997, 148, 267–271. [Google Scholar] [CrossRef]
- Graham, C.S.; Koziel, M.J. Why should hepatitis C affect immune reconstitution in HIV-1-infected patients? Lancet 2000, 356, 1865–1866. [Google Scholar] [CrossRef]
- Taya, N.; Torimoto, Y.; Shindo, M.; Hirai, K.; Hasebe, C.; Kohgo, Y. Fas-mediated apoptosis of peripheral blood mononuclear cells in patients with hepatitis C. Br J Haematol 2000, 110, 89–97. [Google Scholar] [CrossRef]
- Rowland-Jones, S.L.; Pinheiro, S.; Kaul, R.; Hansasuta, P.; Gillespie, G.; Dong, T.; Plummer, F.A.; Bwayo, J.B.; Fidler, S.; Weber, J.; McMichael, A.; Appay, V. How important is the 'quality' of the cytotoxic T lymphocyte (CTL) response in protection against HIV infection? Immunol Lett 2001, 79, 15–20. [Google Scholar] [CrossRef]
- Deeks, S.G.; Kitchen, C.M.; Liu, L.; Guo, H.; Gascon, R.; Narvaez, A.B.; Hunt, P.; Martin, J.N.; Kahn, J.O.; Levy, J.; McGrath, M.S.; Hecht, F.M. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004, 104, 942–947. [Google Scholar] [CrossRef]
- Hazenberg, M.D.; Otto, S.A.; van Benthem, B.H.; Roos, M.T.; Coutinho, R.A.; Lange, J.M.; Hamann, D.; Prins, M.; Miedema, F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS 2003, 17, 1881–1888. [Google Scholar] [CrossRef]
- Rasmussen, A.L.; Wang, I.M.; Shuhart, M.C.; Proll, S.C.; He, Y.; Cristescu, R.; Roberts, C.; Carter, V.S.; Williams, C.M.; Diamond, D.L.; Bryan, J.T.; Ulrich, R.; Korth, M.J.; Thomassen, L.V.; Katze, M.G. Chronic immune activation is a distinguishing feature of liver and PBMC gene signatures from HCV/HIV coinfected patients and may contribute to hepatic fibrogenesis. Virology 2012, 430, 43–52. [Google Scholar] [CrossRef]
- Kottilil, S.; Yan, M.Y.; Reitano, K.N.; Zhang, X.; Lempicki, R.; Roby, G.; Daucher, M.; Yang, J.; Cortez, K.J.; Ghany, M.; Polis, M.A.; Fauci, A.S. Human immunodeficiency virus and hepatitis C infections induce distinct immunologic imprints in peripheral mononuclear cells. Hepatology 2009, 50, 34–45. [Google Scholar] [CrossRef]
- Verani, A.; Gras, G.; Pancino, G. Macrophages and HIV-1: dangerous liaisons. Mol Immunol 2005, 42, 195–212. [Google Scholar]
- Marukian, S.; Jones, C.T.; Andrus, L.; Evans, M.J.; Ritola, K.D.; Charles, E.D.; Rice, C.M.; Dustin, L.B. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008, 48, 1843–1850. [Google Scholar] [CrossRef]
- Bouffard, P.; Hayashi, P.H.; Acevedo, R.; Levy, N.; Zeldis, J.B. Hepatitis C virus is detected in a monocyte/macrophage subpopulation of peripheral blood mononuclear cells of infected patients. J Infect Dis 1992, 166, 1276–1280. [Google Scholar] [CrossRef]
- Laskus, T.; Radkowski, M.; Wang, L.F.; Jang, S.J.; Vargas, H.; Rakela, J. Hepatitis C virus quasispecies in patients infected with HIV-1: correlation with extrahepatic viral replication. Virology 1998, 248, 164–171. [Google Scholar] [CrossRef]
- Swaminathan, S.; Murray, D.D.; Kelleher, A.D. The role of microRNAs in HIV-1 pathogenesis and therapy. AIDS 2012, 26, 1325–1334. [Google Scholar] [CrossRef]
- Sun, G.; Li, H.; Wu, X.; Covarrubias, M.; Scherer, L.; Meinking, K.; Luk, B.; Chomchan, P.; Alluin, J.; Gombart, A.F.; Rossi, J.J. Interplay between HIV-1 infection and host microRNAs. Nucleic Acids Res 2012, 40, 2181–2196. [Google Scholar] [CrossRef]
- Han, Y.; Siliciano, R.F. Keeping quiet: microRNAs in HIV-1 latency. Nat Med 2007, 13, 1138–1140. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med 2007, 13, 1241–1247. [Google Scholar]
- Wang, X.; Ye, L.; Hou, W.; Zhou, Y.; Wang, Y.J.; Metzger, D.S.; Ho, W.Z. Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 2009, 113, 671–674. [Google Scholar] [CrossRef]
- Swaminathan, S.; Zaunders, J.; Wilkinson, J.; Suzuki, K.; Kelleher, A.D. Does the presence of anti-HIV miRNAs in monocytes explain their resistance to HIV-1 infection? Blood 2009, 113, 5029–5030, author reply 5030-5021.. [Google Scholar] [CrossRef]
- Sung, T.L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Chiang, K.; Sung, T.L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J Virol 2012, 86, 3244–3252. [Google Scholar] [CrossRef]
- Goergen, D.; Niepmann, M. Stimulation of Hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res 2012, 167, 343–352. [Google Scholar]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; Reynes, J.; Corbeau, P.; Jeang, K. T.; Benkirane, M. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Manfe, V.; Biskup, E.; Rosbjerg, A.; Kamstrup, M.; Skov, A.G.; Lerche, C.M.; Lauenborg, B.T.; Odum, N.; Gniadecki, R. miR-122 regulates p53/Akt signalling and the chemotherapy-induced apoptosis in cutaneous T-cell lymphoma. PLoS One 2012, 7, e29541. [Google Scholar]
- Ahluwalia, J.K.; Khan, S.Z.; Soni, K.; Rawat, P.; Gupta, A.; Hariharan, M.; Scaria, V.; Lalwani, M.; Pillai, B.; Mitra, D.; Brahmachari, S.K. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 2008, 5, 117. [Google Scholar] [CrossRef]
- Hariharan, M.; Scaria, V.; Pillai, B.; Brahmachari, S.K. Targets for human encoded microRNAs in HIV genes. Biochem Biophys Res Commun 2005, 337, 1214–1218. [Google Scholar] [CrossRef]
- Kaul, D.; Ahlawat, A.; Gupta, S.D. HIV-1 genome-encoded hiv1-mir-H1 impairs cellular responses to infection. Mol Cell Biochem 2009, 323, 143–148. [Google Scholar] [CrossRef]
- Murakami, Y.; Toyoda, H.; Tanaka, M.; Kuroda, M.; Harada, Y.; Matsuda, F.; Tajima, A.; Kosaka, N.; Ochiya, T.; Shimotohno, K. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One 2011, 6, e16081. [Google Scholar]
- Dolganiuc, A.; Petrasek, J.; Kodys, K.; Catalano, D.; Mandrekar, P.; Velayudham, A.; Szabo, G. MicroRNA expression profile in Lieber-DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol Clin Exp Res 2009, 33, 1704–1710. [Google Scholar]
- Romilda, C.; Marika, P.; Alessandro, S.; Enrico, L.; Marina, B.; Andromachi, K.; Umberto, C.; Giacomo, Z.; Claudia, M.; Massimo, R.; Fabio, F. Oxidative DNA damage correlates with cell immortalization and mir-92 expression in hepatocellular carcinoma. BMC Cancer 2012, 12, 177. [Google Scholar]
- Hayes, A.M.; Qian, S.; Yu, L.; Boris-Lawrie, K. Tat RNA silencing suppressor activity contributes to perturbation of lymphocyte miRNA by HIV-1. Retrovirology 2011, 8, 36. [Google Scholar] [CrossRef]
- Swaminathan, S.; Suzuki, K.; Seddiki, N.; Kaplan, W.; Cowley, M.J.; Hood, C.L.; Clancy, J.L.; Murray, D.D.; Mendez, C.; Gelgor, L.; Anderson, B.; Roth, N.; Cooper, D.A.; Kelleher, A.D. Differential regulation of the Let-7 family of microRNAs in CD4+ T cells alters IL-10 expression. J Immunol 2012, 188, 6238–6246. [Google Scholar]
- Russo, A.; Potenza, N. Antiviral effects of human microRNAs and conservation of their target sites. FEBS Lett 2011, 585, 2551–2555. [Google Scholar]
- Wittmann, J.; Jack, H.M. Serum microRNAs as powerful cancer biomarkers. Biochim Biophys Acta 2010, 1806, 200–207. [Google Scholar]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res 2011, 39, 7223–7233. [Google Scholar]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.L.; Schmittgen, T.D.; Nana-Sinkam, S.P.; Jarjoura, D.; Marsh, C.B. Detection of microRNA expression in human peripheral blood microvesicles. PLoS One 2008, 3, e3694. [Google Scholar]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; Tait, J.F.; Tewari, M. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 2011, 108, 5003–5008. [Google Scholar]
- Gupta, A.; Nagilla, P.; Le, H.S.; Bunney, C.; Zych, C.; Thalamuthu, A.; Bar-Joseph, Z.; Mathavan, S.; Ayyavoo, V. Comparative expression profile of miRNA and mRNA in primary peripheral blood mononuclear cells infected with human immunodeficiency virus (HIV-1). PLoS One 2011, 6, e22730. [Google Scholar]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G. J.; Zanesi, N.; Crawford, M.; Ozer, G.H.; Wernicke, D.; Alder, H.; Caligiuri, M.A.; Nana-Sinkam, P.; Perrotti, D.; Croce, C.M. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012, 109, E2110–2116. [Google Scholar]
- Lehmann, S.M.; Kruger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; Habbel, P.; Kalin, R.; Franzoni, E.; Rybak, A.; Nguyen, D.; Veh, R.; Ninnemann, O.; Peters, O.; Nitsch, R.; Heppner, F.L.; Golenbock, D.; Schott, E.; Ploegh, H.L.; Wulczyn, F.G.; Lehnardt, S. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 2012, 15, 827–835. [Google Scholar] [CrossRef]
- Bang, C.; Thum, T. Exosomes: New players in cell-cell communication. Int J Biochem Cell Biol 2012, 44, 2060–2064. [Google Scholar]
- Vickers, K.C.; Remaley, A.T. Lipid-based carriers of microRNAs and intercellular communication. Curr Opin Lipidol 2012, 23, 91–97. [Google Scholar] [CrossRef]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; Milosevic, J.; Tkacheva, O.A.; Divito, S.J.; Jordan, R.; Lyons-Weiler, J.; Watkins, S.C.; Morelli, A.E. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; JH, O. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2012, in press. [Google Scholar]
- Kosaka, N.; Ochiya, T. Unraveling the Mystery of Cancer by Secretory microRNA: Horizontal microRNA Transfer between Living Cells. Front Genet 2011, 2, 97. [Google Scholar]
- Henderson, M.C.; Azorsa, D.O. The genomic and proteomic content of cancer cell-derived exosomes. Front Oncol 2012, 2, 38. [Google Scholar]
- Lages, E.; Ipas, H.; Guttin, A.; Nesr, H.; Berger, F.; Issartel, J.P. MicroRNAs: molecular features and role in cancer. Front Biosci 2012, 17, 2508–2540. [Google Scholar] [CrossRef]
- Chiba, M.; Kimura, M.; Asari, S. Exosomes secreted from human colorectal cancer cell lines contain mRNAs, microRNAs and natural antisense RNAs, that can transfer into the human hepatoma HepG2 and lung cancer A549 cell lines. Oncol Rep 2012, 28, 1551–1558. [Google Scholar]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2012.
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun 2011, 2, 282. [Google Scholar] [CrossRef]
- Pan, Q.; Ramakrishnaiah, V.; Henry, S.; Fouraschen, S.; de Ruiter, P.E.; Kwekkeboom, J.; Tilanus, H.W.; Janssen, H.L.; van der Laan, L.J. Hepatic cell-to-cell transmission of small silencing RNA can extend the therapeutic reach of RNA interference (RNAi). Gut 2012, 61, 1330–1339. [Google Scholar] [CrossRef]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012. [Epub ahead of print]. [Google Scholar]
- Lai, C.P.; Breakefield, X.O. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front Physiol 2012, 3, 228. [Google Scholar]
- Delabranche, X.; Berger, A.; Boisrame-Helms, J.; Meziani, F. Microparticles and infectious diseases. Med Mal Infect 2012, 42, 335–343. [Google Scholar]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci U S A 2010, 107, 20370–20375. [Google Scholar]
- Wurdinger, T.; Gatson, N.N.; Balaj, L.; Kaur, B.; Breakefield, X.O.; Pegtel, D.M. Extracellular vesicles and their convergence with viral pathways. Adv Virol 2012, 2012, 767694. [Google Scholar]
- Kadiu, I.; Narayanasamy, P.; Dash, P.K.; Zhang, W.; Gendelman, H.E. Biochemical and Biologic Characterization of Exosomes and Microvesicles as Facilitators of HIV-1 Infection in Macrophages. J Immunol 2012, 189, 744–754. [Google Scholar] [CrossRef]
- Masciopinto, F.; Giovani, C.; Campagnoli, S.; Galli-Stampino, L.; Colombatto, P.; Brunetto, M.; Yen, T.S.; Houghton, M.; Pileri, P.; Abrignani, S. Association of hepatitis C virus envelope proteins with exosomes. Eur J Immunol 2004, 34, 2834–2842. [Google Scholar] [CrossRef]
- Pegtel, D.M.; van de Garde, M.D.; Middeldorp, J.M. Viral miRNAs exploiting the endosomal-exosomal pathway for intercellular cross-talk and immune evasion. Biochim Biophys Acta 2011, 1809, 715–721. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gupta, A.; Swaminathan, G.; Martin-Garcia, J.; Navas-Martin, S. MicroRNAs, Hepatitis C Virus, and HCV/HIV-1 Co-Infection: New Insights in Pathogenesis and Therapy. Viruses 2012, 4, 2485-2513. https://doi.org/10.3390/v4112485
Gupta A, Swaminathan G, Martin-Garcia J, Navas-Martin S. MicroRNAs, Hepatitis C Virus, and HCV/HIV-1 Co-Infection: New Insights in Pathogenesis and Therapy. Viruses. 2012; 4(11):2485-2513. https://doi.org/10.3390/v4112485
Chicago/Turabian StyleGupta, Archana, Gokul Swaminathan, Julio Martin-Garcia, and Sonia Navas-Martin. 2012. "MicroRNAs, Hepatitis C Virus, and HCV/HIV-1 Co-Infection: New Insights in Pathogenesis and Therapy" Viruses 4, no. 11: 2485-2513. https://doi.org/10.3390/v4112485
APA StyleGupta, A., Swaminathan, G., Martin-Garcia, J., & Navas-Martin, S. (2012). MicroRNAs, Hepatitis C Virus, and HCV/HIV-1 Co-Infection: New Insights in Pathogenesis and Therapy. Viruses, 4(11), 2485-2513. https://doi.org/10.3390/v4112485