TRIM5 Acts as More Than a Retroviral Restriction Factor

Abstract
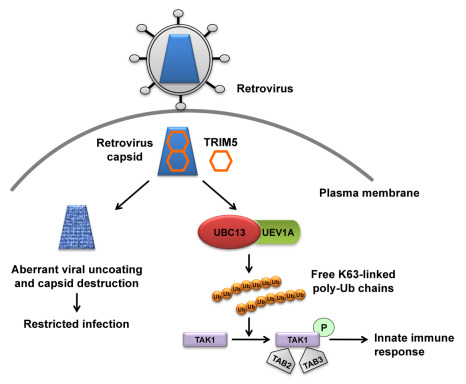
:
{kind=link}
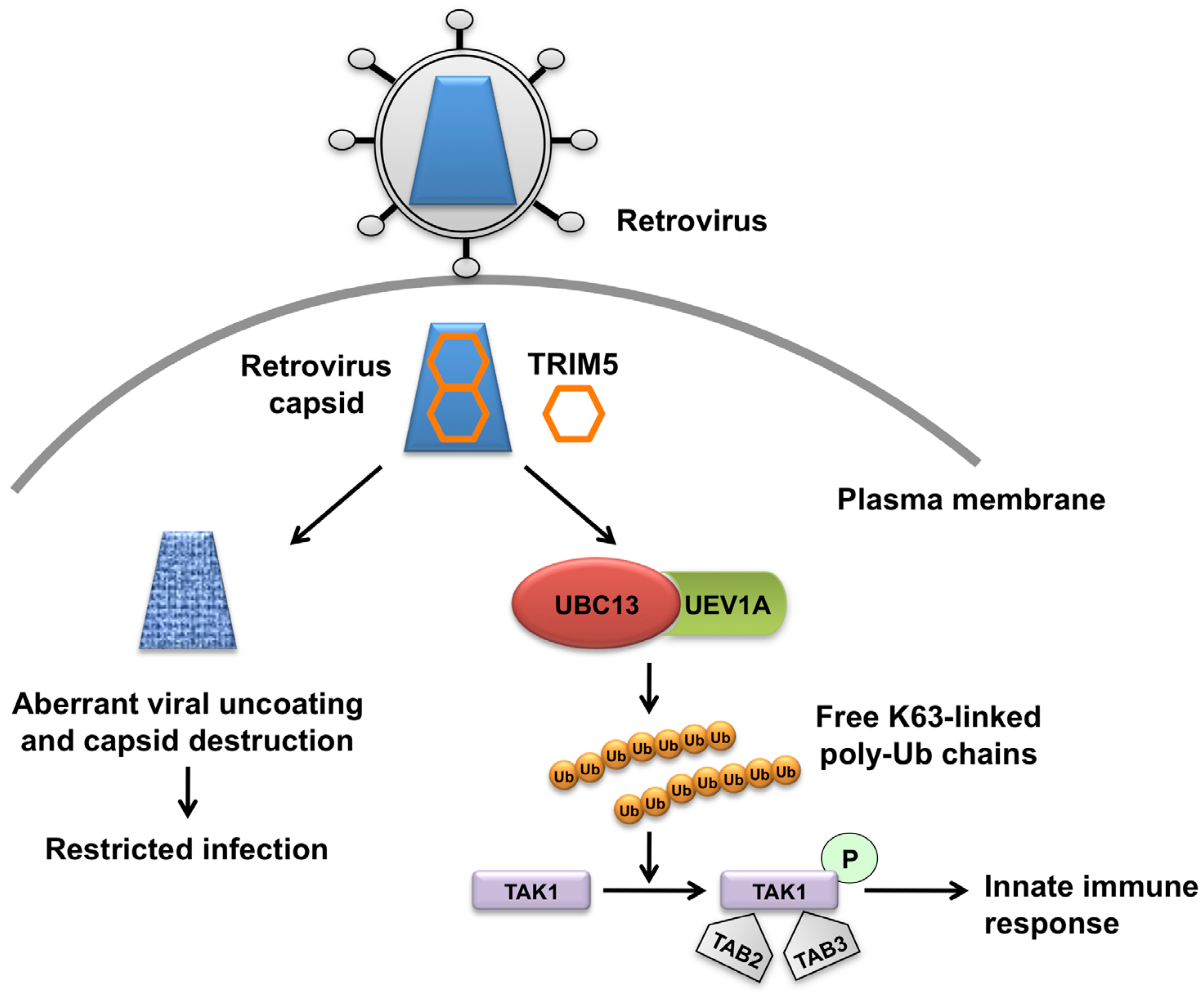{kind=link}
Acknowledgments
Conflict of Interest
References and Notes
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. Trim5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Goff, S.P. Host restriction factors blocking retroviral replication. Annu. Rev. Genet. 2008, 42, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Nisole, S.; Stoye, J.P.; Saib, A. Trim family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Sastri, J.; Campbell, E.M. Recent insights into the mechanism and consequences of TRIM5alpha retroviral restriction. AIDS Res. Hum. Retroviruses 2011, 27, 231–238. [Google Scholar] [CrossRef]
- Sebastian, S.; Luban, J. Trim5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2005, 2, 40. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 534–539. [Google Scholar] [CrossRef]
- Diaz-Griffero, F.; Qin, X.R.; Hayashi, F.; Kigawa, T.; Finzi, A.; Sarnak, Z.; Lienlaf, M.; Yokoyama, S.; Sodroski, J. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 2009, 83, 10737–10751. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Griffero, F.; Kar, A.; Lee, M.; Stremlau, M.; Poeschla, E.; Sodroski, J. Comparative requirements for the restriction of retrovirus infection by TRIM5alpha and TRIMCyp. Virology 2007, 369, 400–410. [Google Scholar] [CrossRef]
- Li, X.; Gold, B.; O'HUigin, C.; Diaz-Griffero, F.; Song, B.; Si, Z.; Li, Y.; Yuan, W.; Stremlau, M.; Mische, C.; et al. Unique features of TRIM5alpha among closely related human TRIM family members. Virology 2007, 360, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J. Virol. 2006, 80, 2855–2862. [Google Scholar] [CrossRef]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin a retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Tareen, S.U.; Emerman, M. Human Trim5alpha has additional activities that are uncoupled from retroviral capsid recognition. Virology 2011, 409, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. TRIM30 alpha negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 2008, 9, 369–377. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Hatziioannou, T.; Zhang, F.; Cowan, S.; Bieniasz, P.D. Restriction of human immunodeficiency virus type 1 by trim-cypa occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 2005, 79, 15567–15572. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic hiv infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Hogstad, B.; Wang, Y.; Levy, D.E.; Unutmaz, D.; Littman, D.R. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 2010, 467, 214–217. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Silva, S.; Wu, L. TRIM5 Acts as More Than a Retroviral Restriction Factor. Viruses 2011, 3, 1204-1209. https://doi.org/10.3390/v3071204
De Silva S, Wu L. TRIM5 Acts as More Than a Retroviral Restriction Factor. Viruses. 2011; 3(7):1204-1209. https://doi.org/10.3390/v3071204
Chicago/Turabian StyleDe Silva, Suresh, and Li Wu. 2011. "TRIM5 Acts as More Than a Retroviral Restriction Factor" Viruses 3, no. 7: 1204-1209. https://doi.org/10.3390/v3071204
APA StyleDe Silva, S., & Wu, L. (2011). TRIM5 Acts as More Than a Retroviral Restriction Factor. Viruses, 3(7), 1204-1209. https://doi.org/10.3390/v3071204