Rev Variation during Persistent Lentivirus Infection


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetic Variation Alters Rev Activity
3. Longitudinal Studies of Rev Variation in Vivo
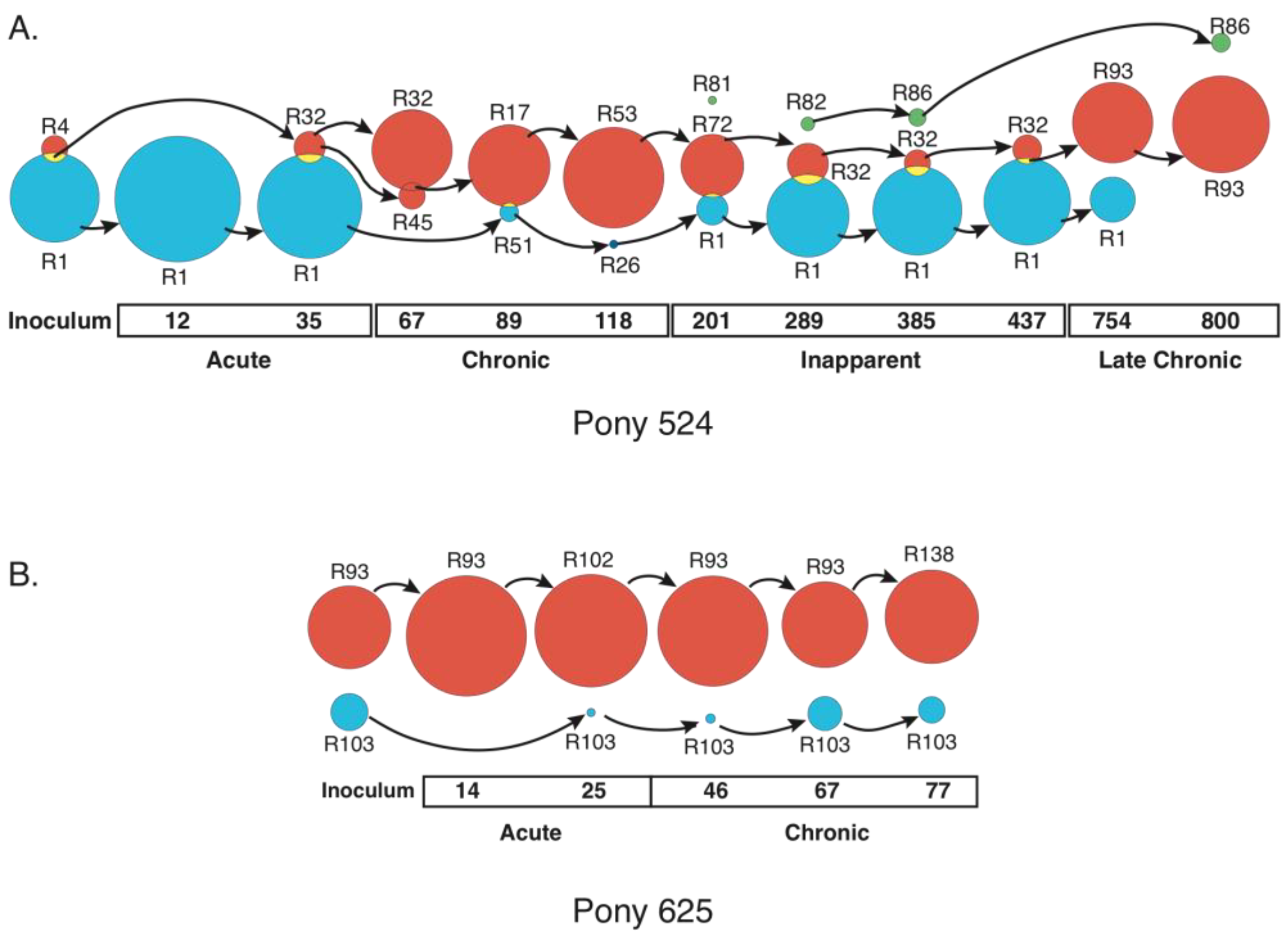
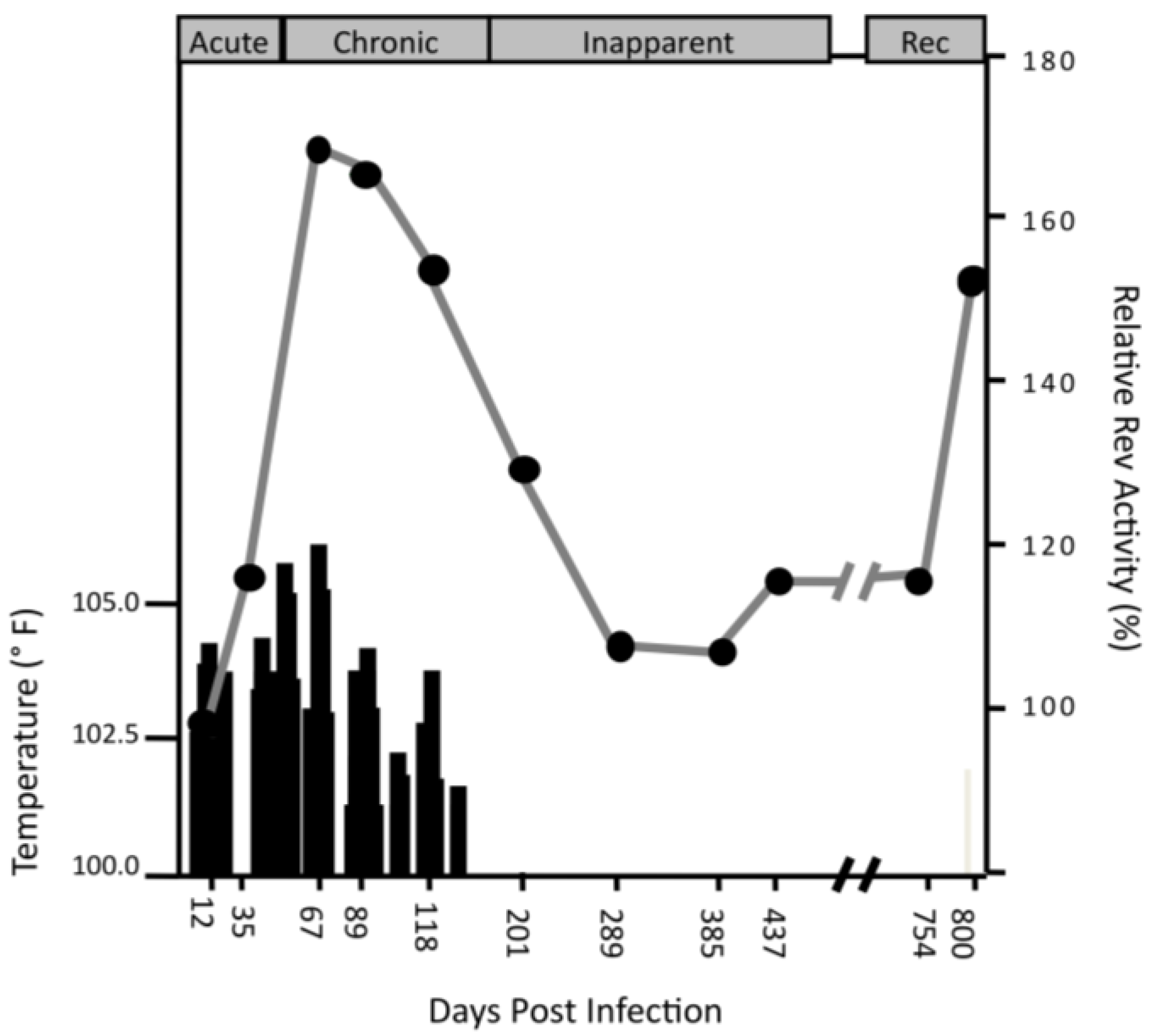
3.1. Rev Subpopulations Evolve During Disease Progression, Sometimes Coexisting or Recurring
3.2. Sub-population Rev Variants Differ in Nuclear Export Activity and Replication Phenotype
4. Selection in an Overlapping Reading Frame
4.1. Model for Estimating Selection in Overlapping Reading Frames
4.2. Evidence for Selection in Both Rev and TM
5. Genetic Determinants of Rev Phenotype
6. Rev Variation and Immune Evasion
7. Summary and Conclusions
Acknowledgements
References and Notes
- Alexandersen, S.; Carpenter, S. Characterization of variable regions in the envelope and S3 open reading frame of equine infectious anemia virus. J. Virol. 1991, 65, 4255–4262. [Google Scholar] [CrossRef] [PubMed]
- Leroux, C.; Issel, C.J.; Montelaro, R.C. Novel and dynamic evolution of equine infectious anemia virus genomic quasispecies associated with sequential disease cycles in an experimentally infected pony. J. Virol. 1997, 71, 9627–9639. [Google Scholar] [CrossRef] [PubMed]
- Belshan, M.; Harris, M.E.; Shoemaker, A.E.; Hope, T.J.; Carpenter, S. Biological characterization of Rev variation in equine infectious anemia virus. J. Virol. 1998, 72, 4421–4426. [Google Scholar] [CrossRef] [PubMed]
- Bobbitt, K.R.; Addo, M.M.; Altfeld, M.; Filzen, T.; Onafuwa, A.A.; Walker, B.D.; Collins, K.L. Rev activity determines sensitivity of HIV-1-infected primary T cells to CTL killing. Immunity 2003, 18, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Addo, M.M.; Altfeld, M.; Rosenberg, E.S.; Eldridge, R.L.; Philips, M.N.; Habeeb, K.; Khatri, A.; Brander, C.; Robbins, G.K.; Mazzara, G.P.; et al. The HIV-1 regulatory proteins Tat and Rev are frequently targeted by cytotoxic T lymphocytes derived from HIV-1-infected individuals. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Mealey, R.H.; Zhang, B.; Leib, S.R.; Littke, M.H.; McGuire, T.C. Epitope specificity is critical for high and moderate avidity cytotoxic T lymphocytes associated with control of viral load and clinical disease in horses with equine infectious anemia virus. Virology 2003, 313, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.; Støvlbaek, J. A maximum-likelihood approach to analyzing nonoverlapping and overlapping reading frames. J. Mol. Evol. 1995, 40, 181–189. [Google Scholar] [CrossRef]
- Belshan, M.; Baccam, P.; Oaks, J.L.; Sponseller, B.A.; Murphy, S.C.; Cornette, J.; Carpenter, S. Genetic and biological variation in equine infectious anemia virus Rev correlates with variable stages of clinical disease in an experimentally infected pony. Virology 2001, 279, 185–200. [Google Scholar] [CrossRef]
- Baccam, P.; Thompson, R.J.; Li, Y.; Sparks, W.O.; Belshan, M.; Dorman, K.S.; Wannemuehler, Y.; Oaks, J.L.; Cornette, J.L.; Carpenter, S. Subpopulations of equine infectious anemia virus Rev coexist in vivo and differ in phenotype. J. Virol. 2003, 77, 12122–12131. [Google Scholar] [CrossRef]
- Sparks, W.O.; Dorman, K.S.; Liu, S.; Carpenter, S. Naturally arising point mutations in non-essential domains of equine infectious anemia virus Rev alter Rev-dependent nuclear-export activity. J. Gen. Virol. 2008, 89, 1043–1048. [Google Scholar] [CrossRef]
- Belshan, M.; Park, G.S.; Bilodeau, P.; Stoltzfus, C.M.; Carpenter, S. Binding of equine infectious anemia virus Rev to an exon splicing enhancer mediates alternative splicing and nuclear export of viral mRNAs. Mol. Cell. Biol. 2000, 20, 3550–3557. [Google Scholar] [CrossRef] [PubMed]
- Baccam, P.; Thompson, R.J.; Fedrigo, O.; Carpenter, S.; Cornette, J.L. PAQ: Partition Analysis of Quasispecies. Bioinformatics 2001, 17, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Yasunaga, T. Molecular evolution of mRNA: A method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J. Mol. Evol. 1980, 16, 23–36. [Google Scholar] [CrossRef]
- Hurst, L.D.; Pal, C. Evidence for purifying selection acting on silent sites in BRCA1. Trends Genet. 2001, 17, 62–65. [Google Scholar] [CrossRef]
- Holmes, E.C.; Lipman, D.J.; Zamarin, D.; Yewdell, J.W. Comment on “Large-scale sequence analysis of avian influenza isolates”. Science 2006, 313, 1573. [Google Scholar] [CrossRef]
- Pavesi, A. Pattern of nucleotide substitution in the overlapping nonstructural genes of influenza A virus and implication for the genetic diversity of the H5N1 subtype. Gene 2007, 402, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Ngandu, N.K.; Scheffler, K.; Moore, P.; Woodman, Z.; Martin, D.; Seoighe, C. Extensive purifying selection acting on synonymous sites in HIV-1 Group M sequences. Virol. J. 2008, 5, 160. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Wu, C.I.; Luo, C.C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol. Biol. Evol. 1985, 2, 150–174. [Google Scholar]
- Pedersen, A.M.; Jensen, J.L. A dependent-rates model and an MCMC-based methodology for the maximum-likelihood analysis of sequences with overlapping reading frames. Mol. Biol. Evol. 2001, 18, 763–776. [Google Scholar] [CrossRef]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Sabath, N.; Landan, G.; Graur, D. A method for the simultaneous estimation of selection intensities in overlapping genes. PLoS One 2008, 3, e3996. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-C.; Dorman, K.S. Twisted Sisters: Disentangling Selection in Overlapping Reading Frames. In Proceedings of the Joint Statistical Meetings, Washington, DC, USA, 4 August 2009. [Google Scholar]
- Lee, J.H.; Murphy, S.C.; Belshan, M.; Sparks, W.O.; Wannemuehler, Y.; Liu, S.; Hope, T.J.; Dobbs, D.; Carpenter, S. Characterization of functional domains of equine infectious anemia virus Rev suggests a bipartite RNA-binding domain. J. Virol. 2006, 80, 3844–3852. [Google Scholar] [CrossRef]
- Fridell, R.A.; Partin, K.M.; Carpenter, S.; Cullen, B.R. Identification of the activation domain of equine infectious anemia virus Rev. J. Virol. 1993, 67, 7317–23. [Google Scholar] [CrossRef]
- Mancuso, V.A.; Hope, T.J.; Zhu, L.; Derse, D.; Phillips, T.; Parslow, T.G. Posttranscriptional effector domains in the Rev proteins of feline immunodeficiency virus and equine infectious anemia virus. J. Virol. 1994, 68, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.E.; Gontarek, R.R.; Derse, D.; Hope, T.J. Differential requirements for alternative splicing and nuclear export functions of equine infectious anemia virus Rev protein. Mol. Cell. Biol. 1998, 18, 3889–3899. [Google Scholar] [CrossRef]
- Iversen, A.K.N.; Shpaer, E.G.; Rodrigo, A.G.; Hirsch, M.S.; Walker, B.D.; Sheppard, H.W.; Merigan, T.C.; Mullins, J.I. Persistence of attenuated Rev genes in a human immunodeficiency virus type 1-infected asymptomatic individual. J. Virol. 1995, 69, 5743–5753. [Google Scholar] [CrossRef]
- Hua, J.; Caffrey, J.J.; Cullen, B.R. Functional consequences of natural sequence variation in the activation domain of HIV-1 Rev. Virology 1996, 222, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Chiavaroli, L.; Wesselingh, S.L.; Gorry, P.R. Persistence of attenuated HIV-1 Rev alleles in an epidemiologically linked cohort of long-term survivors infected with nef-deleted virus. Retrovirology 2007, 4, 43. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2011 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carpenter, S.; Chen, W.-C.; Dorman, K.S. Rev Variation during Persistent Lentivirus Infection. Viruses 2011, 3, 1-11. https://doi.org/10.3390/v3010001
Carpenter S, Chen W-C, Dorman KS. Rev Variation during Persistent Lentivirus Infection. Viruses. 2011; 3(1):1-11. https://doi.org/10.3390/v3010001
Chicago/Turabian StyleCarpenter, Susan, Wei-Chen Chen, and Karin S. Dorman. 2011. "Rev Variation during Persistent Lentivirus Infection" Viruses 3, no. 1: 1-11. https://doi.org/10.3390/v3010001
APA StyleCarpenter, S., Chen, W.-C., & Dorman, K. S. (2011). Rev Variation during Persistent Lentivirus Infection. Viruses, 3(1), 1-11. https://doi.org/10.3390/v3010001