Cidofovir Activity against Poxvirus Infections


Abstract
:
1. Introduction
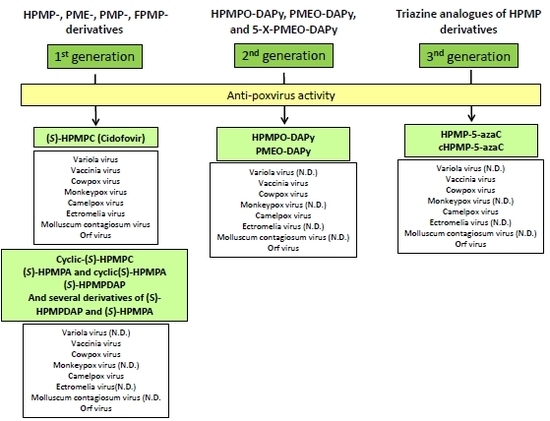
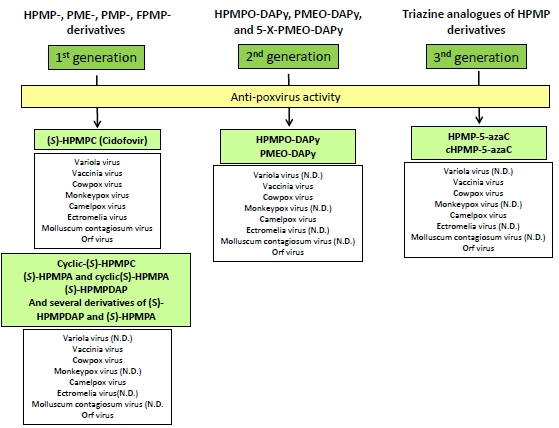
2. Anti-poxvirus Activity in vitro
3. Intracellular Metabolism
3.1. Cellular Uptake
3.2. Activation and Intracellular Half-life
4. Mechanism of Antiviral Activity

5. Mechanism of Resistance
6. In Vivo Efficacy in Animal Models for Poxvirus Infections
7. Dosage and Administration
8. Pharmacology
9. Safety
10. Clinical Efficacy of CDV in the Treatment of Poxvirus Infections
11. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal model | Route of CDV administration | Evidence for Efficacy | Reference |
|---|---|---|---|
| Intranasal or aerosolized CPXV infection in mice | Subcutaneous | One inoculation of 100 mg/kg CDV on day 0, 2, or 4 resulted in 90–100% survival. Treatment on day 0 reduced peak pulmonary virus titers 10- to 100-fold, reduced the severity of viral pneumonitis, and prevented pulmonary hemorrhage. The same dose on day -6 to 2 protected 80%–100% of infected mice, whereas one inoculation on day -16 to -8 or day 3 to 6 was partially protective. | [100] |
| A single dose of CDV 100 mg/kg administered on the day of infection was 80 to 100% protective when given on day 0, 1, 2, 3, or 4 after infection. Lung virus titers (determined on day 4 of the infection) were significantly reduced in groups treated on day 0, 1, or 2. | [101] | ||
| Intranasal | Single treatment of 20 and 40 mg/kg CDV given up to three days after virus inoculation resulted in 80–90% protection. A single 40 mg/kg treatment of infected mice given 1 or 2 days after infection significantly decreased virus titer in lungs and nose/sinus compared to the placebo group. | [102] | |
| Single treatment of 5–40 mg/kg 24 h after virus exposure afforded 80–100% protection from lethal infection and significant reduction in viral titers in the lung tissue. | [103] | ||
| Aerosol | Single treatment of 1-5mg/kg CDV at 1 day before or 2 h after infection showed efficacy as measured by changes in body and lung weight, lung viral titers, pulmonary pathology and survival. | [104] | |
| Treatment with CDV was successful in protecting against lethal intranasal cowpox infection. A dose of drug in the range of 0.5–5 mg/kg was protective when given before (day -1), the day of infection (day 0) or after infection (day +1 or +2); an 80% survival rate was observed when mice were treated 2 days before challenge. | [105] | ||
| Intraperitoneal | Treatment for five consecutive days starting 24 h after infection with CDV at 30 mg/kg per dose was 100% effective in preventing mortality. | [106] | |
| Treatment with CDV at 160, 80 or 40mg/kg as a single dose 24 h after virus exposure afforded 100% protection from lethal infection and significant reduction in viral titers in the lung tissue. | [103] | ||
| Treatment with CDV at 6.7 mg/kg once daily for five days beginning 24 or 48 h after viral inoculation, afforded 100% protection from lethal infection. Even when treatment was started 72 h post-infection, CDV treatment resulted in 66% protection. | [107] | ||
| CDV at a dose of 5 or 10 mg/kg administered daily beginning on day -5, -3, or -1 through day 0 (the viral inoculation day) afforded 93% protection from lethal infection. A single dose of 30 mg/kg CDV -1 before viral inoculation or 1 day post-infection resulted in 100% protection. | [107] | ||
| CDV at 100 mg/kg once a day on days 1 and 2 after infection resulted in 100 protection from lethal infection and significant reduction in viral titers in the lungs. | [108] | ||
| CDV (100 mg/kg/day for two days starting 24 h after virus exposure) led to survival and suppression of tissue virus titers in animals suffering from either a lethal upper respiratory tract infection or both upper and lower respiratory tract infection. | [109] | ||
| Intranasal or aerosolized VACV infection in mice | Intraperitoneal | Treatment with CDV at 160, 80 or 40mg/kg as a single dose 24 h after virus exposure afforded 70% protection from lethal infection. | [103] |
| Treatment with CDV at 5 mg/kg once daily for five days beginning 24, 48, or 72 h after viral inoculation, afforded 73–100% protection from lethal infection. | [107] | ||
| CDV treatment (100 mg/kg/day i.p. for two days) significantly reduced mortality and viral titers in lungs. | [108] | ||
| Intranasal | Single treatment of 5–40 mg/kg 24 h after virus exposure afforded 70–80% protection from lethal infection and significant reduction in viral titers in lung tissue. | [103] | |
| Intraperitoneal CPXV infection in mice | Intraperitoneal | CDV at 100 mg/kg once a day on days 1 and 2 after infection resulted in significant protection from lethal infection and significant reduction in viral titers in the lungs. | [108] |
| CDV at 25 or 100 mg/kg once one day before infection resulted in significant reduction of virus replication in several organs. | [110] | ||
| Intraperitoneal VACV infection in mice | Intraperitoneal | CDV treatment (100 mg/kg/day i.p. for two days) afforded 60% protection from lethal disease and significant reduction in viral titers. | [108] |
| Intravenous, intranasal, or intraperitoneal VACV infection in SCID mice | Subcutaneous | Following administration of CDV, at doses ranging from 1mg/kg/day for five days to 20 mg/kg/twice a week, death could be significantly delayed. | [111] |
| Intranasal CPXV infection in SCID mice | Subcutaneous | Treatment every three days with CDV (100 mg/kg) through day 30 of the infection resulted in significant delay in the time of death but final mortality | [101] |
| Treatment with CDV at 100 mg/kg/dose starting on day 0 and repeating the dose every three days resulted in delay of time of death but not in protection from lethal infection. | [100] | ||
| Cutaneous CPXV infection in hairless mice | Intraperitoneal | Hairless mice treated with 50 mg/kg beginning +24 h after viral inoculation, 3× weekly for one week, had significantly reduced lesion-day AUCs (area under the curve) and mean peak lesion scores. | [112] |
| Cutaneous VACV infection in hairless mice or athymic nude mice | Intraperitoneal / topical | Hairless mice treated with 50 mg/kg of CDV (starting 24 h post-inoculation of the virus once a day for seven days) or topically with 5% CDV 3× a day for seven days) had a significantly lower lesion-day AUCs (area under the curve) and mean peak lesion scores. | [112] |
| Topical treatment with 1% CDV, initiated at the day of infection or at day 1 p.i. during 5 days, completely protected against virus-induced cutaneous lesions and against associated mortality. Systemic treatment with CDV (100 mg/kg 3× or 5× per week initiated at 14 days post-infection caused healing and regression of the lesions. | [113] | ||
| CDV at 100 mg/kg once a day on days 1 and 2 after infection resulted in protection from lethal infection and significant reduction in viral titers in the lungs. | [108] | ||
| Topical treatment with 1%-CDV cream (twice daily for seven days) of immunocompromised mice (hair-less mice treated with cyclophosphamide) was much more effective in reducing the severity of primary lesions and the number of satellite lesions than was systemic CDV treatment (100 mg/kg/day, given every three days). Both forms of treatment delayed death. Topical drug treatment markedly reduced virus titers in the skin and snout, whereas systemic treatment did not. | [114] | ||
| Smallpox vaccine in monkeys | Intravenous | Coadministration of CDV (20 mg/kg) and smallpox vaccine reduced vaccination side effects but interfered with vaccine-elicited immune responses and immunity. | [44] |
| Footpad ECTV inoculation in mice | Intraperitoneal | Mice given 5 mg/kg/dose starting 24 h after infection had mild disease (reduced inflammation and footpad swelling) but showed a 100% recovery. Animals receiving higher doses of CDV (20 or 100 mg/kg/day) had mild footpad swelling and 100% recovery. | [115] |
| Daily treatment with 100 mg/kg/day CDV for five days starting one day after infection with a mouse interleukin-4 (producing virus causing host immune dysfunction and severe disease) delayed but could not prevent death from systemic infection. | [115] | ||
| Intranasal ECTV inoculation in mice | Intraperitoneal | CDV injection at 5 mg/kg on day zero and at 1.25 mg/kg on day three protected 100% of animals from lethality. | [116] |
| Aerosolized MPXV infection in monkeys | Intravenous | A single treatment of 5 mg/kg on the day of infection resulted in significantly reduced mortality and completely protected the animals from clinically and laboratory signs of disease. | [117] |
| Intratracheal MPXV infection in monkeys | Intraperitoneal | A dose of CDV of 5 mg/kg every other day for five days or six doses starting one day after infection resulted in significantly reduced mortality and reduced numbers of cutaneous monkeypox lesions. | [43] |
| Intravenous MPXV infection in monkeys | Intravenous | 5 mg/kg of CDV given before or up to two days after infection led to complete protection with no signs of illness and control of viral replication in blood. | [118,119,120] |
| Intravenous VARV infection in monkeys | Intravenous | 5 mg/kg of CDV given before or up to two days after infection led to complete protection with no signs of illness and control of viral replication in the blood. | [118,119,120] |
| Hind thighs orf virus scarification in lambs | Topical | 1% CDV given for four consecutive days resulted in milder lesions that resolved more quickly than untreated lesions. The scabs of the treated animals contained significantly lower amounts of viable virus meaning there should be less contamination of the environment with virus than would normally occur. | [42] |
| Animals were treated with a paint of 0.5% or 1% CDV + sucralfate 15% (wound healing properties) + NaH2PO4 16% w/w and with sucralfate gel suspension alone as control. The treatment with formulations containing CDV and phosphate salt for four consecutive days resulted in a rapid resolution of the lesions, with scabs containing significantly lower amounts of viable virus when compared with untreated lesions and lesions treated with sucralfate suspension alone. | [121] |
References and Notes
- De Clercq, E.; Holy, A.; Rosenberg, I.; Sakuma, T.; Balzarini, J.; Maudgal, P.C. A novel selective broad-spectrum anti-DNA virus agent. Nature 1986, 323, 464–467. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Sakuma, T.; Baba, M.; Pauwels, R.; Balzarini, J.; Rosenberg, I.; Holy, A. Antiviral activity of phosphonylmethoxyalkyl derivatives of purine and pyrimidines. Antivir. Res. 1987, 8, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Hockova, D.; Holy, A.; Masojidkova, M.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. 5-Substituted-2,4-diamino-6-[2-(phosphonomethoxy)ethoxy]pyrimidines-acycli c nucleoside phosphonate analogues with antiviral activity. J. Med. Chem. 2003, 46, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Andrei, G.; Balzarini, J.; Leyssen, P.; Naesens, L.; Neyts, J.; Pannecouque, C.; Snoeck, R.; Ying, C.; Hockova, D.; Holy, A. Antiviral potential of a new generation of acyclic nucleoside phosphonates, the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines. Nucleos. Nucleot. Nucleic Acids 2005, 24, 331–341. [Google Scholar] [CrossRef]
- Hockova, D.; Holy, A.; Masojidkova, M.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. Synthesis and antiviral activity of 2,4-diamino-5-cyano-6-[2-(phosphonomethoxy)ethoxy]pyrimidine and related compounds. Bioorg. Med. Chem. 2004, 12, 3197–3202. [Google Scholar] [CrossRef]
- Krecmerova, M.; Holy, A.; Pohl, R.; Masojidkova, M.; Andrei, G.; Naesens, L.; Neyts, J.; Balzarini, J.; Clercq, E. D.; Snoeck, R. Ester Prodrugs of Cyclic 1-(S)- [3-Hydroxy-2-(phosphonomethoxy)propyl]-5-azacytosine: Synthesis and Antiviral Activity. J. Med. Chem. 2007, 50, 5765–5772. [Google Scholar] [CrossRef]
- Krecmerova, M.; Holy, A.; Piskala, A.; Masojidkova, M.; Andrei, G.; Naesens, L.; Neyts, J.; Balzarini, J.; De Clercq, E.; Snoeck, R. Antiviral activity of triazine analogues of 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine (cidofovir) and related compounds. J. Med. Chem. 2007, 50, 1069–1077. [Google Scholar] [CrossRef]
- Kern, E.R. In vitro activity of potential anti-poxvirus agents. Antivir. Res. 2003, 57, 35–40. [Google Scholar] [CrossRef]
- De Clercq, E. Cidofovir in the treatment of poxvirus infections. Antivir. Res. 2002, 55, 1–13. [Google Scholar] [CrossRef]
- Baker, R.O.; Bray, M.; Huggins, J.W. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antivir. Res. 2003, 57, 13–23. [Google Scholar] [CrossRef]
- Keith, K.A.; Hitchcock, M.J.; Lee, W.A.; Holy, A.; Kern, E.R. Evaluation of nucleoside phosphonates and their analogs and prodrugs for inhibition of orthopoxvirus replication. Antimicrob. Agents Chemother. 2003, 47, 2193–2198. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, P.F.; Gilray, J.A.; Reid, H.W.; Mercer, A.A. Parapoxviruses are strongly inhibited in vitro by cidofovir. Antivir. Res. 2000, 48, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Vigne, S.; Duraffour, S.; Andrei, G.; Snoeck, R.; Garin, D.; Crance, J.M. Inhibition of vaccinia virus replication by two small interfering RNAs targeting B1R and G7L genes and their synergistic combination with cidofovir. Antimicrob. Agents Chemother. 2009, 53, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Dal Pozzo, F.; Andrei, G.; Holy, A.; van Den, O.J.; Scagliarini, A.; De Clercq, E.; Snoeck, R. Activities of acyclic nucleoside phosphonates against Orf virus in human and ovine cell monolayers and organotypic ovine raft cultures. Antimicrob. Agents Chemother. 2005, 49, 4843–4852. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, R.; De Clercq, E. Role of cidofovir in the treatment of DNA virus infections, other than CMV infections, in immunocompromised patients. Curr. Opin. Investig. Drugs 2002, 3, 1561–1566. [Google Scholar] [PubMed]
- Duraffour, S.; Snoeck, R.; de Vos, R.; van Den Oord, J.J.; Crance, J.M.; Garin, D.; Hruby, D.E.; Jordan, R.; De Clercq, E.; Andrei, G. Activity of the anti-orthopoxvirus compound ST-246 against vaccinia, cowpox and camelpox viruses in cell monolayers and organotypic raft cultures. Antivir. Ther. 2007, 12, 1205–1216. [Google Scholar] [CrossRef]
- Palu, G.; Stefanelli, S.; Rassu, M.; Parolin, C.; Balzarini, J.; De Clercq, E. Cellular uptake of phosphonylmethoxyalkylpurine derivatives. Antivir. Res. 1991, 16, 115–119. [Google Scholar] [CrossRef]
- Connelly, M.C.; Robbins, B.L.; Fridland, A. Mechanism of uptake of the phosphonate analog (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC) in Vero cells. Biochem. Pharmacol. 1993, 46, 1053–1057. [Google Scholar] [CrossRef]
- Cihlar, T.; Chen, M.S. Identification of enzymes catalyzing two-step phosphorylation of cidofovir and the effect of cytomegalovirus infection on their activities in host cells. Mol. Pharmacol. 1996, 50, 1502–1510. [Google Scholar]
- Bronson, J.J.; Ho, H.T.; De Boeck, H.; Woods, K.; Ghazzouli, I.; Martin, J.C.; Hitchcock, M.J. Biochemical pharmacology of acyclic nucleotide analogues. Ann. N. Y. Acad. Sci. 1990, 616, 398–407. [Google Scholar] [CrossRef]
- Ho, H.T.; Woods, K.L.; Bronson, J.J.; De Boeck, H.; Martin, J.C.; Hitchcock, M.J. Intracellular metabolism of the antiherpes agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine. Mol. Pharmacol. 1992, 41, 197–202. [Google Scholar] [PubMed]
- Aduma, P.; Connelly, M.C.; Srinivas, R.V.; Fridland, A. Metabolic diversity and antiviral activities of acyclic nucleoside phosphonates. Mol. Pharmacol. 1995, 47, 816–822. [Google Scholar] [PubMed]
- Xiong, X.; Smith, J.L.; Kim, C.; Huang, E.S.; Chen, M.S. Kinetic analysis of the interaction of cidofovir diphosphate with human cytomegalovirus DNA polymerase. Biochem. Pharmacol. 1996, 51, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, J.M.; Allen, S.J.; McKee, B.H.; Chen, M.S. Kinetic analysis of the interaction between the diphosphate of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, ddCTP, AZTTP, and FIAUTP with human DNA polymerases beta and gamma. Biochem. Pharmacol. 1994, 48, 1986–1988. [Google Scholar] [CrossRef]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Xiong, X.; Smith, J.L.; Chen, M.S. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob. Agents Chemother. 1997, 41, 594–599. [Google Scholar] [CrossRef]
- Hostetler, K. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antivir. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef]
- Magee, W.C.; Aldern, K.A.; Hostetler, K.Y.; Evans, D.H. Cidofovir and (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenine are highly effective inhibitors of vaccinia virus DNA polymerase when incorporated into the template strand. Antimicrob. Agents Chemother. 2008, 52, 586–597. [Google Scholar] [CrossRef]
- Magee, W.C.; Hostetler, K.Y.; Evans, D.H. Mechanism of inhibition of vaccinia virus DNA polymerase by cidofovir diphosphate. Antimicrob. Agents Chemother. 2005, 49, 3153–3162. [Google Scholar] [CrossRef]
- Jesus, D.M.; Costa, L.T.; Goncalves, D.L.; Achete, C.A.; Attias, M.; Moussatche, N.; Damaso, C.R. Cidofovir inhibits genome encapsidation and affects morphogenesis during the replication of vaccinia virus. J. Virol. 2009, 83, 11477–11490. [Google Scholar] [CrossRef]
- Watanabe, T.; Tamaki, K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J. Invest Dermatol. 2008, 128, 1327–1329. [Google Scholar] [CrossRef] [PubMed]
- Andrei, G.; Snoeck, R. Cidofovir. In Kucer’s the Use of Antibiotics; Hodder Arnold: London, UK, 2010; pp. 2403–2428. [Google Scholar]
- Andrei, G.; Gammon, D.B.; Fiten, P.; De Clercq, E.; Opdenakker, G.; Snoeck, R.; Evans, D.H. Cidofovir resistance in vaccinia virus is linked to diminished virulence in mice. J. Virol. 2006, 80, 9391–9401. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.N.; Obraztsova, M.; Kern, E.R.; Quenelle, D.C.; Keith, K.A.; Prichard, M.N.; Luo, M.; Moyer, R.W. Isolation and characterization of cidofovir resistant vaccinia viruses. Virol. J. 2008, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Kornbluth, R.S.; Smee, D.F.; Sidwell, R.W.; Snarsky, V.; Evans, D.H.; Hostetler, K.Y. Mutations in the E9L polymerase gene of cidofovir-resistant vaccinia virus strain WR are associated with the drug resistance phenotype. Antimicrob. Agents Chemother. 2006, 50, 4038–4043. [Google Scholar] [CrossRef] [PubMed]
- Gammon, D.B.; Evans, D.H. The 3'-to-5' exonuclease activity of vaccinia virus DNA polymerase is essential and plays a role in promoting virus genetic recombination. J. Virol. 2009, 83, 4236–4250. [Google Scholar] [CrossRef]
- Farlow, J.; Ichou, M.A.; Huggins, J.; Ibrahim, S. Comparative whole genome sequence analysis of wild-type and cidofovir-resistant monkeypoxvirus. Virol. J. 2010, 7, 110. [Google Scholar] [CrossRef]
- Andrei, G.; Fiten, P.; Duraffour, S.; Opdenakker, G.; Snoeck, R. Characterization of cowpox virus (CPV) mutants arising under pressure with different acyclic nucleoside phosphonates. Presented at the 22nd International Conference on Antiviral Research (22nd ICAR), Miami, FL, USA, May 2009. [Google Scholar]
- Gammon, D.B.; Snoeck, R.; Fiten, P.; Krecmerova, M.; Holy, A.; De Clercq, E.; Opdenakker, G.; Evans, D.H.; Andrei, G. Mechanism of antiviral drug resistance of vaccinia virus: Identification of residues in the viral DNA polymerase conferring differential resistance to antipoxvirus drugs. J. Virol. 2008, 82, 12520–12534. [Google Scholar] [CrossRef]
- Smee, D.F.; Sidwell, R.W. A review of compounds exhibiting anti-orthopoxvirus activity in animal models. Antivir. Res. 2003, 57, 41–52. [Google Scholar] [CrossRef]
- Smee, D.F. Progress in the discovery of compounds inhibiting orthopoxviruses in animal models. Antivir. Chem. Chemother. 2008, 19, 115–124. [Google Scholar] [CrossRef]
- Scagliarini, A.; McInnes, C.J.; Gallina, L.; Dal Pozzo, F.; Scagliarini, L.; Snoeck, R.; Prosperi, S.; Sales, J.; Gilray, J.A.; Nettleton, P.F. Antiviral activity of HPMPC (cidofovir) against orf virus infected lambs. Antivir. Res. 2007, 73, 169–174. [Google Scholar] [CrossRef]
- Stittelaar, K.J.; Neyts, J.; Naesens, L.; van Amerongen, G.; van Lavieren, R.F.; Holy, A.; De Clercq, E.; Niesters, H.G.; Fries, E.; Maas, C.; Mulder, P.G.; van der Zeijst, B.A.; Osterhaus, A.D. Antiviral treatment is more effective than smallpox vaccination upon lethal monkeypox virus infection. Nature 2006, 439, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Huang, D.; Fortman, J.; Wang, R.; Shao, L.; Chen, Z.W. Coadministration of cidofovir and smallpox vaccine reduced vaccination side effects but interfered with vaccine-elicited immune responses and immunity to monkeypox. J. Virol. 2009, 83, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Beadle, J.R.; Hartline, C.; Aldern, K.A.; Rodriguez, N.; Harden, E.; Kern, E.R.; Hostetler, K.Y. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob. Agents Chemother. 2002, 46, 2381–2386. [Google Scholar] [CrossRef]
- Keith, K.A.; Wan, W.B.; Ciesla, S.L.; Beadle, J.R.; Hostetler, K.Y.; Kern, E.R. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Antimicrob. Agents Chemother. 2004, 48, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.B.; Beadle, J.R.; Hartline, C.; Kern, E.R.; Ciesla, S.L.; Valiaeva, N.; Hostetler, K.Y. Comparison of the antiviral activities of alkoxyalkyl and alkyl esters of cidofovir against human and murine cytomegalovirus replication in vitro. Antimicrob. Agents Chemother. 2005, 49, 656–662. [Google Scholar] [CrossRef]
- Hartline, C.B.; Gustin, K.M.; Wan, W.B.; Ciesla, S.L.; Beadle, J.R.; Hostetler, K.Y.; Kern, E.R. Ether lipid-ester prodrugs of acyclic nucleoside phosphonates: activity against adenovirus replication in vitro. J. Infect. Dis. 2005, 191, 396–399. [Google Scholar] [CrossRef]
- Hostetler, K.Y.; Rought, S.; Aldern, K.A.; Trahan, J.; Beadle, J.R.; Corbeil, J. Enhanced antiproliferative effects of alkoxyalkyl esters of cidofovir in human cervical cancer cells in vitro. Mol. Cancer Ther. 2006, 5, 156–159. [Google Scholar] [CrossRef]
- Randhawa, P.; Farasati, N.A.; Shapiro, R.; Hostetler, K.Y. Ether lipid ester derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob. Agents Chemother. 2006, 50, 1564–1566. [Google Scholar] [CrossRef]
- Aldern, K.A.; Ciesla, S.L.; Winegarden, K.L.; Hostetler, K.Y. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol. Pharmacol. 2003, 63, 678–681. [Google Scholar] [CrossRef]
- Bidanset, D.J.; Beadle, J.R.; Wan, W.B.; Hostetler, K.Y.; Kern, E.R. Oral activity of ether lipid ester prodrugs of cidofovir against experimental human cytomegalovirus infection. J. Infect. Dis. 2004, 190, 499–503. [Google Scholar] [CrossRef]
- Kern, E.R.; Collins, D.J.; Wan, W.B.; Beadle, J.R.; Hostetler, K.Y.; Quenelle, D.C. Oral treatment of murine cytomegalovirus infections with ether lipid esters of cidofovir. Antimicrob. Agents Chemother. 2004, 48, 3516–3522. [Google Scholar] [CrossRef] [PubMed]
- Quenelle, D.C.; Collins, D.J.; Wan, W.B.; Beadle, J.R.; Hostetler, K.Y.; Kern, E.R. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob. Agents Chemother. 2004, 48, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ciesla, S.L.; Trahan, J.; Wan, W.B.; Beadle, J.R.; Aldern, K.A.; Painter, G.R.; Hostetler, K.Y. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antivir. Res. 2003, 59, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Painter, G.R.; Hostetler, K.Y. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection. Trends Biotechnol. 2004, 22, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M.; Lietman, P.S.; Lalezari, J.P.; Hitchcock, M.J.; Jaffe, H.S. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.L.; Noble, S. Cidofovir: A review of its use in cytomegalovirus retinitis in patients with AIDS. Drugs 1999, 58, 325–345. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin. Microbiol. Rev. 2003, 16, 569–596. [Google Scholar] [CrossRef]
- Brody, S.R.; Humphreys, M.H.; Gambertoglio, J.G.; Schoenfeld, P.; Cundy, K.C.; Aweeka, F.T. Pharmacokinetics of cidofovir in renal insufficiency and in continuous ambulatory peritoneal dialysis or high-flux hemodialysis. Clin. Pharmacol. Ther. 1999, 65, 21–28. [Google Scholar]
- Snoeck, R.; Wellens, W.; Desloovere, C.; Van Ranst, M.; Naesens, L.; De Clercq, E.; Feenstra, L. Treatment of severe laryngeal papillomatosis with intralesional injections of cidofovir [(S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine]. J. Med. Virol. 1998, 54, 219–225. [Google Scholar] [CrossRef]
- Naiman, A.N.; Ceruse, P.; Coulombeau, B.; Froehlich, P. Intralesional cidofovir and surgical excision for laryngeal papillomatosis. Laryngoscope 2003, 113, 2174–2181. [Google Scholar] [CrossRef]
- Naiman, A.N.; Roger, G.; Gagnieu, M.C.; Bordenave, J.; Mathaut, S.; Ayari, S.; Nicollas, R.; Bour, J.B.; Garabedian, N.; Froehlich, P. Cidofovir plasma assays after local injection in respiratory papillomatosis. Laryngoscope 2004, 114, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Lynch, G.; Lee, W.A. Bioavailability and metabolism of cidofovir following topical administration to rabbits. Antivir. Res. 1997, 35, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Safrin, S.; Cherrington, J.; Jaffe, H.S. Clinical uses of cidofovir. Rev. Med. Virol. 1997, 7, 145–156. [Google Scholar] [CrossRef]
- Li, S.B.; Yang, Z.H.; Feng, J.S.; Fong, C.K.; Lucia, H.L.; Hsiung, G.D. Activity of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC) against guinea pig cytomegalovirus infection in cultured cells and in guinea pigs. Antivir. Res. 1990, 13, 237–252. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Drew, W.L.; Glutzer, E.; James, C.; Miner, D.; Flaherty, J.; Fisher, P.E.; Cundy, K.; Hannigan, J.; Martin, J.C.; et al. (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]cytosine (cidofovir): results of a phase I/II study of a novel antiviral nucleotide analogue. J. Infect. Dis. 1995, 171, 788–796. [Google Scholar] [CrossRef]
- Polis, M.A.; Spooner, K.M.; Baird, B.F.; Manischewitz, J.F.; Jaffe, H.S.; Fisher, P.E.; Falloon, J.; Davey, R.T., Jr.; Kovacs, J.A.; Walker, R.E.; et al. Anticytomegaloviral activity and safety of cidofovir in patients with human immunodeficiency virus infection and cytomegalovirus viruria. Antimicrob. Agents Chemother. 1995, 39, 882–886. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Drew, W.L.; Glutzer, E.; Miner, D.; Safrin, S.; Owen, W.F., Jr.; Davidson, J.M.; Fisher, P.E.; Jaffe, H.S. Treatment with intravenous (S)-1-[3-hydroxy-2-(phosphonylmethoxy)propyl]-cytosine of acyclovir-resistant mucocutaneous infection with herpes simplex virus in a patient with AIDS. J. Infect. Dis. 1994, 170, 570–572. [Google Scholar] [CrossRef]
- Cihlar, T.; Lin, D.C.; Pritchard, J.B.; Fuller, M.D.; Mendel, D.B.; Sweet, D.H. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol. Pharmacol. 1999, 56, 570–580. [Google Scholar] [CrossRef]
- Cihlar, T.; Ho, E.S.; Lin, D.C.; Mulato, A.S. Human renal organic anion transporter 1 (hOAT1) and its role in the nephrotoxicity of antiviral nucleotide analogs. Nucleos. Nucleot. Nucleic Acids 2001, 20, 641–648. [Google Scholar] [CrossRef]
- Ho, E.S.; Lin, D.C.; Mendel, D.B.; Cihlar, T. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J. Am. Soc. Nephrol. 2000, 11, 383–393. [Google Scholar] [CrossRef]
- Uwai, Y.; Ida, H.; Tsuji, Y.; Katsura, T.; Inui, K. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 2007, 24, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Lea, A.P.; Bryson, H.M. Cidofovir. Drugs 1996, 52, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Zabawski, E.J., Jr. A review of topical and intralesional cidofovir. Dermatol. Online. J. 2000, 6, 3. [Google Scholar]
- Zabawski, E.J., Jr.; Cockerell, C.J. Topical and intralesional cidofovir: A review of pharmacology and therapeutic effects. J. Am. Acad. Dermatol. 1998, 39, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Snoeck, R.; Bossens, M.; Parent, D.; Delaere, B.; Degreef, H.; Van Ranst, M.; Noel, J.C.; Wulfsohn, M.S.; Rooney, J.F.; Jaffe, H.S.; De Clercq, E. Phase II double-blind, placebo-controlled study of the safety and efficacy of cidofovir topical gel for the treatment of patients with human papillomavirus infection. Clin. Infect. Dis. 2001, 33, 597–602. [Google Scholar] [CrossRef]
- Calista, D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. A pilot study. J. Eur. Acad. Dermatol. Venereol. 2000, 14, 484–488. [Google Scholar] [CrossRef]
- Bienvenu, B.; Martinez, F.; Devergie, A.; Rybojad, M.; Rivet, J.; Bellenger, P.; Morel, P.; Gluckman, E.; Lebbe, C. Topical use of cidofovir induced acute renal failure. Transplantation 2002, 73, 661–662. [Google Scholar] [CrossRef]
- Redfield, R.R.; Wright, D.C.; James, W.D.; Jones, T.S.; Brown, C.; Burke, D.S. Disseminated vaccinia in a military recruit with human immunodeficiency virus (HIV) disease. N. Engl. J. Med. 1987, 316, 673–676. [Google Scholar] [CrossRef]
- Moore, Z.; Seward, J.F.; Lane, J.M. Smallpox. Lancet 2006, 367, 425–435. [Google Scholar] [CrossRef]
- Marris, E. Dramatic rescue relieves rare case of smallpox infection. Nat. Med. 2007, 13, 517. [Google Scholar] [CrossRef]
- Vora, S.; Damon, I.; Fulginiti, V.; Weber, S.G.; Kahana, M.; Stein, S.L.; Gerber, S.I.; Garcia-Houchins, S.; Lederman, E.; Hruby, D.; et al. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin. Infect. Dis. 2008, 46, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.M.; Anderson, S.A. Qualitative assessment of risk for monkeypox associated with domestic trade in certain animal species, United States. Emerg. Infect. Dis. 2006, 12, 1827–1833. [Google Scholar] [CrossRef]
- Di Giulio, D.B.; Eckburg, P.B. Human monkeypox: An emerging zoonosis. Lancet Infect. Dis. 2004, 4, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kile, J.C.; Fleischauer, A.T.; Beard, B.; Kuehnert, M.J.; Kanwal, R.S.; Pontones, P.; Messersmith, H.J.; Teclaw, R.; Karem, K.L.; Braden, Z.H.; Damon, I.; Khan, A.S.; Fischer, M. Transmission of monkeypox among persons exposed to infected prairie dogs in Indiana in 2003. Arch. Pediatr. Adolesc. Med. 2005, 159, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Campe, H.; Zimmermann, P.; Glos, K.; Bayer, M.; Bergemann, H.; Dreweck, C.; Graf, P.; Weber, B.K.; Meyer, H.; Buttner, M.; Busch, U.; Sing, A. Cowpox virus transmission from pet rats to humans, Germany. Emerg. Infect. Dis. 2009, 15, 777–780. [Google Scholar] [CrossRef]
- Bonnekoh, B.; Falk, K.; Reckling, K.F.; Kenklies, S.; Nitsche, A.; Ghebremedhin, B.; Pokrywka, A.; Franke, I.; Thriene, B.; Konig, W.; Pauli, G.; Gollnick, H. Cowpox infection transmitted from a domestic cat. J. Dtsch. Dermatol. Ges. 2008, 6, 210–213. [Google Scholar] [CrossRef]
- Glatz, M.; Richter, S.; Ginter-Hanselmayer, G.; Aberer, W.; Mullegger, R.R. Human cowpox in a veterinary student. Lancet Infect. Dis. 2010, 10, 288. [Google Scholar] [CrossRef]
- Ninove, L.; Domart, Y.; Vervel, C.; Voinot, C.; Salez, N.; Raoult, D.; Meyer, H.; Capek, I.; Zandotti, C.; Charrel, R.N. Cowpox virus transmission from pet rats to humans, France. Emerg. Infect. Dis. 2009, 15, 781–784. [Google Scholar] [CrossRef]
- Strenger, V.; Muller, M.; Richter, S.; Revilla-Fernandez, S.; Nitsche, A.; Klee, S.R.; Ellerbrok, H.; and Zenz, W.A. 17-year-old girl with a black eschar. Cowpox virus infection. Clin. Infect. Dis. 2009, 48, 91–94. [Google Scholar] [CrossRef]
- Vorou, R.M.; Papavassiliou, V.G.; Pierroutsakos, I.N. Cowpox virus infection: An emerging health threat. Curr. Opin. Infect. Dis. 2008, 21, 153–156. [Google Scholar] [CrossRef]
- Meadows, K.P.; Tyring, S.K.; Pavia, A.T.; Rallis, T.M. Resolution of recalcitrant molluscum contagiosum virus lesions in human immunodeficiency virus-infected patients treated with cidofovir. Arch. Dermatol. 1997, 133, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.G.; Thrasher, A.; Lacey, K.; Harper, J. Topical cidofovir for severe molluscum contagiosum. Lancet 1999, 353, 2042. [Google Scholar] [CrossRef]
- Zabawski, E.J., Jr.; Cockerell, C.J. Topical cidofovir for molluscum contagiosum in children. Pediatr. Dermatol. 1999, 16, 414–415. [Google Scholar] [PubMed]
- Toro, J.R.; Wood, L.V.; Patel, N.K.; Turner, M.L. Topical cidofovir: A novel treatment for recalcitrant molluscum contagiosum in children infected with human immunodeficiency virus 1. Arch. Dermatol. 2000, 136, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Baxter, K.F.; Highet, A.S. Topical cidofovir and cryotherapy—Combination treatment for recalcitrant molluscum contagiosum in a patient with HIV infection. J. Eur. Acad. Dermatol. Venereol. 2004, 18, 230–231. [Google Scholar] [CrossRef]
- Geerinck, K.; Lukito, G.; Snoeck, R.; De Vos, R.; De Clercq, E.; Vanrenterghem, Y.; Degreef, H.; Maes, B. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J. Med. Virol. 2001, 64, 543–549. [Google Scholar] [CrossRef]
- Lederman, E.R.; Green, G.M.; DeGroot, H.E.; Dahl, P.; Goldman, E.; Greer, P.W.; Li, Y.; Zhao, H.; Paddock, C.D.; Damon, I.K. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin. Infect. Dis. 2007, 44, e100–e103. [Google Scholar] [CrossRef]
- Bray, M.; Martinez, M.; Smee, D.F.; Kefauver, D.; Thompson, E.; Huggins, J.W. Cidofovir protects mice against lethal aerosol or intranasal cowpox virus challenge. J. Infect. Dis. 2000, 181, 10–19. [Google Scholar] [CrossRef]
- Smee, D.F.; Sidwell, R.W.; Kefauver, D.; Bray, M.; Huggins, J.W. Characterization of wild-type and cidofovir-resistant strains of camelpox, cowpox, monkeypox, and vaccinia viruses. Antimicrob. Agents Chemother. 2002, 46, 1329–1335. [Google Scholar] [CrossRef]
- Smee, D.F.; Bailey, K.W.; Wong, M.; Sidwell, R.W. Intranasal treatment of cowpox virus respiratory infections in mice with cidofovir. Antivir. Res. 2000, 47, 171–177. [Google Scholar] [CrossRef]
- Smee, D.F.; Bailey, K.W.; Sidwell, R.W. Comparative effects of cidofovir and cyclic HPMPC on lethal cowpox and vaccinia virus respiratory infections in mice. Chemotherapy 2003, 49, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Martinez, M.; Kefauver, D.; West, M.; Roy, C. Treatment of aerosolized cowpox virus infection in mice with aerosolized cidofovir. Antivir. Res. 2002, 54, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.J.; Baker, R.; Washburn, K.; Bray, M. Aerosolized cidofovir is retained in the respiratory tract and protects mice against intranasal cowpox virus challenge. Antimicrob. Agents Chemother. 2003, 47, 2933–2937. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F.; Bailey, K.W.; Sidwell, R.W. Treatment of lethal cowpox virus respiratory infections in mice with 2-amino-7-[(1,3-dihydroxy-2-propoxy)methyl]purine and its orally active diacetate ester prodrug. Antivir. Res. 2002, 54, 113–120. [Google Scholar] [CrossRef]
- Quenelle, D.C.; Collins, D.J.; Kern, E.R. Efficacy of multiple- or single-dose cidofovir against vaccinia and cowpox virus infections in mice. Antimicrob. Agents Chemother. 2003, 47, 3275–3280. [Google Scholar] [CrossRef]
- Knorr, C.W.; Allen, S.D.; Torres, A.R.; Smee, D.F. Effects of cidofovir treatment on cytokine induction in murine models of cowpox and vaccinia virus infection. Antivir. Res. 2006, 72, 125–133. [Google Scholar] [CrossRef]
- Smee, D.F.; Gowen, B.B.; Wandersee, M.K.; Wong, M.H.; Skirpstunas, R.T.; Baldwin, T.J.; Hoopes, J.D.; Sidwell, R.W. Differential pathogenesis of cowpox virus intranasal infections in mice induced by low and high inoculum volumes and effects of cidofovir treatment. Int. J. Antimicrob. Agents 2008, 31, 352–359. [Google Scholar] [CrossRef]
- Goff, A.; Twenhafel, N.; Garrison, A.; Mucker, E.; Lawler, J.; Paragas, J. In vivo imaging of cidofovir treatment of cowpox virus infection. Virus Res. 2007, 128, 88–98. [Google Scholar] [CrossRef]
- Neyts, J.; Sobis, H.; Snoeck, R.; Vandeputte, M.; De Clercq, E. Efficacy of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)-cytosine and 9-(1,3-dihydroxy-2-propoxymethyl)-guanine in the treatment of intracerebral murine cytomegalovirus infections in immunocompetent and immunodeficient mice. Eur. J. Clin. Microbiol. Infect. Dis. 1993, 12, 269–279. [Google Scholar] [CrossRef]
- Quenelle, D.C.; Collins, D.J.; Kern, E.R. Cutaneous infections of mice with vaccinia or cowpox viruses and efficacy of cidofovir. Antivir. Res. 2004, 63, 33–40. [Google Scholar] [CrossRef]
- Neyts, J.; Leyssen, P.; Verbeken, E.; De Clercq, E. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob. Agents Chemother. 2004, 48, 2267–2273. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F.; Bailey, K.W.; Wong, M.H.; Wandersee, M.K.; Sidwell, R.W. Topical cidofovir is more effective than is parenteral therapy for treatment of progressive vaccinia in immunocompromised mice. J. Infect. Dis. 2004, 190, 1132–1139. [Google Scholar] [CrossRef]
- Robbins, S.J.; Jackson, R.J.; Fenner, F.; Beaton, S.; Medveczky, J.; Ramshaw, I.A.; Ramsay, A.J. The efficacy of cidofovir treatment of mice infected with ectromelia (mousepox) virus encoding interleukin-4. Antivir. Res. 2005, 66, 1–7. [Google Scholar] [CrossRef]
- Parker, S.; Touchette, E.; Oberle, C.; Almond, M.; Robertson, A.; Trost, L.C.; Lampert, B.; Painter, G.; Buller, R.M. Efficacy of therapeutic intervention with an oral ether-lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antivir. Res. 2008, 77, 39–49. [Google Scholar] [CrossRef]
- Huggins, J.W.; Baker, R.O.; Martinez, M.; Bray, M. Cidofovir (HPMPC) treatment of monkeypox. Antivir. Res. 1998, 37, A73. [Google Scholar]
- Huggins, J.W.; Martinez, M.J.; Hartman, C.J.; Hensley, L.E.; Jackson, D.L.; Kefauver, D.F.; Kulesh, D.A.; Larsen, T.; Miller, D.M.; Mucker, E.M.; Shamblin, J.D.; Tate, M.K.; Whitehouse, C.A.; Zwiers, S.H.; Jahrling, P.B. Successful cidofovir treatment of smallpox-like disease in variola and monkeypox primate models. Antivir. Res. 2004, 62, A57–A58. [Google Scholar]
- Huggins, J.W.; Zwiers, S.H.; Baker, R.; Hensley, L.; Larsen, T.; Martinez, M.J.; J; Jahrling, L. Cidofovir treatment with vza-ariola (smallpox) in the hemorrhagic smallpox primate model and the IV monkeypox primate model. Antivir. Res. 2003, 57, A78. [Google Scholar]
- Huggins, J.W.; Raymond, J.; Fisher, R.; Jahrling, L.; Hensley, L. Sequential determination of virus in blood and tissues of the variola cynomolgus monkey model of classical smallpox reveals that IV cidofovir can effectively treat monkeys with extensive viral burden. Antivir. Res. 2006, 70, A36–A37. [Google Scholar]
- Sonvico, F.; Colombo, G.; Gallina, L.; Bortolotti, F.; Rossi, A.; McInnes, C.J.; Massimo, G.; Colombo, P.; Scagliarini, A. Therapeutic paint of cidofovir/sucralfate gel combination topically administered by spraying for treatment of orf virus infections. AAPS. J. 2009, 11, 242–249. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2010 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrei, G.; Snoeck, R. Cidofovir Activity against Poxvirus Infections. Viruses 2010, 2, 2803-2830. https://doi.org/10.3390/v2122803
Andrei G, Snoeck R. Cidofovir Activity against Poxvirus Infections. Viruses. 2010; 2(12):2803-2830. https://doi.org/10.3390/v2122803
Chicago/Turabian StyleAndrei, Graciela, and Robert Snoeck. 2010. "Cidofovir Activity against Poxvirus Infections" Viruses 2, no. 12: 2803-2830. https://doi.org/10.3390/v2122803
APA StyleAndrei, G., & Snoeck, R. (2010). Cidofovir Activity against Poxvirus Infections. Viruses, 2(12), 2803-2830. https://doi.org/10.3390/v2122803