Adenoviral Producer Cells

Abstract
:1. Producer cells for non-replicating first generation Ad vectors
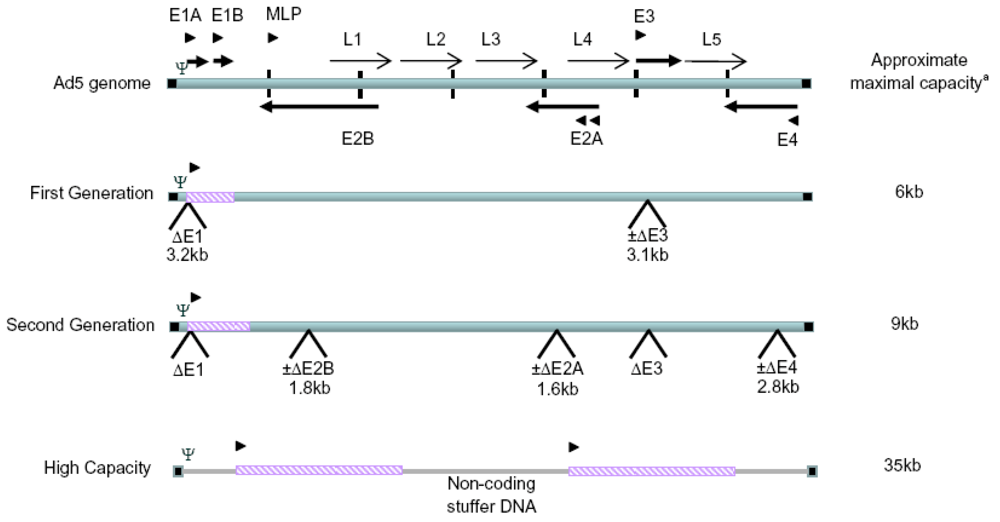

{kind=link}
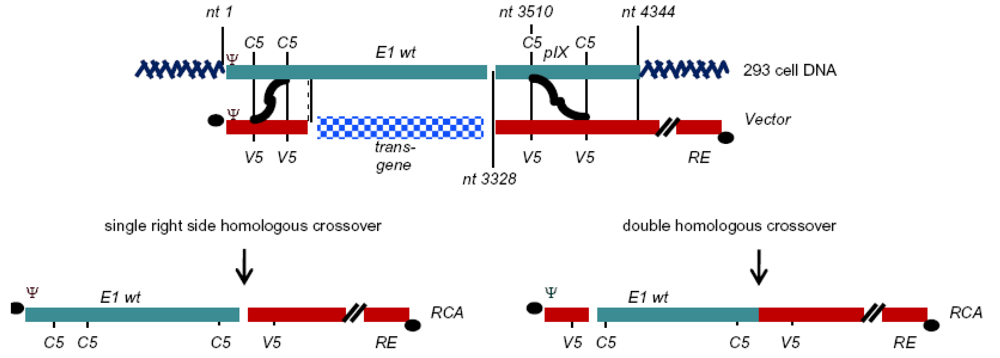
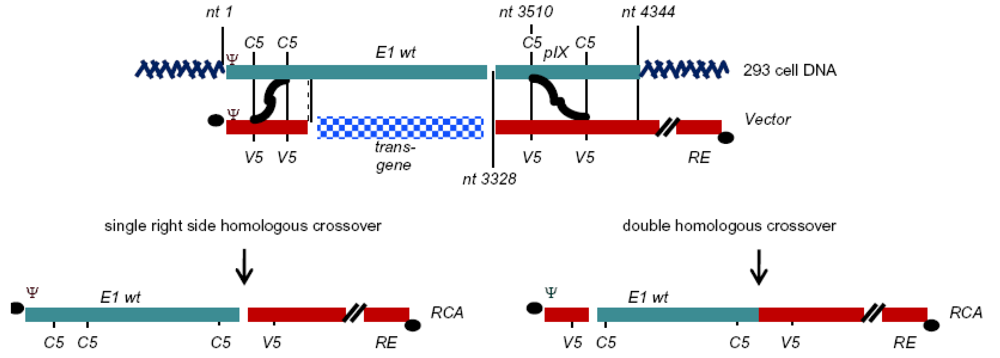{kind=link}
{kind=link}
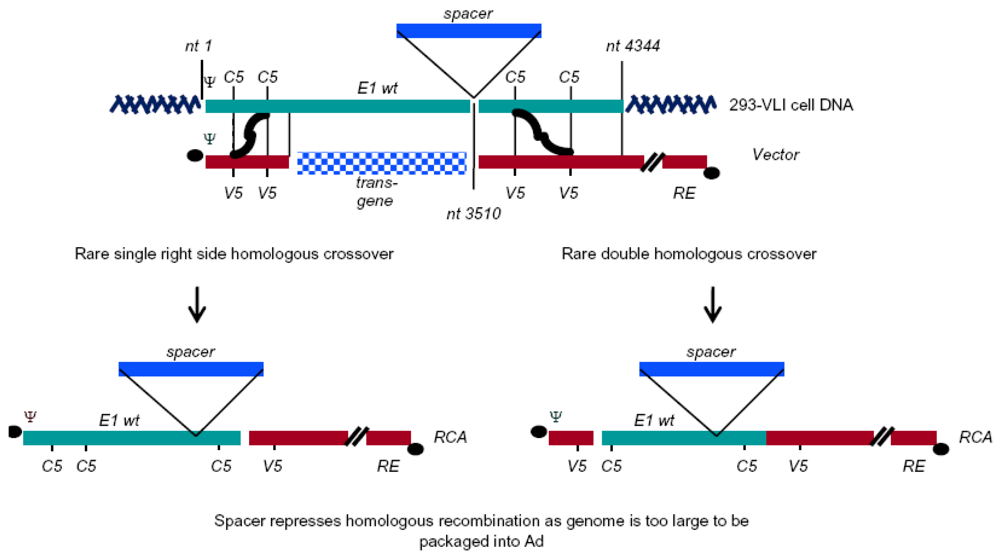{kind=link}
| Cell Line | Parental Cells | Ad Sequence | Promoter Requirement | 3’ End Requirement | Ref |
|---|---|---|---|---|---|
| HEK293 | Human embryonic kidney (HEK) | 1 to 4344 | Not applicable (N/A) | N/A | [2] |
| 911 | Human embryonic retinoblasts (HER) | 79 – 5789 | N/A | N/A | [5] |
| pTG6559 | A549 | 505-4034 | muPGK promoter | Rabbit β-globin gene polyA | [6] |
| PER.C6 | HER | 459-3510 | huPGK promoter | Hepatitis B virus polyA | [7] |
| GH329 | HeLa | 511-3924 | huPGK promoter | Yes | [8] |
| N52.E6 | Primary human amniocytes | 505-3522 | muPGK promoter | SV40 splice acceptor and polyA | [9] |
| HeLa-E1 | HeLa | 542-3526 | CMV | bGH polyA | [10] |
| UR | HEL 299 | 459-3510 | RSV-LTR | TK polyA | [11] |
| VLI-293b | HEK293 | 1 to 4344, insertion of spacer at 3510 | N/A | Hepatitis B virus polyA in insert | [12] |
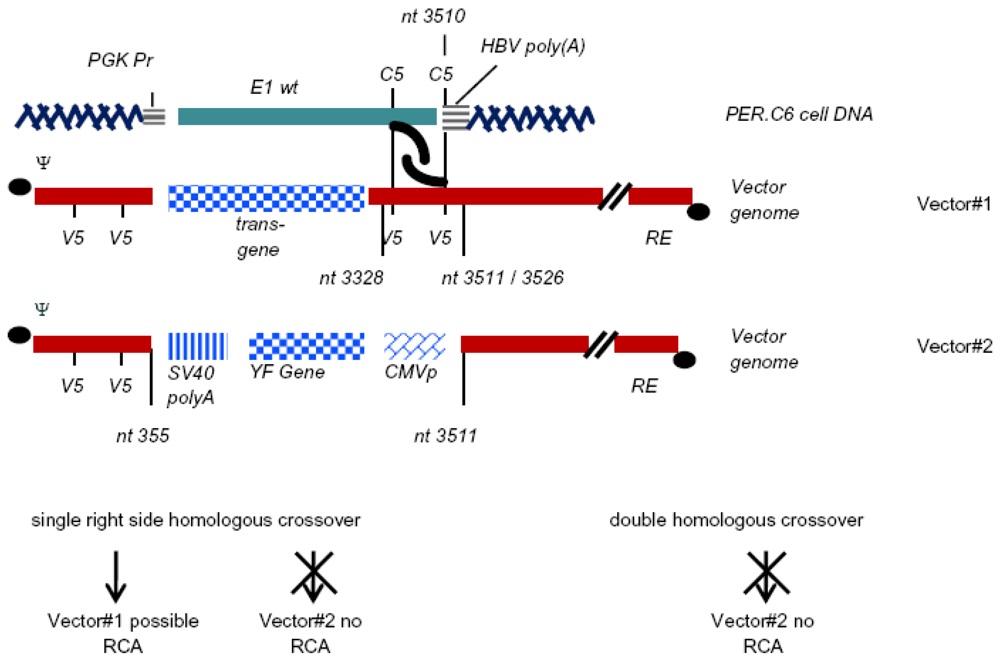
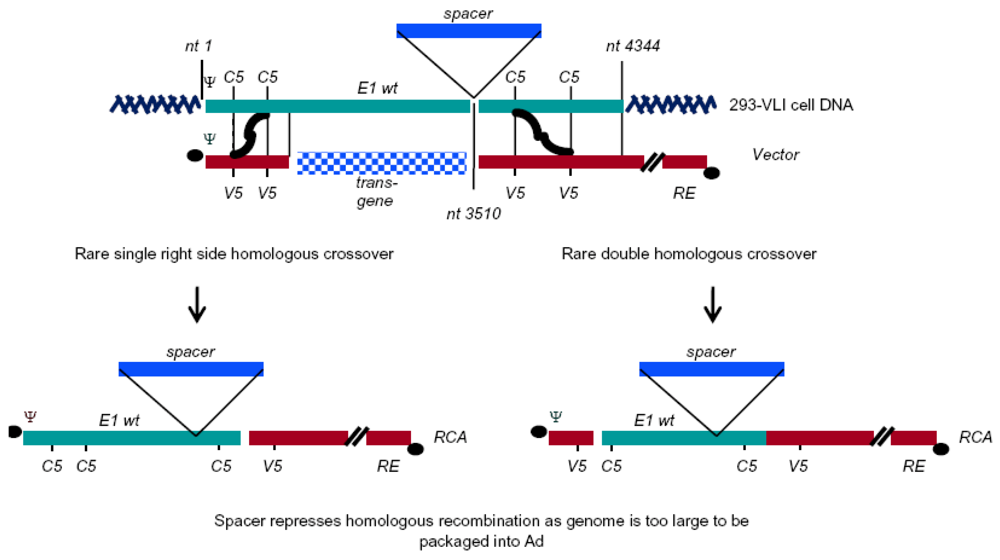
2. Producer cells for non-replicating second generation Ad vectors
| Cell Line | Parental Cells | E2 or E4 Complementation | Viral Products | Inducible System | Refs |
|---|---|---|---|---|---|
| VL2-20 and VK10-9a | HEK293 | E4 | All ORFs | Glucocorticoid | [31] |
| 293-E4 | HEK293 | E4 | All ORFs | cAMP | [49] |
| C7 | LP-293 | E2B | Pol and pTPb | No | [50,57] |
| 293-ORF6 | HEK293 | E4 | ORF6 | Metalc | [51] |
| MT-ORF6 | HEK293 | E4 | ORF6 | Metal | [52] |
| MMTV-ORF6 | HEK293 | E4 | ORF6 | Glucocorticoid | [52] |
| AE1-2a | A549d | E2A | DBP | Glucocorticoid | [53] |
| 293-pTP | HEK293 | E2B | pTP | Tetracycline | [54] |
| IGRP2 | HEK293 | E4 | ORF6 and 7 | Glucocorticoid | [55] |
| 293-C2 | HEK293 | E2A | DBP | No | [56] |
| 911E4 | 911 | E4 | All ORFs | Tetracycline | [58] |
| 293-E2A | HEK293 | E2A | DBP | Glucocorticoid | [59] |
| 293-E4 | HEK293 | E4 | All ORFs | Yese | [59] |
| 293-E4ORF6+7 | HEK293 | E4 | ORF6 and 7 | Tetracycline | [59] |
| A70.S54 | AE1-2a | E2A/E4 | DBP/All ORF | Glucocorticoid | [60] |
| E2T | HEK293 | E2A | DBP | Tetracycline | [61] |
3. Producer cells for high capacity Ad vectors
| Cell Line | Parental Cells | Ad vector complementation | Recombinase System | Refs |
|---|---|---|---|---|
| 293Cre4 | HEK293 | E1 | Cre recombinase | [69] |
| 293cre415a | 293Cre4 | E1 | Cre recombinase | [79] |
| CRE8 | HEK293 | E1 | Cre recombinase | [70] |
| CreE | E2T | E1/E2A | Cre recombinase | [72] |
| 293FLP | HEK293 | E1 | FLPe recombinase | [76] |
| 293CreFLP | 293Cre4 | E1 | eitherb | [76] |
| 293-FLPe6 | HEK293 | E1 | FLPe recombinase | [77] |
| C7-Cre | C7 | E1/E2B | Cre recombinase | [73] |
| 116 | 293N3Sc | E1 | Cre recombinase | [78] |
| PER.C6-Cre | PER.C6 | E1 | Cre recombinase | [75] |
4. Producer cell lines for novel Ad vector serotypes
5. Conclusions
References
- Wold, W.; Tollefson, A.; Hermiston, T. E3 transcription unit of adenovirus. In The Molecular Repertoire of Adenoviruses; Bohm, P., Doerfler, W., Eds.; Springer-Verlag: Berlin, Germany, 1993. [Google Scholar]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. Faseb J. 2002, 16, 869–871. [Google Scholar] [PubMed]
- Louis, N.; Evelegh, C.; Graham, F.L. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology 1997, 233, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Fallaux, F.J.; Kranenburg, O.; Cramer, S.J.; Houweling, A.; Van Ormondt, H.; Hoeben, R.C.; Van Der Eb, A.J. Characterization of 911: a new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum. Gene Ther. 1996, 7, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Imler, J.L.; Chartier, C.; Dreyer, D.; Dieterle, A.; Sainte-Marie, M.; Faure, T.; Pavirani, A.; Mehtali, M. Novel complementation cell lines derived from human lung carcinoma A549 cells support the growth of E1-deleted adenovirus vectors. Gene Ther. 1996, 3, 75–84. [Google Scholar] [PubMed]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; Valerio, D.; Hoeben, R.C. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.P.; Engdahl, R.K.; Wilson, J.M. A cell line for high-yield production of E1-deleted adenovirus vectors without the emergence of replication-competent virus. Hum. Gene Ther. 2000, 11, 213–219. [Google Scholar] [PubMed]
- Schiedner, G.; Hertel, S.; Kochanek, S. Efficient transformation of primary human amniocytes by E1 functions of Ad5: generation of new cell lines for adenoviral vector production. Hum. Gene Ther. 2000, 11, 2105–2116. [Google Scholar] [PubMed]
- Kim, J.S.; Lee, S.H.; Cho, Y.S.; Park, K.; Kim, Y.H.; Lee, J.H. Development of a packaging cell line for propagation of replication-deficient adenovirus vector. Exp. Mol. Med. 2001, 33, 145–149. [Google Scholar] [PubMed]
- Xu, Q.; Arevalo, M.T.; Pichichero, M.E.; Zeng, M. A new complementing cell line for replication-incompetent E1-deleted adenovirus propagation. Cytotechnology 2006, 51, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hedley, S.J. Unpublished work. 2010. [Google Scholar]
- Farson, D.; Tao, L.; Ko, D.; Li, Q.; Brignetti, D.; Segawa, K.; Mittelstaedt, D.; Harding, T.; Yu, D.C.; Li, Y. Development of novel E1-complementary cells for adenoviral production free of replication-competent adenovirus. Mol. Ther. 2006, 14, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Howe, J.A.; Pelka, P.; Antelman, D.; Wilson, C.; Cornell, D.; Hancock, W.; Ramachandra, M.; Avanzini, J.; Horn, M.; Wills, K.; Sutjipto, S.; Ralston, R. Matching complementing functions of transformed cells with stable expression of selected viral genes for production of E1-deleted adenovirus vectors. Virology 2006, 345, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Murakami, P.; Pungor, E.; Files, J.; Do, L.; van Rijnsoever, R.; Vogels, R.; Bout, A.; McCaman, M. A single short stretch of homology between adenoviral vector and packaging cell line can give rise to cytopathic effect-inducing, helper-dependent E1-positive particles. Hum. Gene Ther. 2002, 13, 909–920. [Google Scholar] [PubMed]
- Lochmuller, H.; Jani, A.; Huard, J.; Prescott, S.; Simoneau, M.; Massie, B.; Karpati, G.; Acsadi, G. Emergence of early region 1-containing replication-competent adenovirus in stocks of replication-defective adenovirus recombinants (delta E1 + delta E3) during multiple passages in 293 cells. Hum. Gene Ther. 1994, 5, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Hehir, K.M.; Armentano, D.; Cardoza, L.M.; Choquette, T.L.; Berthelette, P.B.; White, G.A.; Couture, L.A.; Everton, M.B.; Keegan, J.; Martin, J.M.; Pratt, D.A.; Smith, M.P.; Smith, A.E.; Wadsworth, S.C. Molecular characterization of replication-competent variants of adenovirus vectors and genome modifications to prevent their occurrence. J. Virol. 1996, 70, 8459–8467. [Google Scholar] [PubMed]
- Smith, J.G.; Eck, S.L. Molecular characterization of an adenoviral vector resulting from both homologous and nonhomologous recombination. Cancer Gene Ther. 1999, 6, 475–481. [Google Scholar] [PubMed]
- Zhu, J.; Grace, M.; Casale, J.; Chang, A. T.; Musco, M.L.; Bordens, R.; Greenberg, R.; Schaefer, E.; Indelicato, S.R. Characterization of replication-competent adenovirus isolates from large-scale production of a recombinant adenoviral vector. Hum. Gene Ther. 1999, 10, 113–121. [Google Scholar] [PubMed]
- Lusky, M. Good manufacturing practice production of adenoviral vectors for clinical trials. Hum. Gene Ther. 2005, 16, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Murakami, P.; Havenga, M.; Fawaz, F.; Vogels, R.; Marzio, G.; Pungor, E.; Files, J.; Do, L.; Goudsmit, J.; McCaman, M. Common structure of rare replication-deficient E1-positive particles in adenoviral vector batches. J. Virol. 2004, 78, 6200–6208. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.W.; Lardenoije, R.; Ledwith, B.J.; Brouwer, K.; Manam, S.; Vogels, R.; Kaslow, D.; Zuidgeest, D.; Bett, A.J.; Chen, L.; Van Der Kaaden, M.; Galloway, S.M.; Hill, R.B.; Machotka, S.V.; Anderson, C.A.; Lewis, J.; Martinez, D.; Lebron, J.; Russo, C.; Valerio, D.; Bout, A. Propagation of adenoviral vectors: Use of PER.C6 cells. In Adenoviral Vectors for Gene Therapy; Curiel, D.T., Douglas, J.T., Eds.; 2002; Academic Press: San Diego, USA. [Google Scholar]
- Parks, R.J. Adenovirus protein IX: a new look at an old protein. Mol. Ther. 2005, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, J.; Van der Heijdt, S.; Hoeben, R.C. The adenovirus capsid: major progress in minor proteins. J. Gen. Virol. 2005, 86, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, G.; Haj-Ahmad, Y.; Graham, F.L. Protein IX, a minor component of the human adenovirus capsid, is essential for the packaging of full length genomes. Embo J. 1987, 6, 1733–1739. [Google Scholar] [PubMed]
- Sargent, K.L.; Ng, P.; Evelegh, C.; Graham, F.L.; Parks, R.J. Development of a size-restricted pIX-deleted helper virus for amplification of helper-dependent adenovirus vectors. Gene Ther. 2004, 11, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Calatrava, M.; Grave, L.; Puvion-Dutilleul, F.; Chatton, B.; Kedinger, C. Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J. Virol. 2001, 75, 7131–7141. [Google Scholar] [CrossRef] [PubMed]
- Sargent, K.L.; Meulenbroek, R.A.; Parks, R.J. Activation of adenoviral gene expression by protein IX is not required for efficient virus replication. J. Virol. 2004, 78, 5032–5037. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.C.; Zhang, J.; Toro, H.; Shi, Z.; Van Kampen, K.R. Adenovirus as a carrier for the development of influenza virus-free avian influenza vaccines. Exp. Rev. Vaccines 2009, 8, 469–481. [Google Scholar] [CrossRef]
- Colby, W.W.; Shenk, T. Adenovirus type 5 virions can be assembled in vivo in the absence of detectable polypeptide IX. J. Virol. 1981, 39, 977–980. [Google Scholar] [PubMed]
- Krougliak, V.; Graham, F.L. Development of cell lines capable of complementing E1, E4, and protein IX defective adenovirus type 5 mutants. Hum. Gene Ther. 1995, 6, 1575–1586. [Google Scholar] [CrossRef]
- Vellinga, J.; Uil, T.G.; de Vrij, J.; Rabelink, M.J.; Lindholm, L.; Hoeben, R.C. A system for efficient generation of adenovirus protein IX-producing helper cell lines. J. Gene Med. 2006, 8, 147–154. [Google Scholar] [CrossRef]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [PubMed]
- Ginsberg, H.S. The adenoviruses . 1984. [Google Scholar]
- Weitzman, M.D. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front. Biosci. 2005, 10, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Hemstrom, C.; Nordqvist, K.; Pettersson, U.; Virtanen, A. Gene product of region E4 of adenovirus type 5 modulates accumulation of certain viral polypeptides. J. Virol. 1988, 62, 3258–3264. [Google Scholar] [PubMed]
- Bridge, E.; Ketner, G. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 1989, 63, 631–638. [Google Scholar] [PubMed]
- Huang, M.M.; Hearing, P. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J. Virol. 1989, 63, 2605–2615. [Google Scholar] [PubMed]
- Ketner, G.; Bridge, E.; Virtanen, A.; Hemstrom, C.; Pettersson, U. Complementation of adenovirus E4 mutants by transient expression of E4 cDNA and deletion plasmids. Nucleic Acids Res. 1989, 17, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. Mechanism of activation of early viral transcription by the adenovirus E1A gene product. Cell 1981, 26, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, R.B.; Berk, A.J. Cis-acting induction of adenovirus transcription. Cell 1983, 33, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, M.J.; Kao, H.T.; Feldman, L.T.; Nevins, J.R.; Strickland, S. Common control of the heat shock gene and early adenovirus genes: evidence for a cellular E1A-like activity. Mol. Cell. Biol. 1984, 4, 867–874. [Google Scholar] [PubMed]
- Yang, Y.; Nunes, F.A.; Berencsi, K.; Furth, E.E.; Gonczol, E.; Wilson, J.M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4407–4411. [Google Scholar] [CrossRef] [PubMed]
- Gilgenkrantz, H.; Duboc, D.; Juillard, V.; Couton, D.; Pavirani, A.; Guillet, J.G.; Briand, P.; Kahn, A. Transient expression of genes transferred in vivo into heart using first-generation adenoviral vectors: role of the immune response. Hum. Gene Ther. 1995, 6, 1265–1274. [Google Scholar] [CrossRef]
- Yang, Y.; Su, Q.; Wilson, J.M. Role of viral antigens in destructive cellular immune responses to adenovirus vector-transduced cells in mouse lungs. J. Virol. 1996, 70, 7209–7212. [Google Scholar] [PubMed]
- Klessig, D.F.; Grodzicker, T.; Cleghon, V. Construction of human cell lines which contain and express the adenovirus DNA binding protein gene by cotransformation with the HSV-1 tk gene. Virus Res. 1984, 1, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.E.; Cleghon, V.; Klessig, D.F. Construction, characterization, and utilization of cell lines which inducibly express the adenovirus DNA-binding protein. Virology 1992, 190, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.H.; Ketner, G. A cell line that supports the growth of a defective early region 4 deletion mutant of human adenovirus type 2. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 5383–5386. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, X.C.; Finer, M.H. A packaging cell line for propagation of recombinant adenovirus vectors containing two lethal gene-region deletions. Gene Ther. 1995, 2, 775–783. [Google Scholar] [PubMed]
- Amalfitano, A.; Begy, C.R.; Chamberlain, J.S. Improved adenovirus packaging cell lines to support the growth of replication-defective gene-delivery vectors. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 3352–3356. [Google Scholar] [CrossRef] [PubMed]
- Brough, D.E.; Lizonova, A.; Hsu, C.; Kulesa, V.A.; Kovesdi, I. A gene transfer vector-cell line system for complete functional complementation of adenovirus early regions E1 and E4. J. Virol. 1996, 70, 6497–6501. [Google Scholar] [PubMed]
- Gao, G.P.; Yang, Y.; Wilson, J.M. Biology of adenovirus vectors with E1 and E4 deletions for liver-directed gene therapy. J. Virol. 1996, 70, 8934–8943. [Google Scholar] [PubMed]
- Gorziglia, M.I.; Kadan, M.J.; Yei, S.; Lim, J.; Lee, G.M.; Luthra, R.; Trapnell, B.C. Elimination of both E1 and E2 from adenovirus vectors further improves prospects for in vivo human gene therapy. J. Virol. 1996, 70, 4173–4178. [Google Scholar] [PubMed]
- Langer, S.J.; Schaack, J. 293 cell lines that inducibly express high levels of adenovirus type 5 precursor terminal protein. Virology 1996, 221, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.; Dedieu, J.F.; Orsini, C.; Vigne, E.; Denefle, P.; Perricaudet, M. Efficient dual transcomplementation of adenovirus E1 and E4 regions from a 293-derived cell line expressing a minimal E4 functional unit. J. Virol. 1996, 70, 559–565. [Google Scholar] [PubMed]
- Zhou, H.; O'Neal, W.; Morral, N.; Beaudet, A.L. Development of a complementing cell line and a system for construction of adenovirus vectors with E1 and E2a deleted. J. Virol. 1996, 70, 7030–7038. [Google Scholar] [PubMed]
- Amalfitano, A.; Chamberlain, J.S. Isolation and characterization of packaging cell lines that coexpress the adenovirus E1, DNA polymerase, and preterminal proteins: implications for gene therapy. Gene Ther. 1997, 4, 258–263. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Zhou, S.; da Costa, L.T.; Yu, J.; Kinzler, K.W.; Vogelstein, B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 2509–2514. [Google Scholar] [CrossRef] [PubMed]
- Lusky, M.; Christ, M.; Rittner, K.; Dieterle, A.; Dreyer, D.; Mourot, B.; Schultz, H.; Stoeckel, F.; Pavirani, A.; Mehtali, M. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 1998, 72, 2022–2032. [Google Scholar] [PubMed]
- Gorziglia, M.I.; Lapcevich, C.; Roy, S.; Kang, Q.; Kadan, M.; Wu, V.; Pechan, P.; Kaleko, M. Generation of an adenovirus vector lacking E1, E2a, E3, and all of E4 except open reading frame 3. J. Virol. 1999, 73, 6048–6055. [Google Scholar] [PubMed]
- Zhou, H.; Beaudet, A.L. A new vector system with inducible E2a cell line for production of higher titer and safer adenoviral vectors. Virology 2000, 275, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Broker, T.R.; Lewis, J.B. Complex splicing patterns of RNAs from the early regions of adenovirus-2. J. Mol. Biol. 1979, 134, 265–303. [Google Scholar] [CrossRef] [PubMed]
- Schaack, J.; Guo, X.; Ho, W.Y.; Karlok, M.; Chen, C.; Ornelles, D. Adenovirus type 5 precursor terminal protein-expressing 293 and HeLa cell lines. J. Virol. 1995, 69, 4079–4085. [Google Scholar] [PubMed]
- O'Neal, W.K.; Zhou, H.; Morral, N.; Aguilar-Cordova, E.; Pestaner, J.; Langston, C.; Mull, B.; Wang, Y.; Beaudet, A. L.; Lee, B. Toxicological comparison of E2a-deleted and first-generation adenoviral vectors expressing alpha1-antitrypsin after systemic delivery. Hum. Gene Ther. 1998, 9, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Christ, M.; Louis, B.; Stoeckel, F.; Dieterle, A.; Grave, L.; Dreyer, D.; Kintz, J.; Ali Hadji, D.; Lusky, M.; Mehtali, M. Modulation of the inflammatory properties and hepatotoxicity of recombinant adenovirus vectors by the viral E4 gene products. Hum. Gene Ther. 2000, 11, 415–427. [Google Scholar] [PubMed]
- Brough, D.E.; Hsu, C.; Kulesa, V.A.; Lee, G.M.; Cantolupo, L.J.; Lizonova, A.; Kovesdi, I. Activation of transgene expression by early region 4 is responsible for a high level of persistent transgene expression from adenovirus vectors in vivo. J. Virol. 1997, 71, 9206–9213. [Google Scholar] [PubMed]
- Wang, Q.; Greenburg, G.; Bunch, D.; Farson, D.; Finer, M.H. Persistent transgene expression in mouse liver following in vivo gene transfer with a delta E1/delta E4 adenovirus vector. Gene Ther. 1997, 4, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.M.; Alba, R.; Bosch, A.; Chillon, M. Advances in helper-dependent adenoviral vector research. Curr. Gene Ther. 2008, 8, 222–235. [Google Scholar] [CrossRef]
- Parks, R.J.; Chen, L.; Anton, M.; Sankar, U.; Rudnicki, M.A.; Graham, F.L. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13565–13570. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Kitamura, M.; Harris-Stansil, T.; Dai, Y.; Phipps, M.L. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 1997, 71, 1842–1849. [Google Scholar] [PubMed]
- Sandig, V.; Youil, R.; Bett, A.J.; Franlin, L.L.; Oshima, M.; Maione, D.; Wang, F.; Metzker, M.L.; Savino, R.; Caskey, C.T. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhao, T.; Pastore, L.; Nageh, M.; Zheng, W.; Rao, X.M.; Beaudet, A.L. A Cre-expressing cell line and an E1/E2a double-deleted virus for preparation of helper-dependent adenovirus vector. Mol. Ther. 2001, 3, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Barjot, C.; Hartigan-O'Connor, D.; Salvatori, G.; Scott, J.M.; Chamberlain, J.S. Gutted adenoviral vector growth using E1/E2b/E3-deleted helper viruses. J. Gene Med. 2002, 4, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Sakhuja, K.; Ganesh, S.; Yang, L.; Kayda, D.; Brann, T.; Pattison, S.; Golightly, D.; Idamakanti, N.; Pinkstaff, A.; Kaloss, M.; Barjot, C.; Chamberlain, J.S.; Kaleko, M.; Connelly, S. Sustained human factor VIII expression in hemophilia A mice following systemic delivery of a gutless adenoviral vector. Mol. Ther. 2002, 5, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Sakhuja, K.; Reddy, P.S.; Ganesh, S.; Cantaniag, F.; Pattison, S.; Limbach, P.; Kayda, D.B.; Kadan, M.J.; Kaleko, M.; Connelly, S. Optimization of the generation and propagation of gutless adenoviral vectors. Hum. Gene Ther. 2003, 14, 243–254. [Google Scholar] [PubMed]
- Ng, P.; Beauchamp, C.; Evelegh, C.; Parks, R.; Graham, F.L. Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol. Ther. 2001, 3, 809–815. [Google Scholar] [CrossRef]
- Umana, P.; Gerdes, C.A.; Stone, D.; Davis, J.R.; Ward, D.; Castro, M.G.; Lowenstein, P.R. Efficient FLPe recombinase enables scalable production of helper-dependent adenoviral vectors with negligible helper-virus contamination. Nat. Biotechnol. 2001, 19, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.; Ng, P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 2003, 8, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Anton, M.; Graham, F.L. Production and characterization of human 293 cell lines expressing the site-specific recombinase Cre. Somat. Cell. Mol. Genet. 1996, 22, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.; Evelegh, C.; Cummings, D.; Graham, F.L. Cre levels limit packaging signal excision efficiency in the Cre/loxP helper-dependent adenoviral vector system. J. Virol. 2002, 76, 4181–4189. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L. Growth of 293 cells in suspension culture. J. Gen. Virol. 1987, 68, 937–940. [Google Scholar] [CrossRef] [PubMed]
- Chirmule, N.; Propert, K.; Magosin, S.; Qian, Y.; Qian, R.; Wilson, J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999, 6, 1574–1583. [Google Scholar] [CrossRef]
- Seshidhar Reddy, P.; Ganesh, S.; Limbach, M.P.; Brann, T.; Pinkstaff, A.; Kaloss, M.; Kaleko, M.; Connelly, S. Development of adenovirus serotype 35 as a gene transfer vector. Virology 2003, 311, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Vogels, R.; Zuijdgeest, D.; van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; de Bethune, M.P.; Kostense, S.; Penders, G.; Helmus, N.; Koudstaal, W.; Cecchini, M.; Wetterwald, A.; Sprangers, M.; Lemckert, A.; Ophorst, O.; Koel, B.; van Meerendonk, M.; Quax, P.; Panitti, L.; Grimbergen, J.; Bout, A.; Goudsmit, J.; Havenga, M. Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J. Virol. 2003, 77, 8263–8271. [Google Scholar] [CrossRef] [PubMed]
- Nwanegbo, E.; Vardas, E.; Gao, W.; Whittle, H.; Sun, H.; Rowe, D.; Robbins, P. D.; Gambotto, A. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin. Diagn. Lab. Immunol. 2004, 11, 351–357. [Google Scholar] [PubMed]
- Smith, T.A.; White, B.D.; Gardner, J.M.; Kaleko, M.; McClelland, A. Transient immunosuppression permits successful repetitive intravenous administration of an adenovirus vector. Gene Ther. 1996, 3, 496–502. [Google Scholar] [PubMed]
- Ophorst, O.J.; Kostense, S.; Goudsmit, J.; De Swart, R.L.; Verhaagh, S.; Zakhartchouk, A.; Van Meijer, M.; Sprangers, M.; Van Amerongen, G.; Yuksel, S.; Osterhaus, A.D.; Havenga, M.J. An adenoviral type 5 vector carrying a type 35 fiber as a vaccine vehicle: DC targeting, cross neutralization, and immunogenicity. Vaccine 2004, 22, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Lemckert, A.A.; Sumida, S.M.; Holterman, L.; Vogels, R.; Truitt, D.M.; Lynch, D.M.; Nanda, A.; Ewald, B.A.; Gorgone, D.A.; Lifton, M.A.; Goudsmit, J.; Havenga, M.J.; Barouch, D.H. Immunogenicity of heterologous prime-boost regimens involving recombinant adenovirus serotype 11 (Ad11) and Ad35 vaccine vectors in the presence of anti-ad5 immunity. J. Virol. 2005, 79, 9694–9701. [Google Scholar] [CrossRef] [PubMed]
- Sumida, S.M.; Truitt, D.M.; Lemckert, A.A.; Vogels, R.; Custers, J.H.; Addo, M.M.; Lockman, S.; Peter, T.; Peyerl, F.W.; Kishko, M.G.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Essex, M.; Walker, B.D.; Goudsmit, J.; Havenga, M.J.; Barouch, D.H. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J. Immunol. 2005, 174, 7179–7185. [Google Scholar] [PubMed]
- Vlachaki, M.T.; Hernandez-Garcia, A.; Ittmann, M.; Chhikara, M.; Aguilar, L.K.; Zhu, X.; Teh, B.S.; Butler, E.B.; Woo, S.; Thompson, T.C.; Barrera-Saldana, H.; Aguilar-Cordova, E. Impact of preimmunization on adenoviral vector expression and toxicity in a subcutaneous mouse cancer model. Mol. Ther. 2002, 6, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Cichon, G.; Boeckh-Herwig, S.; Schmidt, H.H.; Wehnes, E.; Muller, T.; Pring-Akerblom, P.; Burger, R. Complement activation by recombinant adenoviruses. Gene Ther. 2001, 8, 1794–1800. [Google Scholar] [CrossRef]
- Abrahamsen, K.; Kong, H.L.; Mastrangeli, A.; Brough, D.; Lizonova, A.; Crystal, R.G.; Falck-Pedersen, E. Construction of an adenovirus type 7a E1A- vector. J. Virol. 1997, 71, 8946–8951. [Google Scholar] [PubMed]
- Gao, W.; Robbins, P.D.; Gambotto, A. Human adenovirus type 35: nucleotide sequence and vector development. Gene Ther. 2003, 10, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Peng, B.; Hahn, T.W.; Richardson, E.; Lizonova, A.; Kovesdi, I.; Robert-Guroff, M. Development of an Ad7 cosmid system and generation of an Ad7deltaE1deltaE3HIV(MN) env/rev recombinant virus. Gene Ther. 2003, 10, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Mizuguchi, H.; Yamaguchi, T.; Hayakawa, T. Characterization of in vitro and in vivo gene transfer properties of adenovirus serotype 35 vector. Mol. Ther. 2003, 8, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Holterman, L.; Vogels, R.; van der Vlugt, R.; Sieuwerts, M.; Grimbergen, J.; Kaspers, J.; Geelen, E.; van der Helm, E.; Lemckert, A.; Gillissen, G.; Verhaagh, S.; Custers, J.; Zuijdgeest, D.; Berkhout, B.; Bakker, M.; Quax, P.; Goudsmit, J.; Havenga, M. Novel replication-incompetent vector derived from adenovirus type 11 (Ad11) for vaccination and gene therapy: low seroprevalence and non-cross-reactivity with Ad5. J. Virol. 2004, 78, 13207–13215. [Google Scholar] [CrossRef] [PubMed]
- Sirena, D.; Ruzsics, Z.; Schaffner, W.; Greber, U.F.; Hemmi, S. The nucleotide sequence and a first generation gene transfer vector of species B human adenovirus serotype 3. Virology 2005, 343, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Ni, S.; Li, Z.Y.; Gaggar, A.; DiPaolo, N.; Feng, Q.; Sandig, V.; Lieber, A. Development and assessment of human adenovirus type 11 as a gene transfer vector. J. Virol. 2005, 79, 5090–5104. [Google Scholar] [CrossRef] [PubMed]
- Capone, S.; Meola, A.; Ercole, B.B.; Vitelli, A.; Pezzanera, M.; Ruggeri, L.; Davies, M.E.; Tafi, R.; Santini, C.; Luzzago, A.; Fu, T.M.; Bett, A.; Colloca, S.; Cortese, R.; Nicosia, A.; Folgori, A. A novel adenovirus type 6 (Ad6)-based hepatitis C virus vector that overcomes preexisting anti-ad5 immunity and induces potent and broad cellular immune responses in rhesus macaques. J. Virol. 2006, 80, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Lemckert, A.A.; Grimbergen, J.; Smits, S.; Hartkoorn, E.; Holterman, L.; Berkhout, B.; Barouch, D.H.; Vogels, R.; Quax, P.; Goudsmit, J.; Havenga, M.J. Generation of a novel replication-incompetent adenoviral vector derived from human adenovirus type 49: manufacture on PER.C6 cells, tropism and immunogenicity . J. Gen. Virol. 2006, 87, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Ruzsics, Z.; Wagner, M.; Osterlehner, A.; Cook, J.; Koszinowski, U.; Burgert, H.G. Transposon-assisted cloning and traceless mutagenesis of adenoviruses: Development of a novel vector based on species D. J. Virol. 2006, 80, 8100–8113. [Google Scholar] [CrossRef] [PubMed]
- Ruzsics, Z.; Wagner, M.; Osterlehner, A.; Cook, J.; Koszinowski, U.; Burgert, H.G. Transposon-assisted cloning and traceless mutagenesis of adenoviruses: Development of a novel vector based on species D. J. Virol. 2006, 80, 8100–8113. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Lemckert, A.A.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; O'Brien, K.L.; Carville, A.; Mansfield, K.G.; Goudsmit, J.; Havenga, M.J.; Barouch, D.H. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 2007, 81, 4654–4663. [Google Scholar] [CrossRef] [PubMed]
- Lemiale, F.; Haddada, H.; Nabel, G.J.; Brough, D.E.; King, C.R.; Gall, J.G. Novel adenovirus vaccine vectors based on the enteric-tropic serotype 41. Vaccine 2007, 25, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Klonjkowski, B.; Gilardi-Hebenstreit, P.; Hadchouel, J.; Randrianarison, V.; Boutin, S.; Yeh, P.; Perricaudet, M.; Kremer, E.J. A recombinant E1-deleted canine adenoviral vector capable of transduction and expression of a transgene in human-derived cells and in vivo. Hum. Gene Ther. 1997, 8, 2103–2115. [Google Scholar] [CrossRef]
- Reddy, P.S.; Idamakanti, N.; Chen, Y.; Whale, T.; Babiuk, L.A.; Mehtali, M.; Tikoo, S.K. Replication-defective bovine adenovirus type 3 as an expression vector. J. Virol. 1999, 73, 9137–9144. [Google Scholar] [PubMed]
- Kremer, E.J.; Boutin, S.; Chillon, M.; Danos, O. Canine adenovirus vectors: an alternative for adenovirus-mediated gene transfer. J. Virol. 2000, 74, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Farina, S.F.; Gao, G.P.; Xiang, Z.Q.; Rux, J.J.; Burnett, R.M.; Alvira, M.R.; Marsh, J.; Ertl, H.C.; Wilson, J.M. Replication-defective vector based on a chimpanzee adenovirus. J. Virol. 2001, 75, 11603–11613. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Gao, G.; Lu, Y.; Zhou, X.; Lock, M.; Calcedo, R.; Wilson, J.M. Characterization of a family of chimpanzee adenoviruses and development of molecular clones for gene transfer vectors. Hum. Gene Ther. 2004, 15, 519–530. [Google Scholar] [PubMed]
- Roy, S.; Zhi, Y.; Kobinger, G.P.; Figueredo, J.; Calcedo, R.; Miller, J.R.; Feldmann, H.; Wilson, J.M. Generation of an adenoviral vaccine vector based on simian adenovirus 21. J. Gen. Virol. 2006, 87, 2477–2485. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Segerman, A.; Atkinson, J.P.; Marttila, M.; Dennerquist, V.; Wadell, G.; Arnberg, N. Adenovirus type 11 uses CD46 as a cellular receptor. J. Virol. 2003, 77, 9183–9191. [Google Scholar] [CrossRef] [PubMed]
- Sirena, D.; Lilienfeld, B.; Eisenhut, M.; Kalin, S.; Boucke, K.; Beerli, R.R.; Vogt, L.; Ruedl, C.; Bachmann, M.F.; Greber, U.F.; Hemmi, S. The human membrane cofactor CD46 is a receptor for species B adenovirus serotype 3. J. Virol. 2004, 78, 4454–4462. [Google Scholar] [CrossRef] [PubMed]
- Ornelles, D.A.; Shenk, T. Localization of the adenovirus early region 1B 55-kilodalton protein during lytic infection: association with nuclear viral inclusions requires the early region 4 34-kilodalton protein. J. Virol. 1991, 65, 424–429. [Google Scholar] [PubMed]
- Rubenwolf, S.; Schutt, H.; Nevels, M.; Wolf, H.; Dobner, T. Structural analysis of the adenovirus type 5 E1B 55-kilodalton-E4orf6 protein complex. J. Virol. 1997, 71, 1115–1123. [Google Scholar] [PubMed]
- Weigel, S.; Dobbelstein, M. The nuclear export signal within the E4orf6 protein of adenovirus type 5 supports virus replication and cytoplasmic accumulation of viral mRNA. J. Virol. 2000, 74, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Clawson, D.S.; Lavrukhin, O.; Sandhu, A.; Miller, J.; Wilson, J.M. Rescue of chimeric adenoviral vectors to expand the serotype repertoire. J. Virol. Methods 2007, 141, 14–21. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Kovesdi, I.; Hedley, S.J. Adenoviral Producer Cells. Viruses 2010, 2, 1681-1703. https://doi.org/10.3390/v2081681
Kovesdi I, Hedley SJ. Adenoviral Producer Cells. Viruses. 2010; 2(8):1681-1703. https://doi.org/10.3390/v2081681
Chicago/Turabian StyleKovesdi, Imre, and Susan J. Hedley. 2010. "Adenoviral Producer Cells" Viruses 2, no. 8: 1681-1703. https://doi.org/10.3390/v2081681
APA StyleKovesdi, I., & Hedley, S. J. (2010). Adenoviral Producer Cells. Viruses, 2(8), 1681-1703. https://doi.org/10.3390/v2081681