Lipid Metabolism and HCV Infection

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. HCV epidemiology and outcomes of HCV infection
1.2. Molecular characteristics of HCV
2. Cell organelles and metabolic pathways that contribute to HCV infection
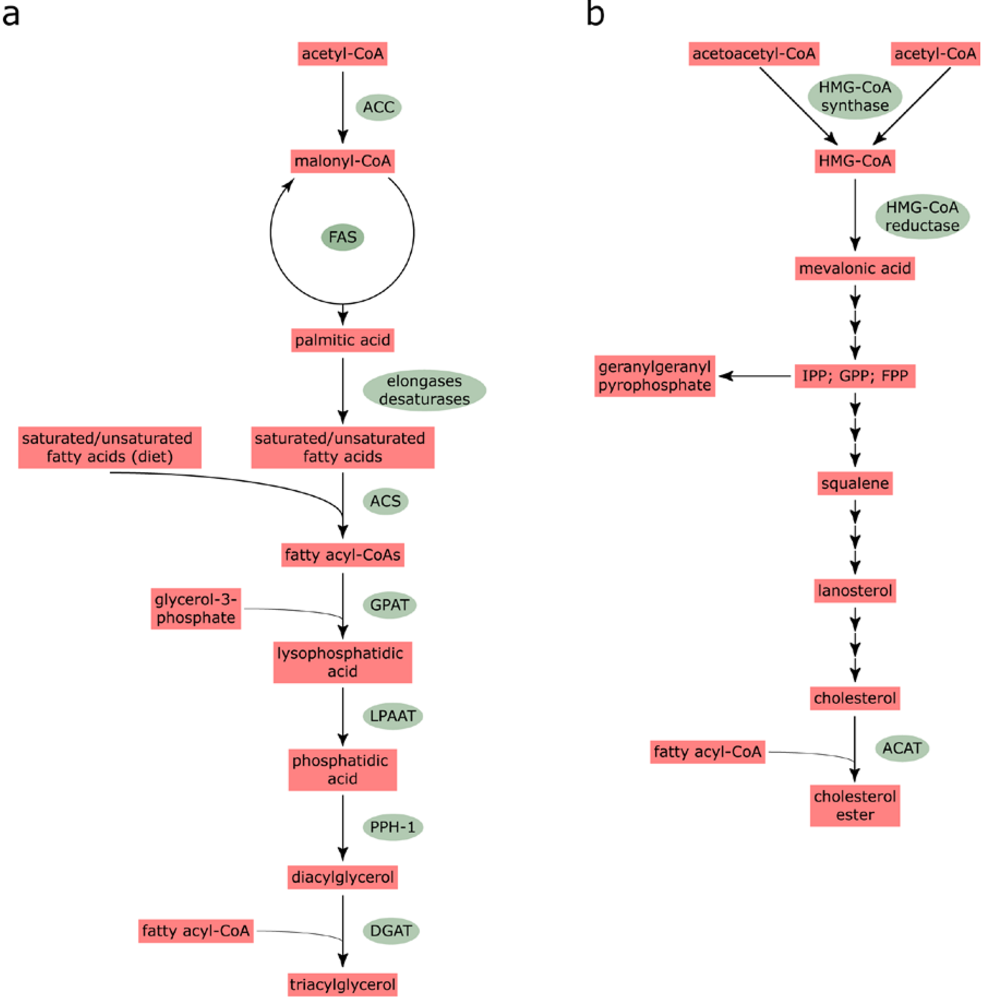
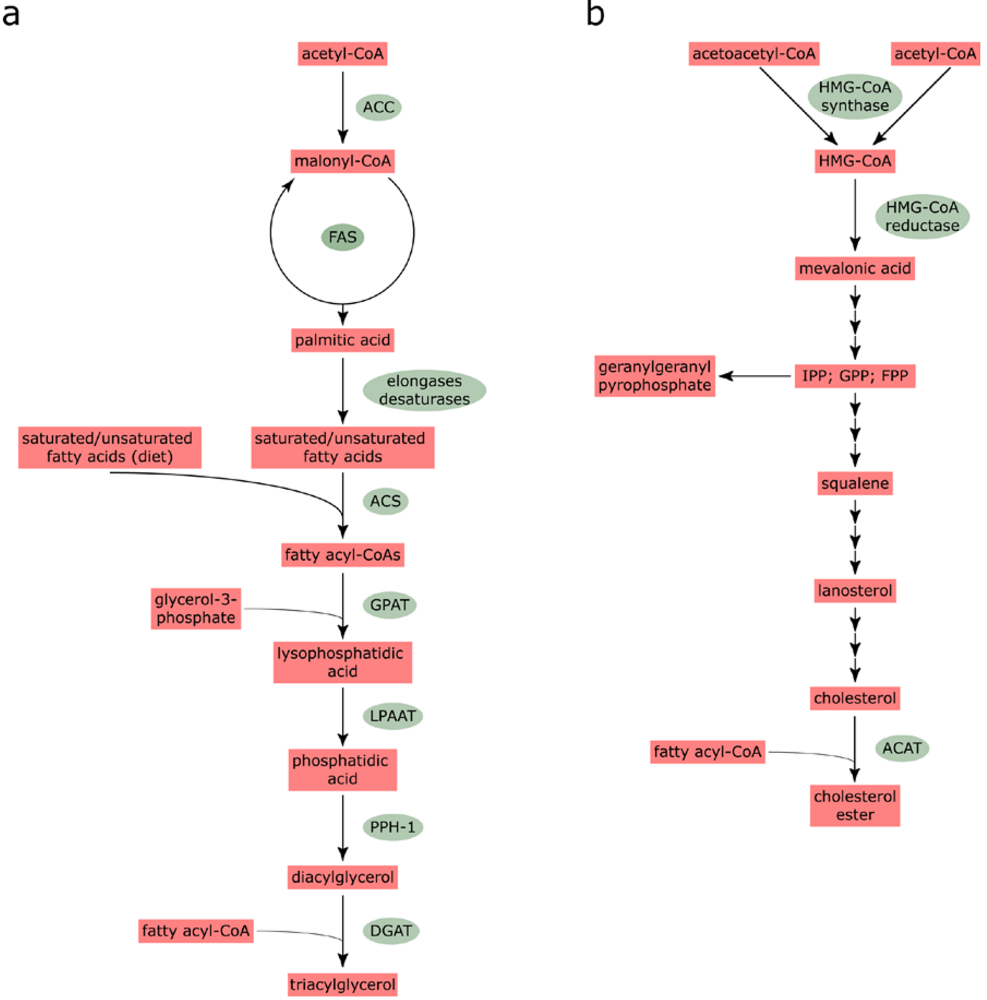
2.1. Fatty acid and cholesterol biosynthesis
2.2. Characteristics of lipid droplets
2.3. Assembly of VLDL
3. HCV proteins that attach to lipid droplets
3.1. Properties of the HCV core protein
3.2. Properties of the HCV NS5A protein
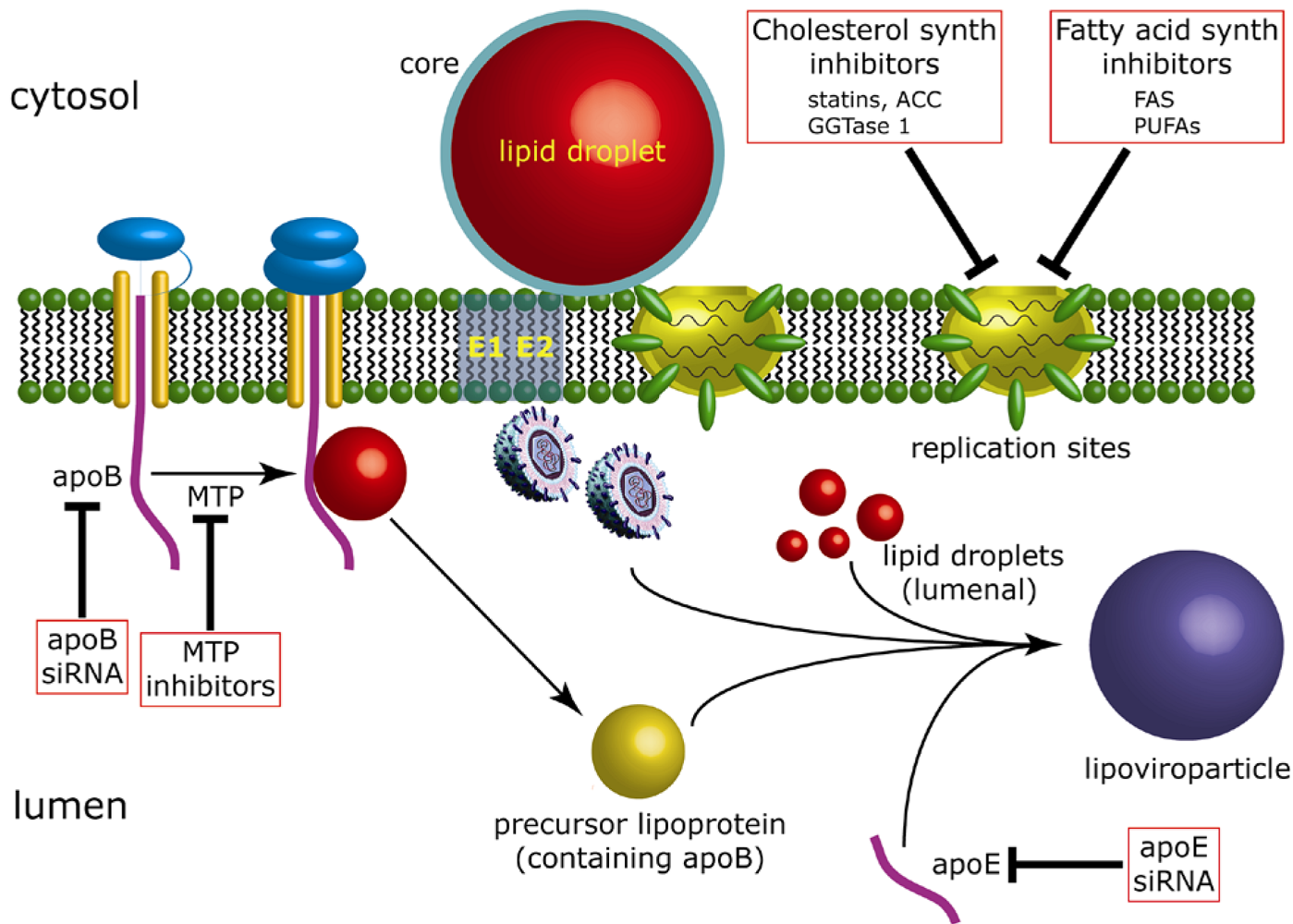
4. The contribution of fatty acid and cholesterol biosynthesis to HCV RNA replication
5. The contribution of lipid droplets to production of infectious HCV
5.1. Lipid droplets are associated with HCV-induced membrane rearrangements
5.2. Virus production is dependent upon recruitment of replication complexes to lipid droplet-associated membranes
5.3. Rationale for targeting replication complexes to lipid droplet-associated regions of the ER membrane
5.4. The link between assembly of HCV and VLDL
6. The role of extracellular factors involved in lipid metabolism that participate in virus entry
6.1. Receptors on the cell surface necessary for virus infection
6.2. Extracellular components that affect virus entry
7. Conclusions and future perspectives

Acknowledgments
References
- The Global Burden of Hepatitis C Working Group. Global burden of disease (GBD) for hepatitis C . J. Clin. Pharmacol. 2004, 44, 20–29. [CrossRef] [PubMed]
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 2007, 13, 2436–2441. [Google Scholar] [PubMed]
- Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board. J. Viral Hepat. 1999, 6, 35–47. [PubMed]
- Di Bisceglie, A.M.; Goodman, Z.D.; Ishak, K.G.; Hoofnagle, J.H.; Melpolder, J.J.; Alter, H.J. Long-term clinical and histopathological follow-up of chronic posttransfusion hepatitis. Hepatology 1991, 14, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M. Hepatitis C. Lancet 1998, 351, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M. Hepatitis C and hepatocellular carcinoma. Hepatology 1997, 26, 34S–38S. [CrossRef] [PubMed]
- Robertson, B.; Myers, G.; Howard, C.; Brettin, T.; Bukh, J.; Gaschen, B.; Gojobori, T.; Maertens, G.; Mizokami, M.; Nainan, O.; Netesov, S.; Nishioka, K.; Shin i, T.; Simmonds, P.; Smith, D.; Stuyver, L.; Weiner, A. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch. Virol. 1998, 143, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deleage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspe, G.; Kuiken, C.; Maertens, G.; Mizokami, M.; Murphy, D.G.; Okamoto, H.; Pawlotsky, J.M.; Penin, F.; Sablon, E.; Shin, I.T.; Stuyver, L.J.; Thiel, H.J.; Viazov, S.; Weiner, A.J.; Widell, A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Holmes, E.C.; Cha, T.A.; Chan, S.W.; McOmish, F.; Irvine, B.; Beall, E.; Yap, P.L.; Kolberg, J.; Urdea, M.S. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J. Gen. Virol. 1993, 74, 2391–2399. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.W.; George, J. Molecular mechanisms of insulin resistance in chronic hepatitis C. World J. Gastroenterol. 2009, 15, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Clement, S. Impact of obesity, steatosis and insulin resistance on progression and response to therapy of hepatitis C. J. Viral Hepat. 2009, 16, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Thomssen, R.; Bonk, S.; Propfe, C.; Heermann, K.H.; Kochel, H.G.; Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. (Berl). 1992, 181, 293–300. [Google Scholar] [CrossRef]
- Thomssen, R.; Bonk, S.; Thiele, A. Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med. Microbiol. Immunol. (Berl). 1993, 182, 329–334. [Google Scholar] [CrossRef]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Schaller, T.; Penin, F.; Bartenschlager, R. From structure to function: new insights into hepatitis C virus RNA replication. J. Biol. Chem. 2006, 281, 9833–9836. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Frese, M.; Pietschmann, T. Novel insights into hepatitis C virus replication and persistence. Adv. Virus. Res. 2004, 63, 71–180. [Google Scholar] [PubMed]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.M.; Patel, A.H.; Targett-Adams, P.; McLauchlan, J. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J. Virol. 2009, 83, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008, 4, e1000032. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 2007, 81, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.P.; Egger, D.; Bienz, K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Witkowski, A.; Joshi, A.K. Structural and functional organization of the animal fatty acid synthase. Prog. Lip. Res. 2003, 42, 289–317. [Google Scholar] [CrossRef]
- Maier, T.; Jenni, S.; Ban, N. Architecture of mammalian fatty acid synthase at 4.5 A resolution. Science 2006, 311, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Guillou, H.; Zadravec, D.; Martin, P.G.; Jacobsson, A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog. Lip. Res. 2010, 49, 186–199. [Google Scholar] [CrossRef]
- Yen, C.L.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lip. Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef]
- Shi, Y.; Cheng, D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am. J. Physiol. 2009, 297, E10–E18. [Google Scholar] [CrossRef]
- McTaggart, S.J. Isoprenylated proteins. Cell Mol. Life Sci. 2006, 63, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. 2009, 297, E1–E9. [Google Scholar] [CrossRef]
- Tauchi-Sato, K.; Ozeki, S.; Houjou, T.; Taguchi, R.; Fujimoto, T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty Acid composition. J. Biol. Chem. 2002, 277, 44507–44512. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Parton, R.G. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell. Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Bickel, P.E.; Tansey, J.T.; Welte, M.A. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim. Biophys. Acta 2009, 1791, 419–440. [Google Scholar] [PubMed]
- Olofsson, S.O.; Bostrom, P.; Andersson, L.; Rutberg, M.; Perman, J.; Boren, J. Lipid droplets as dynamic organelles connecting storage and efflux of lipids. Biochim. Biophys. Acta 2009, 1791, 448–458. [Google Scholar] [PubMed]
- Mahley, R.W.; Innerarity, T.L.; Rall, S.C.; Weisgraber, K.H. Plasma lipoproteins: apolipoprotein structure and function. J. Lip. Res. 1984, 25, 1277–1294. [Google Scholar]
- Kane, J.P. Apolipoprotein B: structural and metabolic heterogeneity. Annu. Rev. Physiol. 1983, 45, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Elovson, J.; Chatterton, J.E.; Bell, G.T.; Schumaker, V.N.; Reuben, M.A.; Puppione, D.L.; Reeve, J.R.; Young, N.L. Plasma very low density lipoproteins contain a single molecule of apolipoprotein. J. Lip. Res. 1988, 29, 1461–1473. [Google Scholar]
- Wetterau, J.R.; Combs, K.A.; Spinner, S.N.; Joiner, B.J. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J. Biol. Chem. 1990, 265, 9800–9807. [Google Scholar] [PubMed]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J. Lip. Res. 2003, 44, 22–32. [Google Scholar] [CrossRef]
- Kulinski, A.; Rustaeus, S.; Vance, J.E. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J. Biol. Chem. 2002, 277, 31516–31525. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Liang, J.-S.; Fisher, E.A.; Ginsberg, H.N. The late addition of core lipids to nascent apolipoprotein B100, resulting in the assembly and secretion of triglyceride-rich lipoproteins, is independent of both microsomal triglyceride transfer protein activity and new triglyceride synthesis. J. Biol. Chem. 2002, 277, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.F.; Wiggins, D.; Brown, A.M.; Hebbachi, A.M. Synthesis and function of hepatic very-low-density lipoprotein. Biochem. Soc. Trans. 2004, 32, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Itabe, H.; Sakai, J.; Makita, M.; Noda, J.; Mori, M.; Higashi, Y.; Kojima, S.; Takano, T. Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim. Biophys. Acta 2004, 1644, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, B.; Asp, L.; Bostrom, P.; Ruiz, M.; Stillemark-Billton, P.; Linden, D.; Boren, J.; Olofsson, S.O. Adipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Lehner, R.; Vance, D.E. Cloning and expression of a cDNA encoding a hepatic microsomal lipase that mobilizes stored triacylglycerol. Biochem. J. 1999, 343, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.; Alam, M.; Sun, F.; Agellon, L.B.; Vance, D.E.; Lehner, R. Apolipoprotein B and triacylglycerol secretion in human triacylglycerol hydrolase transgenic mice. J. Lip. Res. 2007, 48, 2597–2606. [Google Scholar] [CrossRef]
- Yamazaki, T.; Sasaki, E.; Kakinuma, C.; Yano, T.; Miura, S.; Ezaki, O. Increased very low density lipoprotein secretion and gonadal fat mass in mice overexpressing liver DGAT1. J. Biol. Chem. 2005, 280, 21506–21514. [Google Scholar] [CrossRef] [PubMed]
- Buhman, K.F.; Accad, M.; Farese, R.V. Mammalian acyl-CoA:cholesterol acyltransferases. Biochim. Biophys. Acta 2000, 1529, 142–154. [Google Scholar] [PubMed]
- Buhman, K.K.; Accad, M.; Novak, S.; Choi, R.S.; Wong, J.S.; Hamilton, R.L.; Turley, S.; Farese, R.V. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat. Med. 2000, 6, 1341–1347. [Google Scholar] [CrossRef]
- Liang, J.J.; Oelkers, P.; Guo, C.; Chu, P.C.; Dixon, J.L.; Ginsberg, H.N.; Sturley, S.L. Overexpression of human diacylglycerol acyltransferase 1, acyl-coa:cholesterol acyltransferase 1, or acyl-CoA:cholesterol acyltransferase 2 stimulates secretion of apolipoprotein B-containing lipoproteins in McA-RH7777 cells. J. Biol. Chem. 2004, 279, 44938–44944. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.S.; Stone, S.J.; Tietge, U.J.; Tow, B.; Billheimer, J.T.; Wong, J.S.; Hamilton, R.L.; Farese, R.V.; Rader, D.J. Short-term overexpression of DGAT1 or DGAT2 increases hepatic triglyceride but not VLDL triglyceride or apoB production. J. Lip. Res. 2006, 47, 2297–2305. [Google Scholar] [CrossRef]
- Hussy, P.; Langen, H.; Mous, J.; Jacobsen, H. Hepatitis C virus core protein: carboxy-terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. Virology 1996, 224, 93–104. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. Embo J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Santolini, E.; Migliaccio, G.; La Monica, N. Biosynthesis and biochemical properties of the hepatitis C virus core protein. J. Virol. 1994, 68, 3631–3641. [Google Scholar] [PubMed]
- Ogino, T.; Fukuda, H.; Imajoh-Ohmi, S.; Kohara, M.; Nomoto, A. Membrane binding properties and terminal residues of the mature hepatitis C virus capsid protein in insect cells. J. Virol. 2004, 78, 11766–11777. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J. Lipid droplets and hepatitis C virus infection. Biochim. Biophys. Acta 2009, 1791, 552–559. [Google Scholar] [PubMed]
- McLauchlan, J. Properties of the hepatitis C virus core protein: a structural protein that modulates cellular processes. J. Viral Hepat. 2000, 7, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Montserret, R.; Hope, R.G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.P.; Penin, F.; McLauchlan, J. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Vanbelle, C.; Ebel, C.; Penin, F.; Lavergne, J.P. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J. Virol. 2005, 79, 11353–11365. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Gorbalenya, A.E.; Rice, C.M. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 2004, 279, 48576–48587. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Pietschmann, T.; Bartenschlager, R. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 2005, 79, 3187–3194. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Evans, M.J.; Gosert, R.; Yuan, Z.; Blum, H.E.; Goff, S.P.; Lindenbach, B.D.; Rice, C.M. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 2004, 78, 7400–7409. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Foss, K.L.; Treadaway, J.C.; Rice, C.M. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 2008, 82, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Tellinghuisen, T.L.; Marcotrigiano, J.; Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 2005, 435, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Brass, V.; Bieck, E.; Montserret, R.; Wolk, B.; Hellings, J.A.; Blum, H.E.; Penin, F.; Moradpour, D. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2002, 277, 8130–8139. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Brass, V.; Appel, N.; Ramboarina, S.; Montserret, R.; Ficheux, D.; Blum, H.E.; Bartenschlager, R.; Moradpour, D. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 2004, 279, 40835–40843. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.T.; Polyak, S.J.; Tu, H.; Taylor, D.R.; Gretch, D.R.; Lai, M.M. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 2002, 292, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Bigger, C.B.; Brasky, K.M.; Lanford, R.E. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 2001, 75, 7059–7066. [Google Scholar] [CrossRef] [PubMed]
- Bigger, C.B.; Guerra, B.; Brasky, K.M.; Hubbard, G.; Beard, M.R.; Luxon, B.A.; Lemon, S.M.; Lanford, R.E. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J. Virol. 2004, 78, 13779–13792. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; Chisari, F.V. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef]
- Ye, J.; Wang, C.; Sumpter, R.; Brown, M.S.; Goldstein, J.L.; Gale, M. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl. Acad. Sci. USA 2003, 100, 15865–15870. [Google Scholar] [CrossRef]
- Delang, L.; Paeshuyse, J.; Vliegen, I.; Leyssen, P.; Obeid, S.; Durantel, D.; Zoulim, F.; Op de Beeck, A.; Neyts, J. Statins potentiate the in vitro anti-hepatitis C virus activity of selective hepatitis C virus inhibitors and delay or prevent resistance development. Hepatology 2009, 50, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Abe, K.; Yamada, M.; Dansako, H.; Naka, K.; Kato, N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 2006, 44, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Bader, T.; Fazili, J.; Madhoun, M.; Aston, C.; Hughes, D.; Rizvi, S.; Seres, K.; Hasan, M. Fluvastatin inhibits hepatitis C replication in humans. Am. J. Gastroenterol. 2008, 103, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Madhoun, M.F.; Bader, T. Statins improve ALT values in chronic hepatitis C patients with abnormal values. Dig. Dis. Sci. 2010, 55, 870–871. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, L.; Caramma, I.; Mazzali, C.; Cesari, M.; Olivetti, M.; Galli, M.; Antinori, S. Fluvastatin as an adjuvant to pegylated interferon and ribavirin in HIV/hepatitis C virus genotype 1 co-infected patients: an open-label randomized controlled study. J. Antimicrob. Chemother. 2010, 65, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Forde, K.A.; Law, C.; O'Flynn, R.; Kaplan, D.E. Do statins reduce hepatitis C RNA titers during routine clinical use? World J. Gastroenterol. 2009, 15, 5020–5027. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, J.G.; Chan, J.L.; McMahon, C.M.; Chung, R.T. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: a pilot clinical trial. Hepatology 2007, 45, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gale, M.; Keller, B.C.; Huang, H.; Brown, M.S.; Goldstein, J.L.; Ye, J. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 2005, 18, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Leu, G.Z.; Lin, T.Y.; Hsu, J.T. Anti-HCV activities of selective polyunsaturated fatty acids. Biochem. Biophys. Res. Commun. 2004, 318, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, Y.; Ye, J. Inhibition of hepatitis C virus replication by peroxidation of arachidonate and restoration by vitamin E. Proc. Natl. Acad. Sci. USA 2007, 104, 18666–18670. [Google Scholar] [CrossRef]
- Westaway, E.G.; Mackenzie, J.M.; Kenney, M.T.; Jones, M.K.; Khromykh, A.A. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 1997, 71, 6650–6661. [Google Scholar] [PubMed]
- Roingeard, P.; Hourioux, C.; Blanchard, E.; Prensier, G. Hepatitis C virus budding at lipid droplet-associated ER membrane visualized by 3D electron microscopy. Histochem. Cell Biol. 2008, 130, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Boulant, S.; McLauchlan, J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008, 82, 2182–2195. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef] [PubMed]
- Rouille, Y.; Helle, F.; Delgrange, D.; Roingeard, P.; Voisset, C.; Blanchard, E.; Belouzard, S.; McKeating, J.; Patel, A.H.; Maertens, G.; Wakita, T.; Wychowski, C.; Dubuisson, J. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J. Virol. 2006, 80, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; Hope, G.; Boulant, S.; McLauchlan, J. Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J. Biol. Chem. 2008, 283, 16850–16859. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient initiation of HCV RNA replication in cell culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Brass, V.; Penin, F. Function follows form: the structure of the N-terminal domain of HCV NS5A. Hepatology 2005, 42, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Suzuki, R.; Murakami, K.; Aizaki, H.; Ishii, K.; Murayama, A.; Date, T.; Matsuura, Y.; Miyamura, T.; Wakita, T.; Suzuki, T. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 2008, 82, 7964–7976. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Zayas, M.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 2005, 19, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Hwang, J.; Sharma, S.D.; Hargittai, M.R.; Chen, Y.; Arnold, J.J.; Raney, K.D.; Cameron, C.E. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 2005, 280, 36417–36428. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Malcolm, B.A. Trans-complementation of HCV replication by non-structural protein 5A. Virus Res. 2006, 115, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Appel, N.; Herian, U.; Bartenschlager, R. Efficient rescue of hepatitis C virus RNA replication by trans-complementation with nonstructural protein 5A. J. Virol. 2005, 79, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Tanji, Y.; Kaneko, T.; Satoh, S.; Shimotohno, K. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 1995, 69, 3980–3986. [Google Scholar] [PubMed]
- Reed, K.E.; Xu, J.; Rice, C.M. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J. Virol. 1997, 71, 7187–7197. [Google Scholar] [PubMed]
- Reed, K.E.; Rice, C.M. Identification of the major phosphorylation site of the hepatitis C virus H strain NS5A protein as serine 2321. J. Biol. Chem. 1999, 274, 28011–28018. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Rice, C.M.; Goff, S.P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. USA 2004, 101, 13038–13043. [Google Scholar] [CrossRef]
- Neddermann, P.; Quintavalle, M.; Di Pietro, C.; Clementi, A.; Cerretani, M.; Altamura, S.; Bartholomew, L.; De Francesco, R. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 2004, 78, 13306–13314. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Hoffmann, S.; Herian, U.; Penin, F.; Bartenschlager, R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol . 2003, 77, 3007–3019. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2008, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M.; Ye, J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef]
- Chang, K.S.; Jiang, J.; Cai, Z.; Luo, G. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Luo, G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 2009, 83, 12680–12691. [Google Scholar] [CrossRef] [PubMed]
- Benga, W.J.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; McLauchlan, J.; Baumert, T.F.; Schuster, C. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 2010, 51, 43–53. [Google Scholar] [PubMed]
- Andre, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Brechot, C.; Paranhos-Baccala, G.; Lotteau, V. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 2002, 76, 6919–6928. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Dubuisson, J. Recent advances in hepatitis C virus cell entry. Viruses 2010, 2, 692–709. [Google Scholar] [CrossRef]
- Burlone, M.E.; Budkowska, A. Hepatitis C virus cell entry: role of lipoproteins and cellular receptors. J. Gen. Virol. 2009, 90, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Abel, G.; Elfahal, M.; Knight, G.B.; Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 12766–12771. [Google Scholar] [CrossRef]
- Germi, R.; Crance, J.M.; Garin, D.; Guimet, J.; Lortat-Jacob, H.; Ruigrok, R.W.; Zarski, J.P.; Drouet, E. Cellular glycosaminoglycans and low density lipoprotein receptor are involved in hepatitis C virus adsorption. J. Med. Virol. 2002, 68, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Pileri, P.; Uematsu, Y.; Campagnoli, S.; Galli, G.; Falugi, F.; Petracca, R.; Weiner, A.J.; Houghton, M.; Rosa, D.; Grandi, G.; Abrignani, S. Binding of hepatitis C virus to CD81. Science 1998, 282, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. Embo J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; von Hahn, T.; Tscherne, D.M.; Syder, A.J.; Panis, M.; Wolk, B.; Hatziioannou, T.; McKeating, J.A.; Bieniasz, P.D.; Rice, C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 2007, 446, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Evans, M.J.; Gaysinskaya, V.A.; Panis, M.; You, H.; de Jong, Y.P.; Rice, C.M. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 2009, 457, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Beglova, N.; Blacklow, S.C. The LDL receptor: how acid pulls the trigger. Trends Biochem. Sci. 2005, 30, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; Smolarsky, M.; Funaro, A.; Malavasi, F.; Larrey, D.; Coste, J.; Fabre, J.M.; Sa-Cunha, A.; Maurel, P. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.M.; Huang, H.; Ye, J.; Gale, M. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 2009, 394, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Andreo, U.; Maillard, P.; Kalinina, O.; Walic, M.; Meurs, E.; Martinot, M.; Marcellin, P.; Budkowska, A. Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell. Microbiol. 2007, 9, 2445–2456. [Google Scholar] [CrossRef]
- Connelly, M.A.; Williams, D.L. Scavenger receptor BI: a scavenger receptor with a mission to transport high density lipoprotein lipids. Curr. Opin. Lipidol. 2004, 15, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Graziani, R.; von Hahn, T.; Moreau, M.; Huby, T.; Paonessa, G.; Santini, C.; Luzzago, A.; Rice, C.M.; Cortese, R.; Vitelli, A.; Nicosia, A. High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J. Virol. 2007, 81, 8063–8071. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Huby, T.; Stamataki, Z.; Vanwolleghem, T.; Meuleman, P.; Farquhar, M.; Schwarz, A.; Moreau, M.; Owen, J.S.; Leroux-Roels, G.; Balfe, P.; McKeating, J.A. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J. Virol. 2007, 81, 3162–3169. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Barth, H.; Baumert, T.; McKeating, J.A.; Chisari, F.V. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 2007, 81, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Koutsoudakis, G.; Schnober, E.K.; Haberstroh, A.; Blum, H.E.; Cosset, F. L.; Wakita, T.; Jaeck, D.; Doffoel, M.; Royer, C.; Soulier, E.; Schvoerer, E.; Schuster, C.; Stoll-Keller, F.; Bartenschlager, R.; Pietschmann, T.; Barth, H.; Baumert, T.F. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 2007, 46, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Voisset, C.; Callens, N.; Blanchard, E.; Op De Beeck, A.; Dubuisson, J.; Vu-Dac, N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J. Biol. Chem. 2005, 280, 7793–7799. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.; Huby, T.; Andreo, U.; Moreau, M.; Chapman, J.; Budkowska, A. The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-containing lipoproteins. FASEB J. 2006, 20, 735–737. [Google Scholar] [PubMed]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar] [CrossRef]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; Baryza, J.; Tallarico, J.; Joberty, G.; Bantscheff, M.; Schirle, M.; Bouwmeester, T.; Mathy, J.E.; Lin, K.; Compton, T.; Labow, M.; Wiedmann, B.; Gaither, L.A. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.H.; Pilote, L.; Cartier, M.; Lippens, J.; Liuzzi, M.; Bethell, R.C.; Cordingley, M.G.; Kukolj, G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009, 387, 5–10. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Targett-Adams, P.; Boulant, S.; Douglas, M.W.; McLauchlan, J. Lipid Metabolism and HCV Infection. Viruses 2010, 2, 1195-1217. https://doi.org/10.3390/v2051195
Targett-Adams P, Boulant S, Douglas MW, McLauchlan J. Lipid Metabolism and HCV Infection. Viruses. 2010; 2(5):1195-1217. https://doi.org/10.3390/v2051195
Chicago/Turabian StyleTargett-Adams, Paul, Steeve Boulant, Mark W. Douglas, and John McLauchlan. 2010. "Lipid Metabolism and HCV Infection" Viruses 2, no. 5: 1195-1217. https://doi.org/10.3390/v2051195
APA StyleTargett-Adams, P., Boulant, S., Douglas, M. W., & McLauchlan, J. (2010). Lipid Metabolism and HCV Infection. Viruses, 2(5), 1195-1217. https://doi.org/10.3390/v2051195