Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles

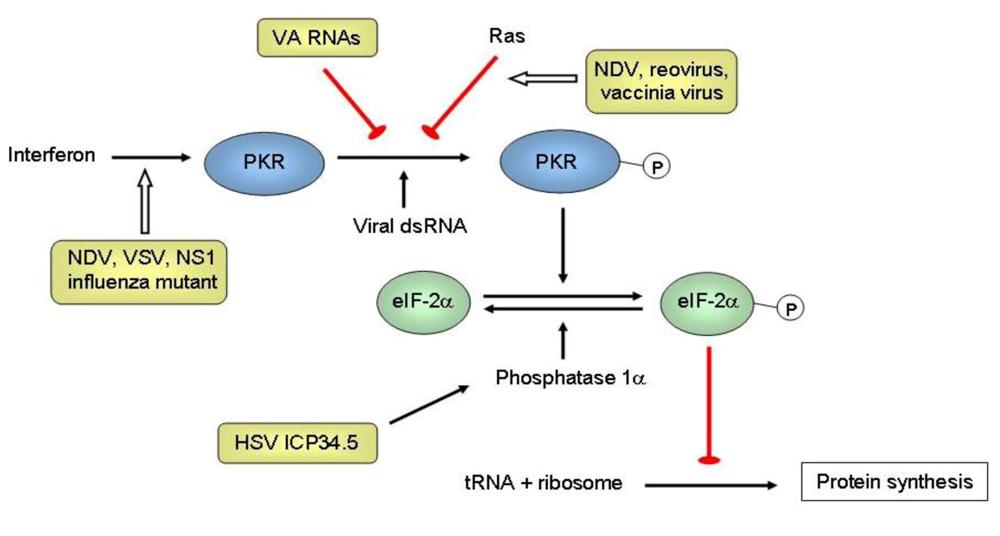
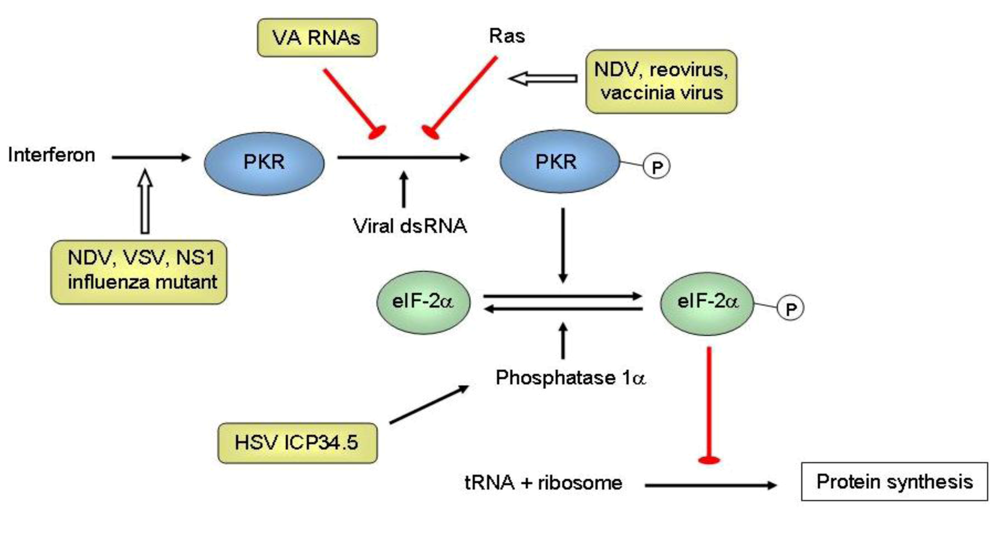{kind=link}
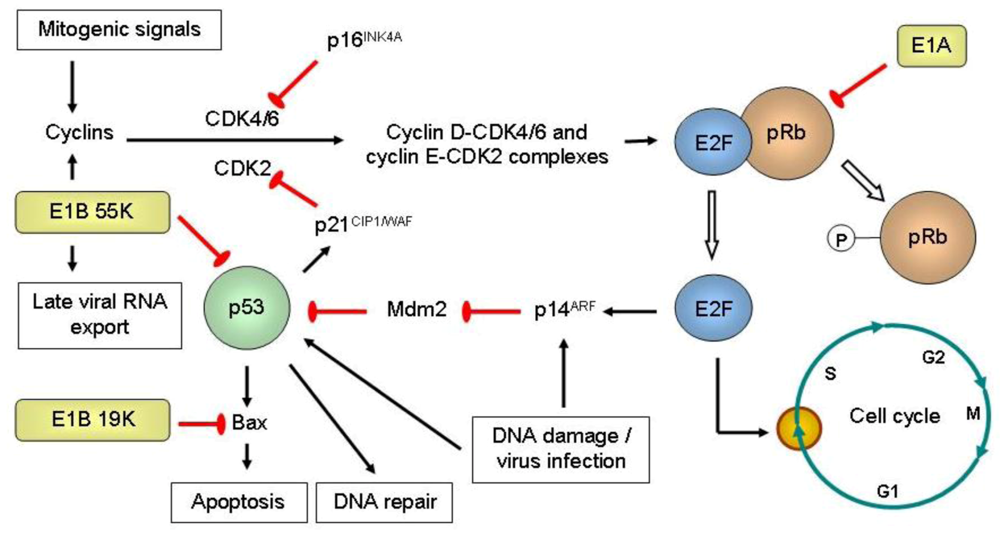{kind=link}
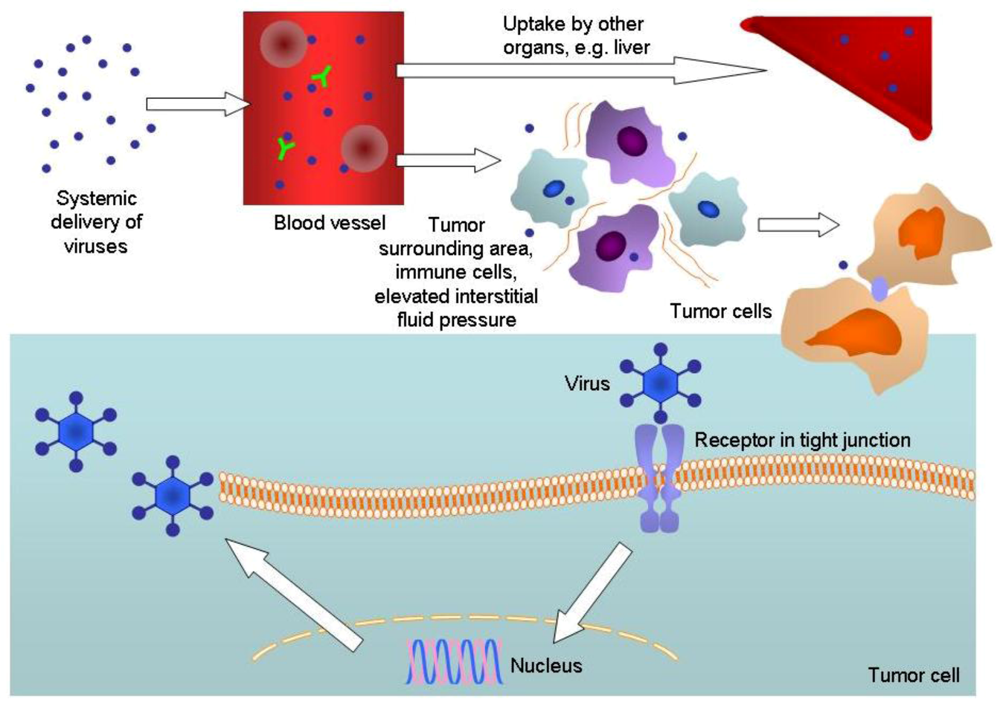{kind=link}
Abstract
1. Introduction
Mechanisms of tumor selectivity
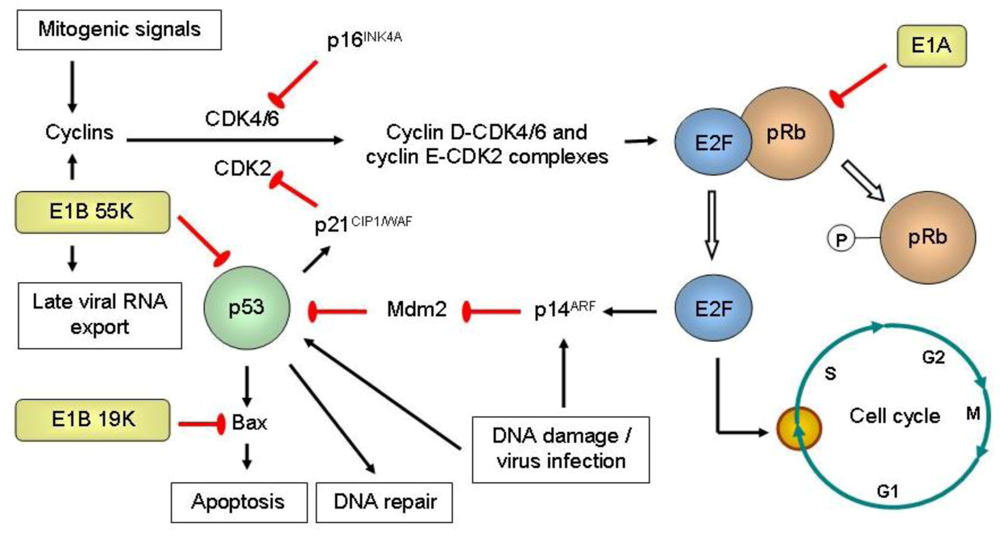
Optimizing oncolytic viruses for improved anti-tumoral potency
Arming oncolytic viruses with therapeutic genes
The tumor environment and oncolytic viruses
Modification of the host immune response in favor of oncolytic viruses
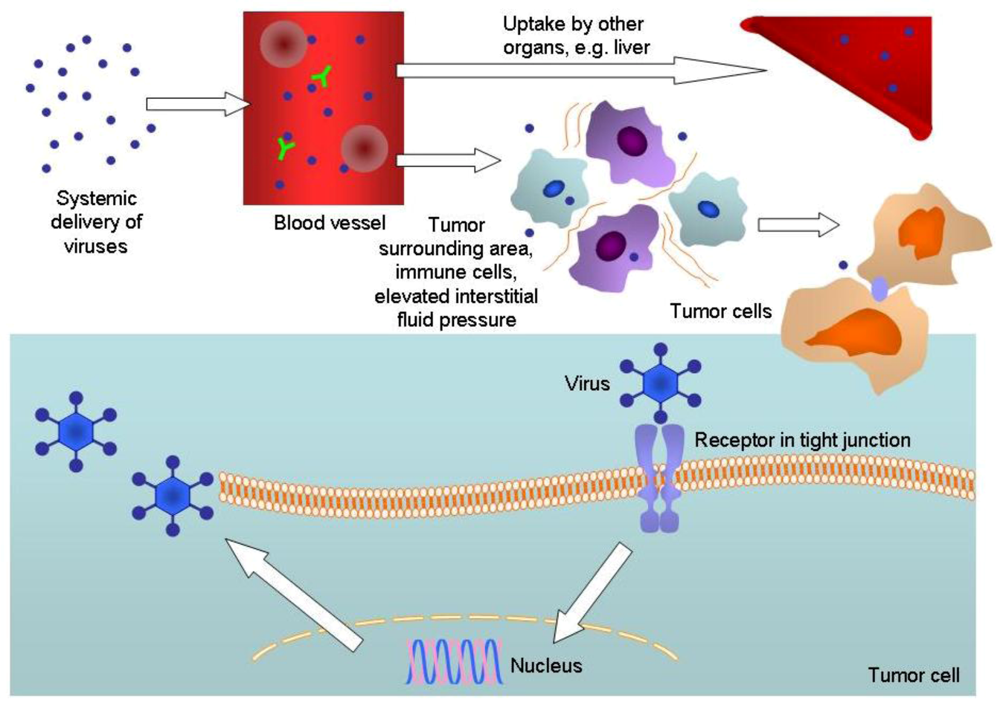
2. Conclusions
3. Competing interests
Acknowledgments
References
- Kelly, E.; Russell, S.J. History of oncolytic viruses: genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [PubMed]
- Garber, K. China approves world's first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [PubMed]
- Thorne, S.H.; Hwang, T.H.; O'Gorman, W.E.; Bartlett, D.L.; Sei, S.; Kanji, F.; Brown, C.; Werier, J.; Cho, J.H.; Lee, D.E.; Wang, Y.; Bell, J.; Kirn, D.H. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J. Clin. Invest. 2007, 117, 3350–3358. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gros, A.; Jose, A.; Gonzalez, J.R.; Alemany, R.; Fillat, C. Urokinase-type plasminogen activator receptor transcriptionally controlled adenoviruses eradicate pancreatic tumors and liver metastasis in mouse models. Neoplasia 2009, 11, 518–528. [Google Scholar] [PubMed]
- Pan, W.; Bodempudi, V.; Esfandyari, T.; Farassati, F. Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One 2009, 4, e6514. [Google Scholar] [CrossRef] [PubMed]
- Cafferata, E.G.; Maccio, D.R.; Lopez, M.V.; Viale, D.L.; Carbone, C.; Mazzolini, G.; Podhajcer, O.L. A novel A33 promoter-based conditionally replicative adenovirus suppresses tumor growth and eradicates hepatic metastases in human colon cancer models. Clin. Cancer Res. 2009, 15, 3037–3049. [Google Scholar] [CrossRef]
- Hsieh, J.L.; Lee, C.H.; Teo, M.L.; Lin, Y.J.; Huang, Y.S.; Wu, C.L.; Shiau, A.L. Transthyretin-driven oncolytic adenovirus suppresses tumor growth in orthotopic and ascites models of hepatocellular carcinoma. Cancer Sci. 2009, 100, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, O.; Matsunaga, A.; Ichimaru, D.; Urata, Y.; Fujiwara, T.; Kawakami, K. Telomerase-specific virotherapy in an animal model of human head and neck cancer. Mol. Cancer Ther. 2009, 8, 171–177. [Google Scholar] [CrossRef]
- Doloff, J.C.; Waxman, D.J.; Jounaidi, Y. Human telomerase reverse transcriptase promoter-driven oncolytic adenovirus with E1B-19 kDa and E1B-55 kDa gene deletions. Hum. Gene Ther. 2008, 19, 1383–1400. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.F.; Wu, C.L.; Huang, S.C.; Hsieh, J.L.; Huang, Y.S.; Chen, Y.F.; Shen, M.R.; Chung, W.J.; Chou, C.Y.; Shiau, A.L. Conditionally replicating E1B-deleted adenovirus driven by the squamous cell carcinoma antigen 2 promoter for uterine cervical cancer therapy. Cancer Gene Ther. 2008, 15, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Shafren, D.R.; Dorahy, D.J.; Ingham, R.A.; Burns, G.F.; Barry, R.D. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J. Virol. 1997, 71, 4736–4743. [Google Scholar] [PubMed]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Yoshida, K.; Miura, Y.; Kobayashi, A.; Hara, H.; Ohnami, S.; Kurisu, K.; Yoshida, T.; Aoki, K. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther. 2009, 16, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.; Braidwood, L.; Brown, S.M. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther. 2008, 15, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Vallath, S.; Saha, A.; Flak, M.; McNeish, I.A.; Vassaux, G.; Marshall, J.F.; Hart, I.R.; Thomas, G.J. In vivo retargeting of adenovirus type 5 to alphavbeta6 integrin results in reduced hepatotoxicity and improved tumor uptake following systemic delivery. J. Virol. 2009, 83, 6416–6428. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.M.; Rodrigues, M.S.; Phadke, A.P.; Butcher, L.D.; Starling, C.; Chen, S.; Chang, D.; Hernandez-Alcoceba, R.; Newman, J.T.; Stone, M.J.; Tong, A.W. Antitumor activity of an oncolytic adenoviral-CD40 ligand (CD154) transgene construct in human breast cancer cells. Clin. Cancer Res. 2009, 15, 1317–1325. [Google Scholar] [CrossRef]
- Piao, Y.; Jiang, H.; Alemany, R.; Krasnykh, V.; Marini, F.C.; Xu, J.; Alonso, M.M.; Conrad, C.A.; Aldape, K.D.; Gomez-Manzano, C.; Fueyo, J. Oncolytic adenovirus retargeted to Delta-EGFR induces selective antiglioma activity. Cancer Gene Ther. 2009, 16, 256–265. [Google Scholar] [PubMed]
- Morrison, J.; Briggs, S.S.; Green, N.; Fisher, K.; Subr, V.; Ulbrich, K.; Kehoe, S.; Seymour, L.W. Virotherapy of ovarian cancer with polymer-cloaked adenovirus retargeted to the epidermal growth factor receptor. Mol. Ther. 2008, 16, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Paraskevakou, G.; Iankov, I.; Giannini, C.; Schroeder, M.; Sarkaria, J.; Puri, R.K.; Russell, S.J.; Galanis, E. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol. Ther. 2008, 16, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Lorence, R.M.; Katubig, B.B.; Reichard, K.W.; Reyes, H.M.; Phuangsab, A.; Sassetti, M.D.; Walter, R.J.; Peeples, M.E. Complete regression of human fibrosarcoma xenografts after local Newcastle disease virus therapy. Cancer Res. 1994, 54, 6017–6021. [Google Scholar] [PubMed]
- Coffey, M.C.; Strong, J.E.; Forsyth, P.A.; Lee, P.W. Reovirus therapy of tumors with activated Ras pathway. Science 1998, 282, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Whitley, R.J.; Kern, E.R.; Chatterjee, S.; Chou, J.; Roizman, B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Invest. 1993, 91, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Muster, T.; Rajtarova, J.; Sachet, M.; Unger, H.; Fleischhacker, R.; Romirer, I.; Grassauer, A.; Url, A.; Garcia-Sastre, A.; Wolff, K.; Pehamberger, H.; Bergmann, M. Interferon resistance promotes oncolysis by influenza virus NS1-deletion mutants. Int. J. Cancer 2004, 110, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Cascallo, M.; Capella, G.; Mazo, A.; Alemany, R. Ras-dependent oncolysis with an adenovirus VAI mutant. Cancer Res. 2003, 63, 5544–5550. [Google Scholar] [PubMed]
- Wang, Y.; Xue, S.A.; Hallden, G.; Francis, J.; Yuan, M.; Griffin, B.E.; Lemoine, N.R. Virus-associated RNA I-deleted adenovirus, a potential oncolytic agent targeting EBV-associated tumors. Cancer Res. 2005, 65, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef]
- Liu, T.C.; Hallden, G.; Wang, Y.; Brooks, G.; Francis, J.; Lemoine, N.; Kirn, D. An E1B-19 kDa gene deletion mutant adenovirus demonstrates tumor necrosis factor-enhanced cancer selectivity and enhanced oncolytic potency. Mol. Ther. 2004, 9, 786–803. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; McCormick, F. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Rao, X.M.; Gomez-Gutierrez, J.G.; Hao, H.; McMasters, K.M.; Zhou, H.S. Adenovirus E1B55K region is required to enhance cyclin E expression for efficient viral DNA replication. J. Virol. 2008, 82, 3415–3427. [Google Scholar] [CrossRef] [PubMed]
- O'Shea, C.C.; Johnson, L.; Bagus, B.; Choi, S.; Nicholas, C.; Shen, A.; Boyle, L.; Pandey, K.; Soria, C.; Kunich, J.; Shen, Y.; Habets, G.; Ginzinger, D.; McCormick, F. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 2004, 6, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Ylosmaki, E.; Hakkarainen, T.; Hemminki, A.; Visakorpi, T.; Andino, R.; Saksela, K. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific MicroRNA. J. Virol. 2008, 82, 11009–11015. [Google Scholar] [CrossRef] [PubMed]
- Cawood, R.; Chen, H.H.; Carroll, F.; Bazan-Peregrino, M.; van Rooijen, N.; Seymour, L.W. Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 2009, 5, e1000440. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Rennie, P.S.; Jia, W.W. MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin. Cancer Res. 2009, 15, 5126–5135. [Google Scholar] [CrossRef] [PubMed]
- Edge, R.E.; Falls, T.J.; Brown, C.W.; Lichty, B.D.; Atkins, H.; Bell, J.C. A let-7 MicroRNA-sensitive vesicular stomatitis virus demonstrates tumor-specific replication. Mol. Ther. 2008, 16, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.J.; Hadac, E.M.; Greiner, S.; Russell, S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008, 14, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Gurlevik, E.; Woller, N.; Schache, P.; Malek, N.P.; Wirth, T.C.; Zender, L.; Manns, M.P.; Kubicka, S.; Kuhnel, F. p53-dependent antiviral RNA-interference facilitates tumor-selective viral replication. Nucleic Acids Res. 2009, 37, e84. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.J.; Douglas, J.T. Armed replicating adenoviruses for cancer virotherapy. Cancer Gene Ther. 2009, 16, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Coukos, G. Biological therapy with oncolytic herpesvirus. Adv. Exp. Med. Biol. 2008, 622, 221–233. [Google Scholar] [PubMed]
- Blackford, A.N.; Grand, R.J. Adenovirus E1B 55-kilodalton protein: multiple roles in viral infection and cell transformation. J. Virol. 2009, 83, 4000–4012. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Kirn, D.; Eckhardt, G.; Rodriguez, G.I.; Soutar, D.S.; Otto, R.; Robertson, A.G.; Park, O.; Gulley, M.L.; Heise, C.; Von Hoff, D.D.; Kaye, S.B. A phase I study of Onyx-015, an E1B attenuated adenovirus, administered intratumorally to patients with recurrent head and neck cancer. Clin. Cancer Res. 2000, 6, 798–806. [Google Scholar]
- Nemunaitis, J.; Ganly, I.; Khuri, F.; Arseneau, J.; Kuhn, J.; McCarty, T.; Landers, S.; Maples, P.; Romel, L.; Randlev, B.; Reid, T.; Kaye, S.; Kirn, D. Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 2000, 60, 6359–6366. [Google Scholar] [PubMed]
- Nemunaitis, J.; Khuri, F.; Ganly, I.; Arseneau, J.; Posner, M.; Vokes, E.; Kuhn, J.; McCarty, T.; Landers, S.; Blackburn, A.; Romel, L.; Randlev, B.; Kaye, S.; Kirn, D. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J. Clin. Oncol. 2001, 19, 289–298. [Google Scholar] [PubMed]
- Mulvihill, S.; Warren, R.; Venook, A.; Adler, A.; Randlev, B.; Heise, C.; Kirn, D. Safety and feasibility of injection with an E1B-55 kDa gene-deleted, replication-selective adenovirus (ONYX-015) into primary carcinomas of the pancreas: a phase I trial. Gene Ther. 2001, 8, 308–315. [Google Scholar] [CrossRef]
- Hecht, J.R.; Bedford, R.; Abbruzzese, J.L.; Lahoti, S.; Reid, T.R.; Soetikno, R.M.; Kirn, D.H.; Freeman, S.M. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin. Cancer Res. 2003, 9, 555–561. [Google Scholar] [PubMed]
- Nemunaitis, J.; Cunningham, C.; Buchanan, A.; Blackburn, A.; Edelman, G.; Maples, P.; Netto, G.; Tong, A.; Randlev, B.; Olson, S.; Kirn, D. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001, 8, 746–759. [Google Scholar] [CrossRef]
- Thomas, M.A.; Broughton, R.S.; Goodrum, F.D.; Ornelles, D.A. E4orf1 limits the oncolytic potential of the E1B-55K deletion mutant adenovirus. J. Virol. 2009, 83, 2406–2416. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Mymryk, J.S. Adenovirus-5 E1A: paradox and paradigm. Nat. Rev. Mol. Cell. Biol. 2002, 3, 441–452. [Google Scholar] [CrossRef]
- Liu, T.C.; Wang, Y.; Hallden, G.; Brooks, G.; Francis, J.; Lemoine, N.R.; Kirn, D. Functional interactions of antiapoptotic proteins and tumor necrosis factor in the context of a replication-competent adenovirus. Gene Ther. 2005, 12, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, T.; Vijayalingam, S.; Chinnadurai, G. Genetic identification of adenovirus type 5 genes that influence viral spread. J. Virol. 2006, 80, 2000–2012. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Modha, D.; White, E. Interaction of E1B 19K with Bax is required to block Bax-induced loss of mitochondrial membrane potential and apoptosis. Oncogene 1998, 17, 2993–3005. [Google Scholar] [PubMed]
- Han, J.; Sabbatini, P.; Perez, D.; Rao, L.; Modha, D.; White, E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996, 10, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, R.; White, E. E1B 19K blocks Bax oligomerization and tumor necrosis factor alpha-mediated apoptosis. J. Virol. 2001, 75, 7506–7516. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.; White, E. E1B 19K inhibits Fas-mediated apoptosis through FADD-dependent sequestration of FLICE. J. Cell Biol. 1998, 141, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Vijayalingam, S.; Subramanian, T.; Ryerse, J.; Varvares, M.; Chinnadurai, G. Down-regulation of multiple cell survival proteins in head and neck cancer cells by an apoptogenic mutant of adenovirus type 5. Virology 2009, 392, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.J.; Safer, B.; Munemitsu, S.M.; Samuel, C.E.; Shenk, T. Adenovirus VAI RNA prevents phosphorylation of the eukaryotic initiation factor 2 alpha subunit subsequent to infection. Proc. Natl. Acad. Sci. USA 1985, 82, 4321–4325. [Google Scholar] [CrossRef]
- Schneider, R.J.; Weinberger, C.; Shenk, T. Adenovirus VAI RNA facilitates the initiation of translation in virus-infected cells. Cell 1984, 37, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Ryerse, J.S.; Scaria, A.; Hermiston, T.W.; Wold, W.S. The E3-11.6-kDa adenovirus death protein (ADP) is required for efficient cell death: characterization of cells infected with adp mutants. Virology 1996, 220, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.; Atencio, I.; Huang, W.M.; Horn, M.; Ramachandra, M. Overexpression of adenovirus E3-11.6K protein induces cell killing by both caspase-dependent and caspase-independent mechanisms. Virology 2004, 326, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Toth, K.; Kuppuswamy, M.; Krajcsi, P.; Tollefson, A.E.; Wold, W.S. Overexpression of the ADP (E3-11.6K) protein increases cell lysis and spread of adenovirus. Virology 2003, 305, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene. Ther. 2005, 16, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; He, X.; Wang, W.; Huang, Y.; Chen, L.; Cong, W.; Gu, J.; Hu, H.; Shi, J.; Li, L.; Su, C. E2F promoter-regulated oncolytic adenovirus with p16 gene induces cell apoptosis and exerts antitumor effect on gastric cancer. Dig. Dis. Sci. 2009, 54, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, C.; Cao, H.; Li, K.; Chen, J.; Jiang, L.; Zhang, Q.; Wu, X.; Jia, X.; Liu, Y.; Wang, W.; Liu, X.; Wu, M.; Qian, Q. A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol. Cancer Ther. 2008, 7, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; Hong, S.M.; Fu, B.; Lin, M.T.; Calhoun, E.S.; Kamiyama, M.; Walter, K.; Nikolskaya, T.; Nikolsky, Y.; Hartigan, J.; Smith, D.R.; Hidalgo, M.; Leach, S.D.; Klein, A.P.; Jaffee, E.M.; Goggins, M.; Maitra, A.; Iacobuzio-Donahue, C.; Eshleman, J.R.; Kern, S.E.; Hruban, R.H.; Karchin, R.; Papadopoulos, N.; Parmigiani, G.; Vogelstein, B.; Velculescu, V.E.; Kinzler, K.W. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Robbins, J.S.; Pister, A.; Zafar, M.B.; Zhang, Z.W.; Gupta, J.; Lee, K.J.; Neuman, K.; Yun, C.O.; Guise, T.; Seth, P. A modified hTERT promoter-directed oncolytic adenovirus replication with concurrent inhibition of TGFbeta signaling for breast cancer therapy. Cancer Gene Ther. 2009. [Google Scholar]
- Jin, J.; Liu, H.; Yang, C.; Li, G.; Liu, X.; Qian, Q.; Qian, W. Effective gene-viral therapy of leukemia by a new fiber chimeric oncolytic adenovirus expressing TRAIL: in vitro and in vivo evaluation. Mol. Cancer Ther. 2009, 8, 1387. [Google Scholar] [CrossRef]
- Chen, L.; Chen, D.; Gong, M.; Na, M.; Li, L.; Wu, H.; Jiang, L.; Qian, Y.; Fang, G.; Xue, X. Concomitant use of Ad5/35 chimeric oncolytic adenovirus with TRAIL gene and taxol produces synergistic cytotoxicity in gastric cancer cells. Cancer Lett. 2009, 284, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, Y.; Newman, K.; Gu, J.; Zhang, X.; Wu, H.; Zhao, M.; Xianyu, Z.; Liu, X. Reexpression of human somatostatin receptor gene 2 gene mediated by oncolytic adenovirus increases antitumor activity of tumor necrosis factor-related apoptosis-inducing ligand against pancreatic cancer. Clin. Cancer Res. 2009, 15, 5154–5160. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.N.; Pei, D.S.; Sun, F.H.; Zhang, B.F.; Liu, X.Y.; Gu, J.F.; Liu, Y.H.; Hu, X.L.; Mao, L.J.; Wen, R.M.; Liu, J.J.; Li, W. Inhibition of renal cancer cell growth by oncolytic adenovirus armed short hairpin RNA targeting hTERT gene. Cancer Biol. Ther. 2009, 8, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.N.; Pei, D.S.; Mao, L.J.; Liu, X.Y.; Mei, D.D.; Zhang, B.F.; Shi, Z.; Wen, R.M.; Sun, X.Q. Inhibition of renal cancer cell growth in vitro and in vivo with oncolytic adenovirus armed short hairpin RNA targeting Ki-67 encoding mRNA. Cancer Gene Ther. 2009, 16, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, C.Y.; Wang, X.H.; Fu, Z.X. Oncolytic adenovirus mediated Survivin knockdown by RNA interference suppresses human colorectal carcinoma growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2009, 28, 81. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Gu, J.; Sun, L.; Qian, Q.; Qian, C.; Liu, X. Oncolytic adenovirus-mediated shRNA against Apollon inhibits tumor cell growth and enhances antitumor effect of 5-fluorouracil. Gene Ther. 2008, 15, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Kojima, T.; Kuroda, S.; Endo, Y.; Sakai, R.; Hioki, M.; Kishimoto, H.; Uno, F.; Kagawa, S.; Watanabe, Y.; Hashimoto, Y.; Urata, Y.; Tanaka, N.; Fujiwara, T. A novel antiangiogenic effect for telomerase-specific virotherapy through host immune system. J. Immunol. 2009, 182, 1763–1769. [Google Scholar] [PubMed]
- Kurozumi, K.; Hardcastle, J.; Thakur, R.; Shroll, J.; Nowicki, M.; Otsuki, A.; Chiocca, E.A.; Kaur, B. Oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates CYR61. Mol. Ther. 2008, 16, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Tysome, J.R.; Lemoine, N.R.; Wang, Y. Combination of anti-angiogenic therapy and virotherapy: arming the oncolytic viruses with anti-angiogenic genes. Curr. Opin. Mol. Ther. 2010, in press. [Google Scholar]
- Tysome, J.R.; Briat, A.; Alusi, G.; Cao, F.; Gao, D.; Yu, J.; Wang, P.; Yang, S.; Dong, Z.; Wang, S.; Deng, L.; Francis, J.; Timiryasova, T.; Fodor, I.; Lemoine, N.R.; Wang, Y. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 2009, 16, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Na, M.; Chen, J.; Wang, X.; Liu, Y.; Wang, W.; Zhang, Q.; Li, L.; Long, J.; Liu, X.; Wu, M.; Fan, X.; Qian, Q. Gene-viral cancer therapy using dual-regulated oncolytic adenovirus with antiangiogenesis gene for increased efficacy. Mol. Cancer Res. 2008, 6, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Pu, Y.Y.; Hu, X.C.; Sun, L.J.; Luo, H.M.; Pan, S.K.; Gu, J.Z.; Cao, X.R.; Su, C.Q. Antiangiogenesis gene armed tumor-targeting adenovirus yields multiple antitumor activities in human HCC xenografts in nude mice. Hepatol. Res. 2009. [Google Scholar]
- Zheng, J.N.; Pei, D.S.; Mao, L.J.; Liu, X.Y.; Sun, F.H.; Zhang, B.F.; Liu, Y.Q.; Liu, J.J.; Li, W.; Han, D. Oncolytic adenovirus expressing interleukin-18 induces significant antitumor effects against melanoma in mice through inhibition of angiogenesis. Cancer Gene Ther. 2010, 17, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.N.; Pei, D.S.; Sun, F.H.; Liu, X.Y.; Mao, L.J.; Zhang, B.F.; Wen, R.M.; Xu, W.; Shi, Z.; Liu, J.J.; Li, W. Potent antitumor efficacy of interleukin-18 delivered by conditionally replicative adenovirus vector in renal cell carcinoma-bearing nude mice via inhibition of angiogenesis. Cancer Biol. Ther. 2009, 8, 599–606. [Google Scholar] [CrossRef]
- He, X.P.; Su, C.Q.; Wang, X.H.; Pan, X.; Tu, Z.X.; Gong, Y.F.; Gao, J.; Liao, Z.; Jin, J.; Wu, H.Y.; Man, X.H.; Li, Z.S. E1B-55kD-deleted oncolytic adenovirus armed with canstatin gene yields an enhanced anti-tumor efficacy on pancreatic cancer. Cancer Lett. 2009, 285, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Castelo-Branco, P.; Rabkin, S.D.; Martuza, R.L. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol. Ther. 2008, 16, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kim, J.H.; Kim, J.; Huang, J.H.; Zhang, S.N.; Kang, Y.A.; Kim, H.; Yun, C.O. Short hairpin RNA-expressing oncolytic adenovirus-mediated inhibition of IL-8: effects on antiangiogenesis and tumor growth inhibition. Gene Ther. 2008, 15, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Frentzen, A.; Yu, Y.A.; Chen, N.; Zhang, Q.; Weibel, S.; Raab, V.; Szalay, A.A. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 12915–12920. [Google Scholar] [CrossRef]
- Guse, K.; Diaconu, I.; Rajecki, M.; Sloniecka, M.; Hakkarainen, T.; Ristimaki, A.; Kanerva, A.; Pesonen, S.; Hemminki, A. Ad5/3-9HIF-Delta24-VEGFR-1-Ig, an infectivity enhanced, dual-targeted and antiangiogenic oncolytic adenovirus for kidney cancer treatment. Gene Ther. 2009, 16, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.A.; Shin, H.C.; Yoo, J.Y.; Kim, J.H.; Kim, J.S.; Yun, C.O. Novel cancer antiangiotherapy using the VEGF promoter-targeted artificial zinc-finger protein and oncolytic adenovirus. Mol. Ther. 2008, 16, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- McNally, L.R.; Rosenthal, E.L.; Zhang, W.; Buchsbaum, D.J. Therapy of head and neck squamous cell carcinoma with replicative adenovirus expressing tissue inhibitor of metalloproteinase-2 and chemoradiation. Cancer Gene Ther. 2009, 16, 246–255. [Google Scholar] [PubMed]
- Mahller, Y.Y.; Vaikunth, S.S.; Ripberger, M.C.; Baird, W.H.; Saeki, Y.; Cancelas, J.A.; Crombleholme, T.M.; Cripe, T.P. Tissue inhibitor of metalloproteinase-3 via oncolytic herpesvirus inhibits tumor growth and vascular progenitors. Cancer Res. 2008, 68, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Lee, S.J.; Li, X.; Jimenez, J.A.; Zhang, Y.P.; Bae, K.H.; Mohammadi, Y.; Kao, C.; Gardner, T.A. Enhanced combined tumor-specific oncolysis and suicide gene therapy for prostate cancer using M6 promoter. Cancer Gene Ther. 2009, 16, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Abei, M.; Ugai, H.; Kawashima, R.; Seo, E.; Wakayama, M.; Murata, T.; Endo, S.; Hamada, H.; Hyodo, I.; Yokoyama, K.K. E1A, E1B double-restricted replicative adenovirus at low dose greatly augments tumor-specific suicide gene therapy for gallbladder cancer. Cancer Gene Ther. 2009, 16, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.Q.; Xu, Y.; Yang, R.J.; Wu, B.; Tan, X.H.; Qin, Y.D.; Zhang, Q.W. Combination effect of oncolytic adenovirus therapy and herpes simplex virus thymidine kinase/ganciclovir in hepatic carcinoma animal models. Acta Pharmacol. Sin. 2009, 30, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Braidwood, L.; Dunn, P.D.; Hardy, S.; Evans, T.R.; Brown, S.M. Antitumor activity of a selectively replication competent herpes simplex virus (HSV) with enzyme prodrug therapy. Anticancer Res. 2009, 29, 2159–2166. [Google Scholar] [PubMed]
- Chalikonda, S.; Kivlen, M.H.; O'Malley, M.E.; Eric Dong, X.D.; McCart, J.A.; Gorry, M.C.; Yin, X.Y.; Brown, C.K.; Zeh 3rd, H.J.; Guo, Z.S.; Bartlett, D.L. Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer Gene Ther. 2008, 15, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, J.; Kintz, J.; Futin, N.; Findeli, A.; Cordier, P.; Schlesinger, Y.; Hoffmann, C.; Tosch, C.; Balloul, J.M.; Erbs, P. Targeted delivery of a suicide gene to human colorectal tumors by a conditionally replicating vaccinia virus. Gene Ther. 2008, 15, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin-Gal, D.; Zamir, G.; Edden, Y.; Pikarsky, E.; Pikarsky, A.; Haim, H.; Haviv, Y.S.; Panet, A. Herpes simplex virus type 1 preferentially targets human colon carcinoma: role of extracellular matrix. J. Virol. 2008, 82, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Gonzalez-Edick, M.; Gibbons, D.; Van Roey, M.; Jooss, K. Intratumoral coadministration of hyaluronidase enzyme and oncolytic adenoviruses enhances virus potency in metastatic tumor models. Clin. Cancer Res. 2008, 14, 3933–3941. [Google Scholar] [CrossRef] [PubMed]
- Nagano, S.; Perentes, J.Y.; Jain, R.K.; Boucher, Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res. 2008, 68, 3795–3802. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Bazan-Peregrino, M.; Carlisle, R.C.; Purdie, L.; Seymour, L.W. Factors influencing retention of adenovirus within tumours following direct intratumoural injection. Gene Ther. 2008, 15, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.V.; Viale, D.L.; Cafferata, E.G.; Bravo, A.I.; Carbone, C.; Gould, D.; Chernajovsky, Y.; Podhajcer, O.L. Tumor associated stromal cells play a critical role on the outcome of the oncolytic efficacy of conditionally replicative adenoviruses. PLoS One 2009, 4, e5119. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.H.; Hermiston, T.W. Effect of hypoxia on Ad5 infection, transgene expression and replication. Gene Ther. 2005, 12, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.H.; Bauzon, M.; Hermiston, T.W. The effect of hypoxia on the uptake, replication and lytic potential of group B adenovirus type 3 (Ad3) and type 11p (Ad11p). Gene Ther. 2006, 13, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Hiley, C.T.; Yuan, M.; Lemoine, N.R.; Wang, Y. Lister strain vaccinia virus, a potential therapeutic vector for hypoxic tumours. Gene Ther. 2009. [Google Scholar]
- Aghi, M.K.; Liu, T.C.; Rabkin, S.; Martuza, R.L. Hypoxia enhances the replication of oncolytic herpes simplex virus. Mol. Ther. 2009, 17, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Burch, A.D.; Britton, A. Hypoxia enhances the replication of oncolytic herpes simplex virus in p53- breast cancer cells. Cell Cycle 2009, 8, 2194–2197. [Google Scholar] [CrossRef] [PubMed]
- Anders, M.; Christian, C.; McMahon, M.; McCormick, F.; Korn, W.M. Inhibition of the Raf/MEK/ERK pathway up-regulates expression of the coxsackievirus and adenovirus receptor in cancer cells. Cancer Res. 2003, 63, 2088–2095. [Google Scholar] [PubMed]
- O'Prey, J.; Wilkinson, S.; Ryan, K.M. Tumor antigen LRRC15 impedes adenoviral infection: implications for virus-based cancer therapy. J. Virol. 2008, 82, 5933–5939. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Fishelson, Z.; Donin, N.; Zell, S.; Schultz, S.; Kirschfink, M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 2003, 40, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, N.; Higashi, T.; Nouso, K.; Nakatsukasa, H.; Kobayashi, Y.; Ishizaki, M.; Toshikuni, N.; Yoshida, K.; Uematsu, S.; Tsuji, T. Expression of membrane cofactor protein (MCP, CD46) in human liver diseases. Br. J. Cancer 1999, 80, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.P.; Mathure, S.; Kaul, R.; Khan, S.; Carson, L.F.; Twiggs, L.B.; Martens, M.G.; Kaul, A. Expression of complement regulatory proteins-CD 35, CD 46, CD 55, and CD 59-in benign and malignant endometrial tissue. Gynecol. Oncol. 2000, 76, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Ulasov, I.V.; Rolle, C.E.; Han, Y.; Lesniak, M.S. A chimeric adenovirus with an Ad 3 fiber knob modification augments glioma virotherapy. J. Gene Med. 2009, 11, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Gonzalez-Edick, M.; Gibbons, D.; Ge, Y.; VanRoey, M.; Robinson, M.; Jooss, K. Combination therapy with radiation or cisplatin enhances the potency of Ad5/35 chimeric oncolytic adenovirus in a preclinical model of head and neck cancer. Cancer Gene Ther. 2009, 16, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, G.; Liu, H.; Yang, C.; Yang, X.; Jin, J.; Liu, X.; Qian, Q.; Qian, W. E1B 55-kDa deleted, Ad5/F35 fiber chimeric adenovirus, a potential oncolytic agent for B-lymphocytic malignancies. J. Gene Med. 2009, 11, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.B.; Lu, B.; Park, M.; Makhija, S.K.; Numnum, T.M.; Kendrick, J.E.; Wang, M.; Tsuruta, Y.; Fisher, P.; Alvarez, R.D.; Zhou, F.; Siegal, G.P.; Wu, H.; Curiel, D.T. Development of an optimized conditionally replicative adenoviral agent for ovarian cancer. Int. J. Oncol. 2008, 32, 1179–1188. [Google Scholar] [PubMed]
- Shashkova, E.V.; May, S.M.; Barry, M.A. Characterization of human adenovirus serotypes 5, 6, 11, and 35 as anticancer agents. Virology 2009, 394, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, L.; Papareddy, P.; Silver, J.; Bergh, A.; Mei, Y.F. Replication-competent Ad11p vector (RCAd11p) efficiently transduces and replicates in hormone-refractory metastatic prostate cancer cells. Hum. Gene Ther. 2009, 20, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Liu, Y.; Li, Z.Y.; Tuve, S.; Strauss, R.; Lieber, A. Comparison of adenoviruses from species B, C, E, and F after intravenous delivery. Mol. Ther. 2007, 15, 2146–2153. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Shimozato, O.; Li, Q.; Kawamura, K.; Ma, G.; Namba, M.; Ogawa, T.; Kaiho, I.; Tagawa, M. Adenovirus type 5 substituted with type 11 or 35 fiber structure increases its infectivity to human cells enabling dual gene transfer in CD46-dependent and -independent manners. Anticancer Res. 2007, 27, 2311–2316. [Google Scholar] [PubMed]
- Stone, D.; Ni, S.; Li, Z.Y.; Gaggar, A.; DiPaolo, N.; Feng, Q.; Sandig, V.; Lieber, A. Development and assessment of human adenovirus type 11 as a gene transfer vector. J. Virol. 2005, 79, 5090–5104. [Google Scholar] [CrossRef] [PubMed]
- Tuve, S.; Wang, H.; Ware, C.; Liu, Y.; Gaggar, A.; Bernt, K.; Shayakhmetov, D.; Li, Z.; Strauss, R.; Stone, D.; Lieber, A. A new group B adenovirus receptor is expressed at high levels on human stem and tumor cells. J. Virol. 2006, 80, 12109–12120. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Akitomo, K.; Kawabata, K.; Hayakawa, T.; Mizuguchi, H. Downregulation of human CD46 by adenovirus serotype 35 vectors. Gene Ther. 2007, 14, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.; Sova, P.; Liu, Y.; Li, Z.Y.; Tuve, S.; Pritchard, D.; Brinkkoetter, P.; Moller, T.; Wildner, O.; Pesonen, S.; Hemminki, A.; Urban, N.; Drescher, C.; Lieber, A. Epithelial phenotype confers resistance of ovarian cancer cells to oncolytic adenoviruses. Cancer Res. 2009, 69, 5115–5125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gangeswaran, R.; Zhao, X.; Wang, P.; Tysome, J.; Bhakta, V.; Yuan, M.; Chikkanna-Gowda, C.P.; Jiang, G.; Gao, D.; Cao, F.; Francis, J.; Yu, J.; Liu, K.; Yang, H.; Zhang, Y.; Zang, W.; Chelala, C.; Dong, Z.; Lemoine, N. CEACAM6 attenuates adenovirus infection by antagonizing viral trafficking in cancer cells. J. Clin. Invest. 2009, 119, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Shiina, M.; Lacher, M.D.; Christian, C.; Korn, W.M. RNA interference-mediated knockdown of p21(WAF1) enhances anti-tumor cell activity of oncolytic adenoviruses. Cancer Gene Ther. 2009, 16, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Hofstetter, W.; Guo, W.; Li, H.; Pataer, A.; Peng, H.H.; Guo, Z.S.; Bartlett, D.L.; Lin, A.; Swisher, S.G.; Fang, B. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008, 15, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; Lang, F.F.; Fueyo, J. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J. Natl. Cancer Inst 2007, 99, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Guse, K.; Bauerschmitz, G.; Virkkunen, P.; Tarkkanen, M.; Tanner, M.; Hakkarainen, T.; Kanerva, A.; Desmond, R.A.; Pesonen, S.; Hemminki, A. Oncolytic adenoviruses kill breast cancer initiating CD44+CD24-/low cells. Mol. Ther. 2007, 15, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Mahller, Y.Y.; Williams, J.P.; Baird, W.H.; Mitton, B.; Grossheim, J.; Saeki, Y.; Cancelas, J.A.; Ratner, N.; Cripe, T.P. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One 2009, 4, e4235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Komaki, R.; Wang, L.; Fang, B.; Chang, J.Y. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin. Cancer Res. 2008, 14, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.C.; Francis, J.; Brooks, G.; Lemoine, N.; Kirn, D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Johnson, P.R.; Knipe, D.M.; Chiocca, E.A. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003, 10, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Wolff, G.; Falck-Pedersen, E.; Crystal, R.G. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum. Gene Ther. 1997, 8, 37–44. [Google Scholar] [CrossRef]
- Zamarin, D.; Martinez-Sobrido, L.; Kelly, K.; Mansour, M.; Sheng, G.; Vigil, A.; Garcia-Sastre, A.; Palese, P.; Fong, Y. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol. Ther. 2009, 17, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Lyons, M.; Onion, D.; Green, N.K.; Aslan, K.; Rajaratnam, R.; Bazan-Peregrino, M.; Phipps, S.; Hale, S.; Mautner, V.; Seymour, L.W.; Fisher, K.D. Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol. Ther. 2006, 14, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Fulci, G.; Tyminski, E.; Chiocca, E.A. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Ther. 2004, 11, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; Chiocca, E.A. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef]
- Qiao, J.; Wang, H.; Kottke, T.; White, C.; Twigger, K.; Diaz, R.M.; Thompson, J.; Selby, P.; de Bono, J.; Melcher, A.; Pandha, H.; Coffey, M.; Vile, R.; Harrington, K. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin. Cancer Res. 2008, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zeng, Z.; Fu, X.; Zhang, X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res. 2007, 67, 7850–7855. [Google Scholar] [CrossRef] [PubMed]
- Ungerechts, G.; Springfeld, C.; Frenzke, M.E.; Lampe, J.; Parker, W.B.; Sorscher, E.J.; Cattaneo, R. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol. Ther. 2007, 15, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Coffey, M.C.; Thompson, B.G.; Yoon, C.S.; Waisman, D.M.; Lee, P.W. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003, 63, 348–353. [Google Scholar] [PubMed]
- Thomas, M.A.; Spencer, J.F.; Toth, K.; Sagartz, J.E.; Phillips, N.J.; Wold, W.S. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol. Ther. 2008, 16, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, K.; Hardcastle, J.; Thakur, R.; Yang, M.; Christoforidis, G.; Fulci, G.; Hochberg, F.H.; Weissleder, R.; Carson, W.; Chiocca, E.A.; Kaur, B. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J. Natl. Cancer Inst. 2007, 99, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Ichikawa, T.; Wakimoto, H.; Silver, J.S.; Deisboeck, T.S.; Finkelstein, D.; Harsh, G.R.t.; Louis, D.N.; Bartus, R.T.; Hochberg, F.H.; Chiocca, E.A. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat. Med. 1999, 5, 881–887. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, D.C.; Charlton, D.; Henderson, D.R. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of CN706 in the nude mouse LNCaP xenograft model: implications and proposals for human therapy. Hum. Gene Ther. 2000, 11, 1553–1567. [Google Scholar] [PubMed]
- Tsai, V.; Johnson, D.E.; Rahman, A.; Wen, S.F.; LaFace, D.; Philopena, J.; Nery, J.; Zepeda, M.; Maneval, D.C.; Demers, G.W.; Ralston, R. Impact of human neutralizing antibodies on antitumor efficacy of an oncolytic adenovirus in a murine model. Clin. Cancer Res. 2004, 10, 7199–7206. [Google Scholar] [CrossRef]
- Herrlinger, U.; Kramm, C.M.; Aboody-Guterman, K.S.; Silver, J.S.; Ikeda, K.; Johnston, K.M.; Pechan, P.A.; Barth, R.F.; Finkelstein, D.; Chiocca, E.A.; Louis, D.N.; Breakefield, X.O. Pre-existing herpes simplex virus 1 (HSV-1) immunity decreases, but does not abolish, gene transfer to experimental brain tumors by a HSV-1 vector. Gene Ther. 1998, 5, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Spencer, J.F.; Toth, K.; Wold, W.S. Effect of preexisting immunity on oncolytic adenovirus vector INGN 007 antitumor efficacy in immunocompetent and immunosuppressed Syrian hamsters. J. Virol. 2009, 83, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Spencer, J.F.; Toth, K.; Wold, W.S. Pre-existing immunity and passive immunity to adenovirus 5 prevents toxicity caused by an oncolytic adenovirus vector in the Syrian hamster model. Mol. Ther. 2009, 17, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Kostense, S.; Koudstaal, W.; Sprangers, M.; Weverling, G.J.; Penders, G.; Helmus, N.; Vogels, R.; Bakker, M.; Berkhout, B.; Havenga, M.; Goudsmit, J. Adenovirus types 5 and 35 seroprevalence in AIDS risk groups supports type 35 as a vaccine vector. Aids 2004, 18, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Holterman, L.; Vogels, R.; van der Vlugt, R.; Sieuwerts, M.; Grimbergen, J.; Kaspers, J.; Geelen, E.; van der Helm, E.; Lemckert, A.; Gillissen, G.; Verhaagh, S.; Custers, J.; Zuijdgeest, D.; Berkhout, B.; Bakker, M.; Quax, P.; Goudsmit, J.; Havenga, M. Novel replication-incompetent vector derived from adenovirus type 11 (Ad11) for vaccination and gene therapy: low seroprevalence and non-cross-reactivity with Ad5. J. Virol. 2004, 78, 13207–13215. [Google Scholar] [CrossRef] [PubMed]
- Seshidhar Reddy, P.; Ganesh, S.; Limbach, M.P.; Brann, T.; Pinkstaff, A.; Kaloss, M.; Kaleko, M.; Connelly, S. Development of adenovirus serotype 35 as a gene transfer vector. Virology 2003, 311, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Sumida, S.M.; Truitt, D.M.; Lemckert, A.A.; Vogels, R.; Custers, J.H.; Addo, M.M.; Lockman, S.; Peter, T.; Peyerl, F.W.; Kishko, M.G.; Jackson, S.S.; Gorgone, D.A.; Lifton, M.A.; Essex, M.; Walker, B.D.; Goudsmit, J.; Havenga, M.J.; Barouch, D.H. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J. Immunol. 2005, 174, 7179–7185. [Google Scholar] [PubMed]
- Komarova, S.; Kawakami, Y.; Stoff-Khalili, M.A.; Curiel, D.T.; Pereboeva, L. Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Mol. Cancer Ther. 2006, 5, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Hakkarainen, T.; Sarkioja, M.; Lehenkari, P.; Miettinen, S.; Ylikomi, T.; Suuronen, R.; Desmond, R.A.; Kanerva, A.; Hemminki, A. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum. Gene Ther. 2007, 18, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Sonabend, A.M.; Ulasov, I.V.; Tyler, M.A.; Rivera, A.A.; Mathis, J.M.; Lesniak, M.S. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells 2008, 26, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Iankov, I.D.; Blechacz, B.; Liu, C.; Schmeckpeper, J.D.; Tarara, J.E.; Federspiel, M.J.; Caplice, N.; Russell, S.J. Infected cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol. Ther. 2007, 15, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Munguia, A.; Ota, T.; Miest, T.; Russell, S.J. Cell carriers to deliver oncolytic viruses to sites of myeloma tumor growth. Gene Ther. 2008, 15, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Coukos, G.; Makrigiannakis, A.; Kang, E.H.; Caparelli, D.; Benjamin, I.; Kaiser, L.R.; Rubin, S.C.; Albelda, S.M.; Molnar-Kimber, K.L. Use of carrier cells to deliver a replication-selective herpes simplex virus-1 mutant for the intraperitoneal therapy of epithelial ovarian cancer. Clin. Cancer Res. 1999, 5, 1523–1537. [Google Scholar] [PubMed]
- Raykov, Z.; Balboni, G.; Aprahamian, M.; Rommelaere, J. Carrier cell-mediated delivery of oncolytic parvoviruses for targeting metastases. Int. J. Cancer 2004, 109, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kottke, T.; Willmon, C.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Ryno, P.; Barber, G.N.; Chester, J.; Selby, P.; Harrington, K.; Melcher, A.; Vile, R.G. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; Melcher, A.A. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Schirrmacher, V. Cross-infection of tumor cells by contact with T lymphocytes loaded with Newcastle disease virus. Int. J. Oncol. 2009, 34, 951–962. [Google Scholar] [PubMed]
- Ong, H.T.; Hasegawa, K.; Dietz, A.B.; Russell, S.J.; Peng, K.W. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007, 14, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Su, Y.; Zhou, S.; Xiao, W.; Ling, W.; Hu, B.; Liu, Y.; Qi, Y. Immune analysis on mtHSV mediated tumor therapy in HSV-1 seropositive mice. Cancer Biol. Ther. 2007, 6, 724–731. [Google Scholar] [PubMed]
- Kangasniemi, L.; Koskinen, M.; Jokinen, M.; Toriseva, M.; Ala-Aho, R.; Kahari, V.M.; Jalonen, H.; Yla-Herttuala, S.; Moilanen, H.; Stenman, U.H.; Diaconu, I.; Kanerva, A.; Pesonen, S.; Hakkarainen, T.; Hemminki, A. Extended release of adenovirus from silica implants in vitro and in vivo. Gene Ther. 2009, 16, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Huard, J.; Lochmuller, H.; Acsadi, G.; Jani, A.; Massie, B.; Karpati, G. The route of administration is a major determinant of the transduction efficiency of rat tissues by adenoviral recombinants. Gene Ther. 1995, 2, 107–115. [Google Scholar] [PubMed]
- Alemany, R.; Suzuki, K.; Curiel, D.T. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000, 81, 2605–2609. [Google Scholar] [PubMed]
- Hollon, T. Researchers and regulators reflect on first gene therapy death. Nat. Med. 2000, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.; Greig, J.A.; Denby, L.; Custers, J.; Morita, T.; Francischetti, I.M.; Monteiro, R.Q.; Barouch, D.H.; van Rooijen, N.; Napoli, C.; Havenga, M.J.; Nicklin, S.A.; Baker, A.H. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; Doronin, K.; Senac, J.S.; Barry, M.A. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res. 2008, 68, 5896–5904. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Shashkova, E.V.; May, S.M.; Hofherr, S.E.; Barry, M.A. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum. Gene Ther. 2009, 20, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; May, S.M.; Doronin, K.; Barry, M.A. Expanded anticancer therapeutic window of hexon-modified oncolytic adenovirus. Mol. Ther. 2009, 17, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.; Fisher, K.D.; Finberg, R.W.; Seymour, L.W. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood 2009, 113, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Liu, H.L.; Zhang, X.R.; Xu, J.P.; Hu, W.K.; Liang, M.; Chen, S.Y.; Hu, F.; Chu, D.T. A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther. 2009, 16, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ye, X.; Fang, C.; Lu, Q.; Zhao, Y.; Liu, F.; Liang, M.; Hu, F.; Chen, H.Z. Intratumor injection of oncolytic adenovirus expressing HSP70 prolonged survival in melanoma B16 bearing mice by enhanced immune response. Cancer Biol. Ther. 2008, 7, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, N.; Aldrich, M.; Weksberg, D.; Rollins, L.; Goltsova, T.; Chen, S.Y.; Huang, X.F. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J. Immunother. 2009, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Willmon, C.L.; Saloura, V.; Fridlender, Z.G.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Kottke, T.; Federspiel, M.; Barber, G.; Albelda, S.M.; Vile, R.G. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009, 69, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Lei, N.; Shen, F.B.; Chang, J.H.; Wang, L.; Li, H.; Yang, C.; Li, J.; Yu, D.C. An oncolytic adenovirus expressing granulocyte macrophage colony-stimulating factor shows improved specificity and efficacy for treating human solid tumors. Cancer Gene Ther. 2009, 16, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Roh, M.S.; Lee, Y.K.; Kim, M.K.; Han, J.Y.; Park, B.H.; Trown, P.; Kirn, D.H.; Hwang, T.H. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Ther. 2009. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Zhao, X.; Wu, X.; Guo, Y.; Guo, H.; Cao, J.; Lou, D.; Yu, D.; Li, J. A Phase I study of KH901, a conditionally replicating granulocyte-macrophage colony-stimulating factor: armed oncolytic adenovirus for the treatment of head and neck cancers. Cancer Biol. Ther. 2009, 8, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Bortolanza, S.; Bunuales, M.; Otano, I.; Gonzalez-Aseguinolaza, G.; Ortiz-de-Solorzano, C.; Perez, D.; Prieto, J.; Hernandez-Alcoceba, R. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol. Ther. 2009, 17, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.J.; Wang, Y.G.; Cao, X.; Zhong, S.Y.; Wei, R.C.; Wu, Y.M.; Yue, X.T.; Li, G.C.; Liu, X.Y. Potent antitumor effect of interleukin-24 gene in the survivin promoter and retinoblastoma double-regulated oncolytic adenovirus. Hum. Gene Ther. 2009, 20, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xia, Q.; Zhang, R.; Lv, C.; Zhang, W.; Wang, Y.; Cui, Q.; Liu, L.; Cai, R.; Qian, C. Treatment of cancer with a novel dual-targeted conditionally replicative adenovirus armed with mda-7/IL-24 gene. Clin. Cancer Res. 2008, 14, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Wang, Y.; Le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007, 4, e353. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; Kuppuswamy, M.N.; Wold, W.S.; Doronin, K. Anticancer activity of oncolytic adenovirus vector armed with IFN-alpha and ADP is enhanced by pharmacologically controlled expression of TRAIL. Cancer Gene Ther. 2008, 15, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Haralambieva, I.; Iankov, I.; Hasegawa, K.; Harvey, M.; Russell, S.J.; Peng, K.W. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol. Ther. 2007, 15, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Wu, L.; Meseck, M.; Chen, L.; Ebert, O.; Garcia-Sastre, A.; Fallon, J.; Mandeli, J.; Woo, S.L. Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther. 2009, 16, 266–278. [Google Scholar] [PubMed]
- Endo, Y.; Sakai, R.; Ouchi, M.; Onimatsu, H.; Hioki, M.; Kagawa, S.; Uno, F.; Watanabe, Y.; Urata, Y.; Tanaka, N.; Fujiwara, T. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene 2008, 27, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, N.; Aldrich, M.; Rollins, L.; Ren, W.; Goltsova, T.; Chen, S.Y.; Huang, X.F. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Mol. Ther. 2009, 17, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, E.; Woller, N.; Mundt, B.; Knocke, S.; Gurlevik, E.; Saborowski, M.; Malek, N.; Manns, M.P.; Wirth, T.; Kuhnel, F.; Kubicka, S. Antitumoral immune response by recruitment and expansion of dendritic cells in tumors infected with telomerase-dependent oncolytic viruses. Cancer Res. 2009, 69, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.M.; Monie, A.; Wu, A.; Pai, S.I.; Hung, C.F. Combination of viral oncolysis and tumor-specific immunity to control established tumors. Clin. Cancer Res. 2009, 15, 4581–4588. [Google Scholar] [CrossRef]
- Huang, J.H.; Zhang, S.N.; Choi, K.J.; Choi, I.K.; Kim, J.H.; Lee, M.; Kim, H.; Yun, C.O. Therapeutic and tumor-specific immunity induced by combination of dendritic cells and oncolytic adenovirus expressing IL-12 and 4-1BBL. Mol. Ther. 2009. [Google Scholar]
- Robinson, M.; Ge, Y.; Ko, D.; Yendluri, S.; Laflamme, G.; Hawkins, L.; Jooss, K. Comparison of the E3 and L3 regions for arming oncolytic adenoviruses to achieve a high level of tumor-specific transgene expression. Cancer Gene Ther. 2008, 15, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bortolanza, S.; Bunuales, M.; Alzuguren, P.; Lamas, O.; Aldabe, R.; Prieto, J.; Hernandez-Alcoceba, R. Deletion of the E3-6.7K/gp19K region reduces the persistence of wild-type adenovirus in a permissive tumor model in Syrian hamsters . Cancer Gene Ther. 2009, 16, 703–712. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Burgert, H.G.; Owen, D.P.; Stanton, R.J.; Prod'homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; Little, A.M.; Wang, E.C.; Tomasec, P.; Wilkinson, G.W. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.M.; Bennink, J.R.; Yewdell, J.W.; Brodsky, F.M. Cutting edge: adenovirus E19 has two mechanisms for affecting class I MHC expression. J. Immunol. 1999, 162, 5049–5052. [Google Scholar] [PubMed]
- Hermiston, T.W.; Tripp, R.A.; Sparer, T.; Gooding, L.R.; Wold, W.S. Deletion mutation analysis of the adenovirus type 2 E3-gp19K protein: identification of sequences within the endoplasmic reticulum lumenal domain that are required for class I antigen binding and protection from adenovirus-specific cytotoxic T lymphocytes. J. Virol. 1993, 67, 5289–5298. [Google Scholar] [PubMed]
- Reddy, P.S.; Burroughs, K.D.; Hales, L.M.; Ganesh, S.; Jones, B.H.; Idamakanti, N.; Hay, C.; Li, S.S.; Skele, K.L.; Vasko, A.J.; Yang, J.; Watkins, D.N.; Rudin, C.M.; Hallenbeck, P.L. Seneca Valley virus, a systemically deliverable oncolytic picornavirus, and the treatment of neuroendocrine cancers. J. Natl. Cancer Inst. 2007, 99, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.; Yang, W.; Alain, T.; Shi, Z.Q.; Muzik, H.; Barrett, J.W.; McFadden, G.; Bell, J.; Hamilton, M.G.; Senger, D.L.; Forsyth, P.A. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005, 65, 9982–9990. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Uzawa, K.; Kasamatsu, A.; Shinozuka, K.; Sakuma, K.; Yamatoji, M.; Shiiba, M.; Shino, Y.; Shirasawa, H.; Tanzawa, H. Oncolytic activity of Sindbis virus in human oral squamous carcinoma cells. Br. J. Cancer 2009, 101, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Vaha-Koskela, M.J.; Kallio, J.P.; Jansson, L.C.; Heikkila, J.E.; Zakhartchenko, V.A.; Kallajoki, M.A.; Kahari, V.M.; Hinkkanen, A.E. Oncolytic capacity of attenuated replicative semliki forest virus in human melanoma xenografts in severe combined immunodeficient mice. Cancer Res. 2006, 66, 7185–7194. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78-106. https://doi.org/10.3390/v2010078
Wong HH, Lemoine NR, Wang Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses. 2010; 2(1):78-106. https://doi.org/10.3390/v2010078
Chicago/Turabian StyleWong, Han Hsi, Nicholas R. Lemoine, and Yaohe Wang. 2010. "Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles" Viruses 2, no. 1: 78-106. https://doi.org/10.3390/v2010078
APA StyleWong, H. H., Lemoine, N. R., & Wang, Y. (2010). Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses, 2(1), 78-106. https://doi.org/10.3390/v2010078