RNA Editing and its Control in Hepatitis Delta Virus Replication

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
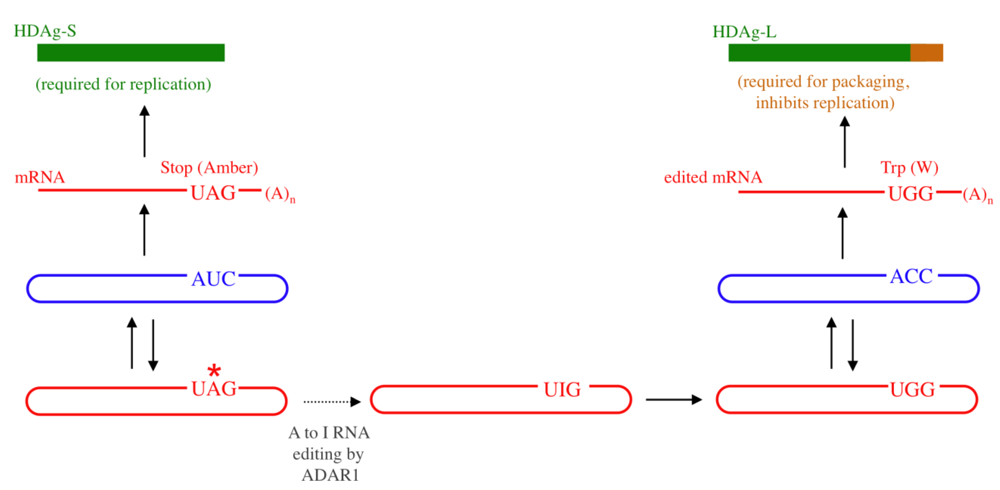
:1. Introduction
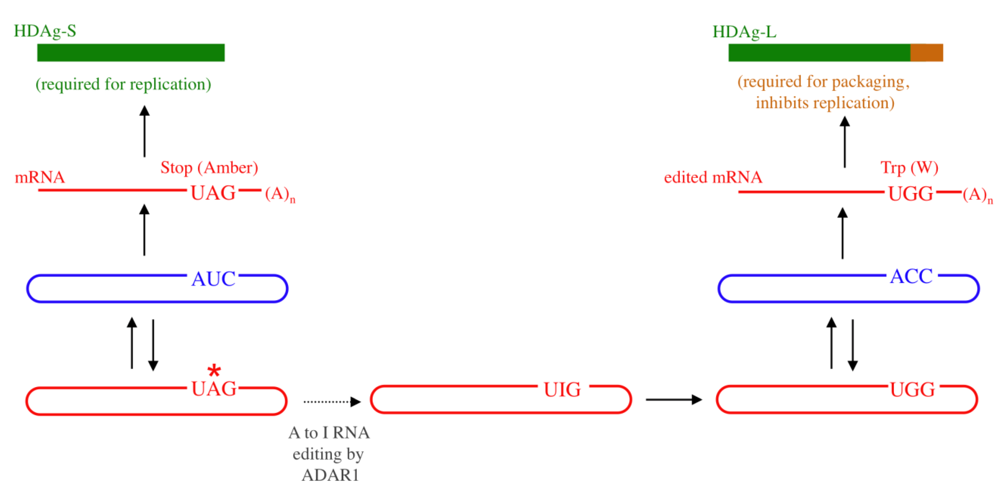
2. RNA Structural Requirements for Editing
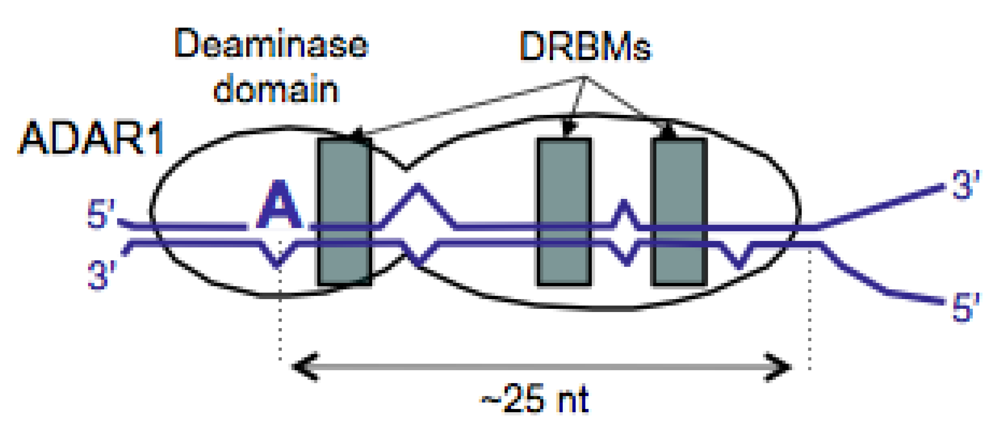
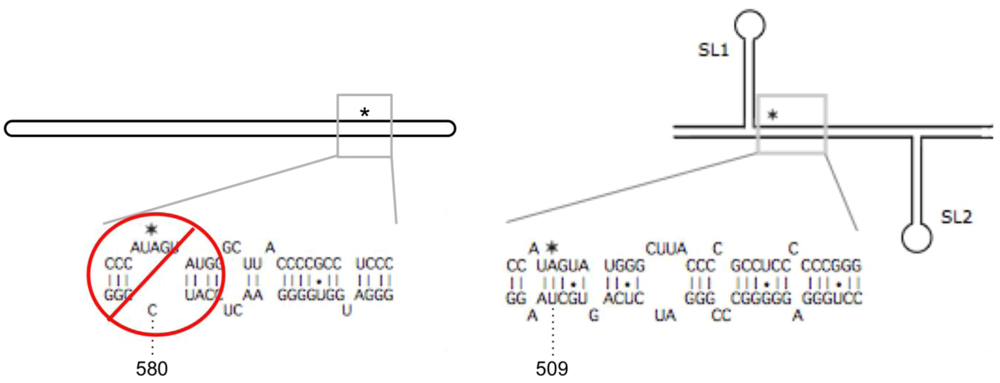
2.1. Structural Requirements for Editing the HDV Genotype 1 amber/W Site
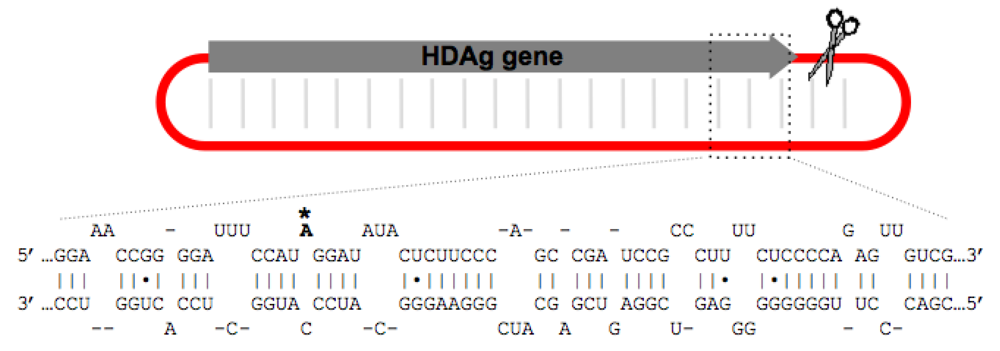
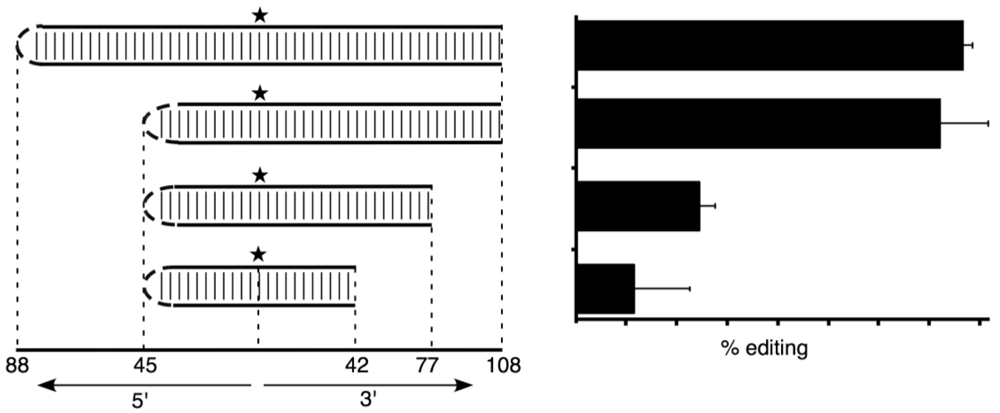
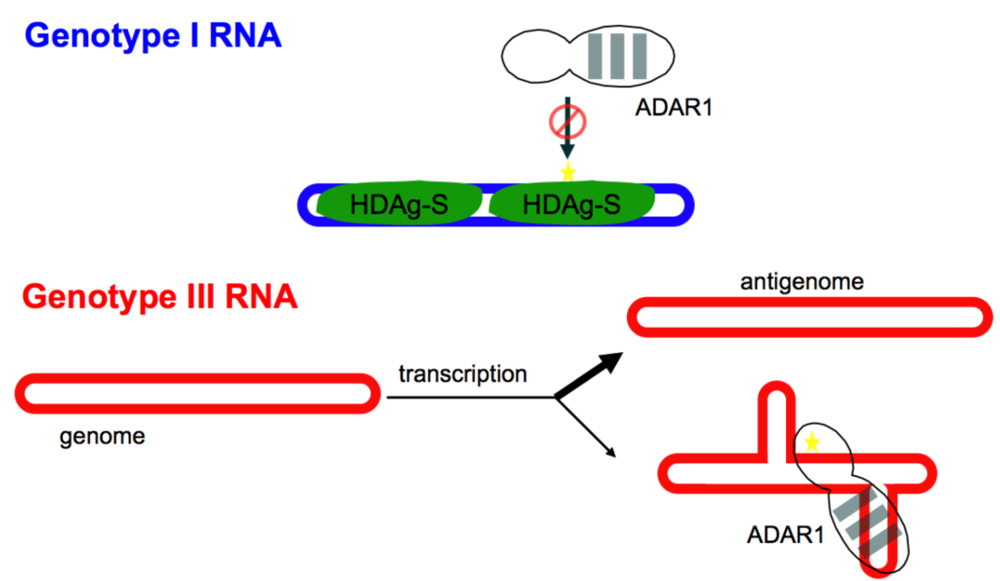
2.2. Editing the HDV Type 3 amber/W Site Requires a Branched Structure
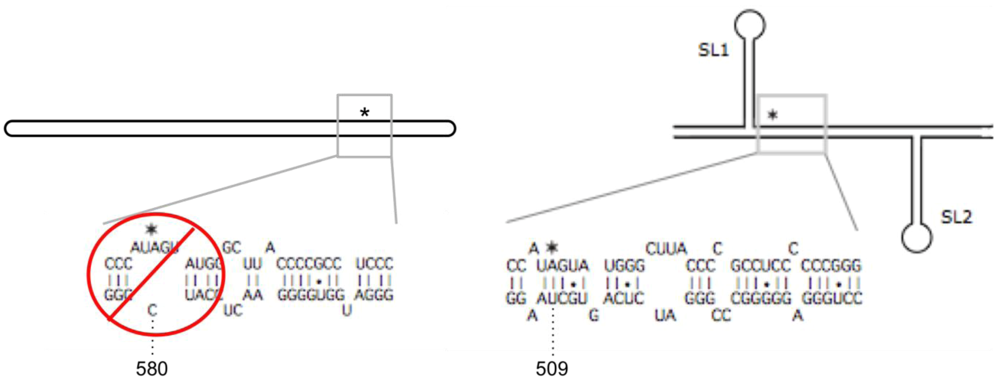
3. Control of HDV RNA Editing
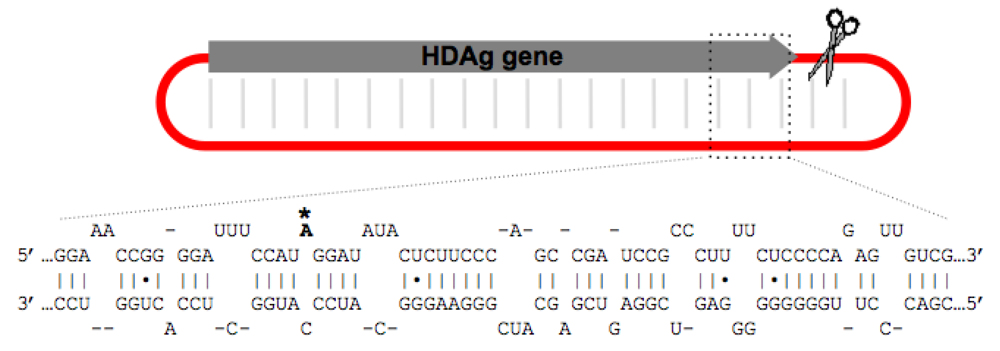
3.1. Restriction of Editing to the amber/W Site
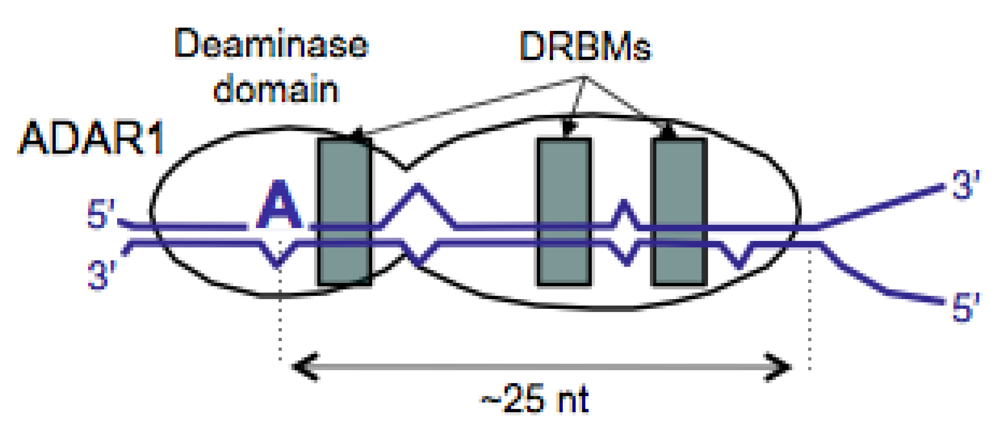
3.2. Mechanisms for Controlling Levels of amber/W Site Editing
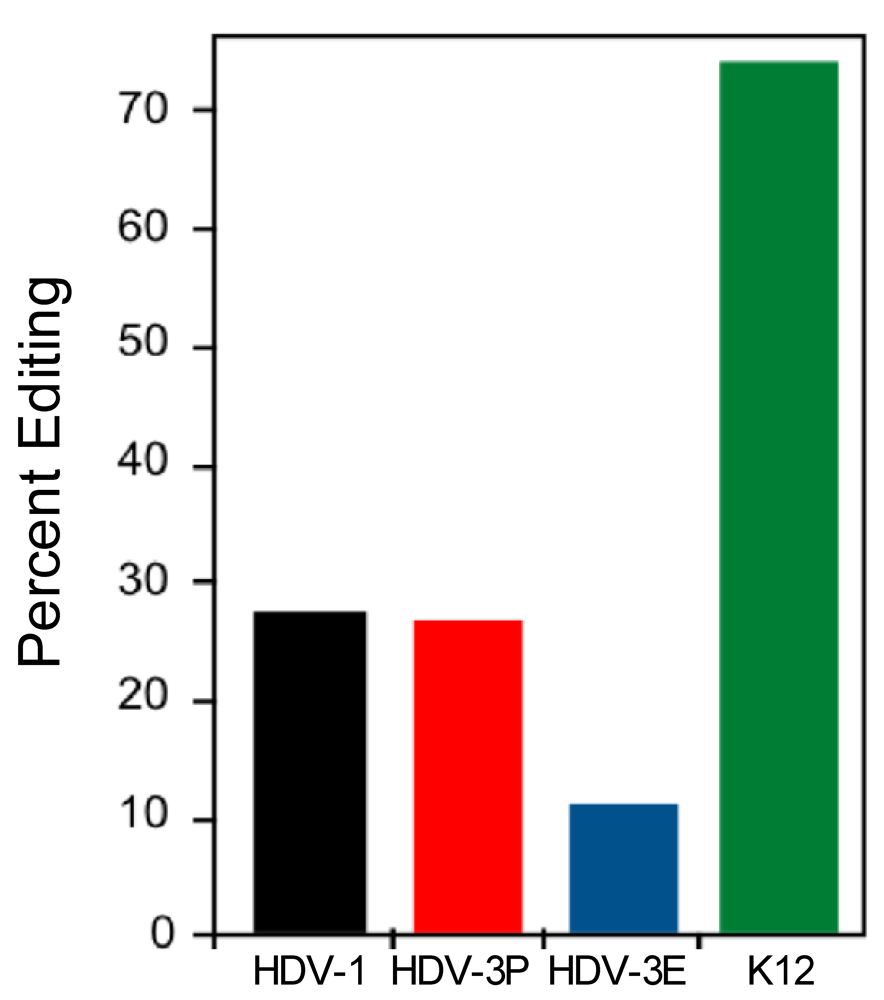
3.3. The Type 1 and Type 3 amber/W Sites are Sub-optimal for Editing by ADAR1
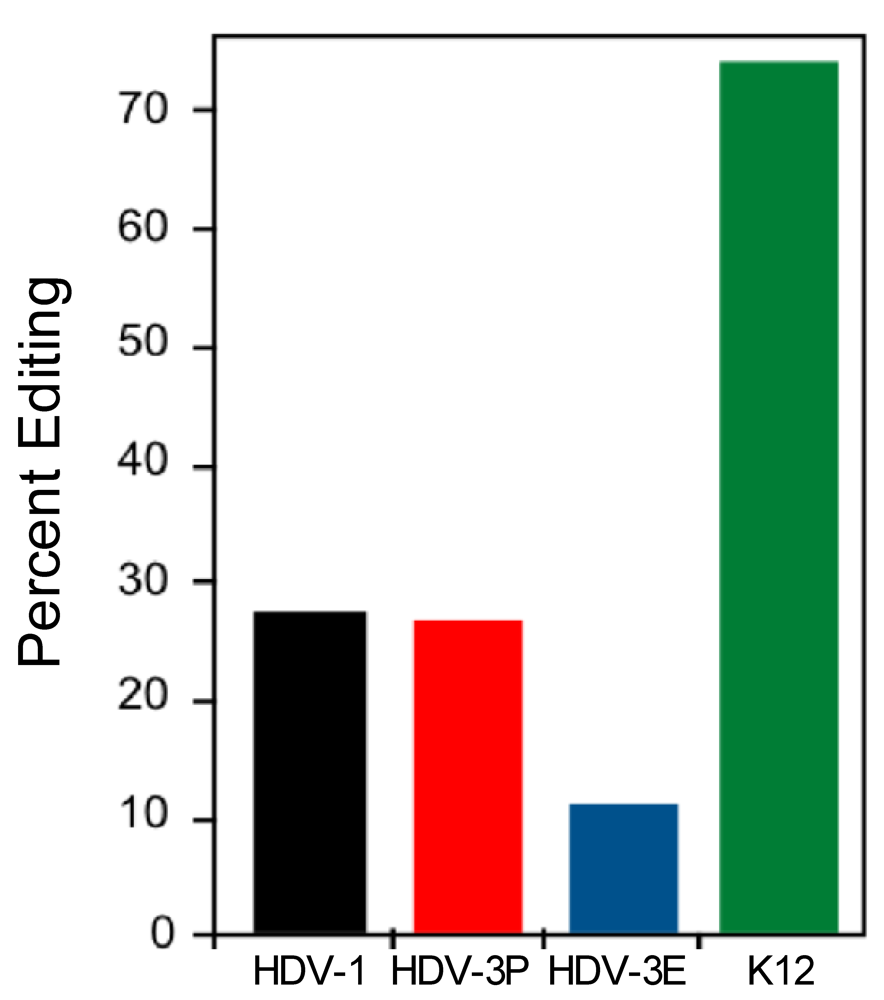
3.4. Control of Editing by Limiting Substrate Availability—Different Mechanisms for Different Structures
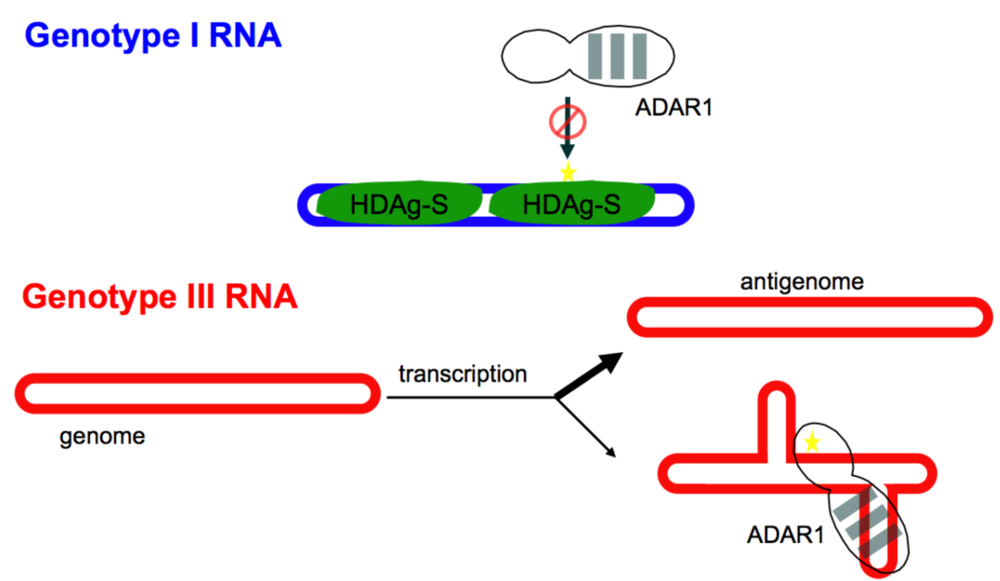
3.5. Negative Feedback Regulation of Editing
4. Summary and Perspectives
Acknowledgments
References
- Rizzetto, M. The delta agent. Hepatology 1983, 3, 729–737. [Google Scholar] [PubMed]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The large form of hepatitis delta antigen is crucial for assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef]
- Hwang, S.B.; Lee, C.Z.; Lai, M.M. Hepatitis delta antigen expressed by recombinant baculoviruses: comparison of biochemical properties and post-translational modifications between the large and small forms. Virology 1992, 190, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta virus large antigen. Science 1992, 256, 1331–1333. [Google Scholar] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [PubMed]
- Chao, M.; Hsieh, S.Y.; Taylor, J. Role of two forms of hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome replication. J. Virol. 1990, 64, 5066–5069. [Google Scholar] [PubMed]
- Jayan, G.C.; Casey, J.L. Inhibition of hepatitis delta virus RNA editing by short inhibitory RNA-mediated knockdown of Adar1 but not Adar2 expression. J. Virol. 2002, 76 in press. [Google Scholar]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef]
- Polson, A.G.; Bass, B.L.; Casey, J.L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature 1996, 380, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Jayan, G.C.; Casey, J.L. Effects of conserved RNA secondary structures on hepatitis delta virus genotype I RNA editing, replication, and virus production. J. Virol. 2005, 79, 11187–11193. [Google Scholar] [CrossRef] [PubMed]
- Jayan, G.C.; Casey, J.L. Increased RNA editing and inhibition of hepatitis delta virus replication by high-level expression of ADAR1 and ADAR2. J. Virol. 2002, 76, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Cornillez-Ty, C.; Lazinski, D.W. By inhibiting replication, the large hepatitis delta antigen can indirectly regulate amber/W editing and its own expression. J. Virol. 2004, 78, 8120–8134. [Google Scholar] [CrossRef] [PubMed]
- Linnstaedt, S.D.; Kasprzak, W.K.; Shapiro, B.A.; Casey, J.L. The fraction of RNA that folds into the correct branched secondary structure determines hepatitis delta virus type 3 RNA editing levels. RNA 2009, 15, 1177–1187. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Drakas, R.; Nishikura, K. Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J. Biol. Chem. 1995, 270, 17098–17105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Samuel, C.E. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA- specific adenosine deaminase. J. Virol. 1996, 70, 1961–1968. [Google Scholar] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Keegan, L.P.; Leroy, A.; Sproul, D.; O'Connell, M.A. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome. Biol. 2004, 5, 209. [Google Scholar] [CrossRef]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Levanon, E.Y.; Yoon, J.K.; Aach, J.; Xie, B.; Leproust, E.; Zhang, K.; Gao, Y.; Church, G.M. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009, 324, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Bass, B.L. Preferential selection of adenosines for modification by double- stranded RNA adenosine deaminase. Embo. J. 1994, 13, 5701–5711. [Google Scholar] [PubMed]
- Lehmann, K.A.; Bass, B.L. The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 1999, 291, 1–13. [Google Scholar] [CrossRef]
- Liu, Y.; Herbert, A.; Rich, A.; Samuel, C.E. Double-stranded RNA-specific adenosine deaminase: nucleic acid binding properties. Methods 1998, 15, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Ohman, M.; Kallman, A.M.; Bass, B.L. In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA 2000, 6, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.; Melcher, T.; Herb, A.; Seeburg, P.H.; Keller, W.; Krause, S.; Higuchi, M.; O'Connell, M.A. Structural requirements for RNA editing in glutamate receptor pre-mRNAs by recombinant double-stranded RNA adenosine deaminase. J. Biol. Chem. 1996, 271, 12221–12226. [Google Scholar] [CrossRef] [PubMed]
- Jaikaran, D.C.; Collins, C.H.; MacMillan, A.M. Adenosine to inosine editing by ADAR2 requires formation of a ternary complex on the GluR-B R/G site. J. Biol. Chem. 2002, 277, 37624–37629. [Google Scholar] [CrossRef] [PubMed]
- Aruscavage, P.J.; Bass, B.L. A phylogenetic analysis reveals an unusual sequence conservation within introns involved in RNA editing. RNA 2000, 6, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L. RNA Editing in Hepatitis Delta Virus Genotype III Requires a Branched Double-Hairpin RNA Structure. J. Virol. 2002, 76, 7385–7397. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Gerin, J.L. Hepatitis D virus RNA editing: specific modification of adenosine in the antigenomic RNA. J. Virol. 1995, 69, 7593–7600. [Google Scholar] [PubMed]
- Casey, J.L.; Bergmann, K.F.; Brown, T.L.; Gerin, J.L. Structural requirements for RNA editing in hepatitis delta virus: evidence for a uridine-to-cytidine editing mechanism. Proc. Natl. Acad. Sci. USA 1992, 89, 7149–7153. [Google Scholar] [CrossRef]
- Wong, S.K.; Sato, S.; Lazinski, D.W. Substrate recognition by ADAR1 and ADAR2. RNA 2001, 7, 846–858. [Google Scholar] [CrossRef]
- Lomeli, H.; Mosbacher, J.; Melcher, T.; Hoger, T.; Geiger, J.R.; Kuner, T.; Monyer, H.; Higuchi, M.; Bach, A.; Seeburg, P.H. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 1994, 266, 1709–1713. [Google Scholar] [PubMed]
- Herb, A.; Higuchi, M.; Sprengel, R.; Seeburg, P.H. Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 1875–1880. [Google Scholar] [CrossRef]
- Sato, S.; Wong, S.K.; Lazinski, D.W. Hepatitis delta virus minimal substrates competent for editing by adar1 and adar2. J. Virol. 2001, 75, 8547–8555. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.; Rich, A. The role of binding domains for dsRNA and Z-DNA in the in vivo editing of minimal substrates by ADAR1. Proc. Natl. Acad. Sci. USA 2001, 98, 12132–12137. [Google Scholar] [CrossRef]
- Linnstaedt, S.D.; Kasprzak, W.K.; Shapiro, B.A.; Casey, J.L. The role of a metastable RNA secondary structure in hepatitis delta virus genotype III RNA editing. RNA 2006, 12, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.P.; Lai, M.M. Oligomerization of hepatitis delta antigen is required for both the trans-activating and trans-dominant inhibitory activities of the delta antigen. J. Virol. 1992, 66, 6641–6648. [Google Scholar] [PubMed]
- Polson, A.G.; Ley 3rd, H.L.; Bass, B.L.; Casey, J.L. Hepatitis delta virus RNA editing is highly specific for the amber/W site and is suppressed by hepatitis delta antigen. Mol. Cell. Biol. 1998, 18, 1919–1926. [Google Scholar] [PubMed]
- Netter, H.J.; Wu, T.T.; Bockol, M.; Cywinski, A.; Ryu, W.S.; Tennant, B.C.; Taylor, J.M. Nucleotide sequence stability of the genome of hepatitis delta virus. J. Virol. 1995, 69, 1687–1692. [Google Scholar] [PubMed]
- Casey, J.L.; Tennant, B.C.; Gerin, J.L. Genetic changes in hepatitis delta virus from acutely and chronically infected woodchucks. J. Virol. 2006, 80, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Gudima, S.O.; Taylor, J.M. Evolution of hepatitis delta virus RNA genome following long-term replication in cell culture. J. Virol. 2005, 79, 13310–13316. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.Z.; Linnstaedt, S.D.; Muralidhar, S.; Cashman, K.A.; Rosenthal, L.J.; Casey, J.L. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. J. Virol. 2007, 81, 13544–13551. [Google Scholar] [CrossRef] [PubMed]
- Defenbaugh, D.A.; Johnson, M.; Chen, R.; Zheng, Y.Y.; Casey, J.L. Hepatitis delta antigen requires a minimum length of the hepatitis delta virus unbranched rod RNA structure for binding. J. Virol. 2009, 83, 4548–4556. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Bih, F.Y.; Chao, Y.C.; Govindarajan, S.; Lai, M.M. Evolution of hepatitis delta virus RNA during chronic infection. Virology 1992, 188, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Chang, M.F.; Baker, S.C.; Govindarajan, S.; Lai, M.M. Characterization of hepatitis delta antigen: specific binding to hepatitis delta virus RNA. J. Virol. 1990, 64, 4051–4058. [Google Scholar] [PubMed]
- Cheng, Q.; Jayan, G.C.; Casey, J.L. Differential inhibition of RNA editing in hepatitis delta virus genotype III by the short and long forms of hepatitis delta antigen. J. Virol. 2003, 77, 7786–7795. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Polson, A.G.; Bass, B.L.; Gerin, J.L. Hepatitis delta virus genetic variations and RNA editing. Rizzetto, M., Purcell, R.H., Gerin, J.L., Verme, G., Eds.; Edizioni minerva Medica: Turin, Italy, 1997; pp. 320–326. [Google Scholar]
- Hsu, S.C.; Syu, W.J.; Sheen, I.J.; Liu, H.T.; Jeng, K.S.; Wu, J.C. Varied assembly and RNA editing efficiencies between genotypes I and II hepatitis D virus and their implications. Hepatology 2002, 35, 665–672. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Chen, R.; Linnstaedt, S.D.; Casey, J.L. RNA Editing and its Control in Hepatitis Delta Virus Replication. Viruses 2010, 2, 131-146. https://doi.org/10.3390/v2010131
Chen R, Linnstaedt SD, Casey JL. RNA Editing and its Control in Hepatitis Delta Virus Replication. Viruses. 2010; 2(1):131-146. https://doi.org/10.3390/v2010131
Chicago/Turabian StyleChen, Renxiang, Sarah D. Linnstaedt, and John L. Casey. 2010. "RNA Editing and its Control in Hepatitis Delta Virus Replication" Viruses 2, no. 1: 131-146. https://doi.org/10.3390/v2010131
APA StyleChen, R., Linnstaedt, S. D., & Casey, J. L. (2010). RNA Editing and its Control in Hepatitis Delta Virus Replication. Viruses, 2(1), 131-146. https://doi.org/10.3390/v2010131