Abstract
Background: While enteroviruses (EVs) are recognized causes of diverse illnesses, little is known about the epidemiology and molecular characteristics of uncommon enterovirus C (EV-C) types, including EV-C104, EV-C105, and EV-C109. Methods: We conducted genomic surveillance of EVs at the Johns Hopkins Health System between 2018 and 2024 (a total of 3715 samples), identifying EV-C104, EV-C105, and EV-C109 in respiratory samples. VP4-VP2 and whole-genome sequencing were used to assess genetic diversity and intra-host evolution. Results: Five EV-C105 infections were identified primarily in pediatric patients, presenting with a range of clinical features including fever, gastrointestinal symptoms, and cerebellitis. Prolonged EV-C104 and EV-C109 infections were identified in two immunocompromised adults. EV-C104 persisted for over five months and showed evidence of viral genomic changes (intra-host evolution). EV-C109 was detected over a four-month period. Phylogenetic analysis revealed a novel EV-C105 clade (C3) closely related to recent USA strains. EV-C104 genomes aligned with genotype B sequences from the USA and Europe, while EV-C109 sequences were similar to 2014–2015 strains from the Netherlands. Conclusions: These findings highlight the emergence, persistence, and genetic evolution of uncommon EV-C types in Maryland, especially among immunocompromised hosts, emphasizing the importance of continued genomic surveillance and clinical correlations.
1. Introduction
Enteroviruses (EVs) are a diverse group of RNA viruses belonging to the Picornaviridae family, known to cause a wide range of clinical manifestations, from mild respiratory infections to severe systemic diseases such as meningitis, myocarditis, and acute flaccid paralysis [1]. While enteroviruses such as EV-D68 and EV-A71 have been studied due to their association with outbreaks and severe disease, less is known about the uncommon and emerging enterovirus types within the species enterovirus C (EV-C), including EV-C104, EV-C105, and EV-C109 [2,3,4,5,6]. Notably, poliovirus, the most well-recognized EV to cause acute flaccid paralysis (poliomyelitis), belongs to Enterovirus species C [7], highlighting the clinical importance of this group. Emerging EV-C types have been sporadically reported worldwide, often associated with respiratory and neurological disease, but their epidemiology and molecular features remain poorly understood [5,8,9].
EV-C105, first identified in 2010 in the Democratic Republic of Congo, has been linked to acute flaccid paralysis and respiratory infections [9]. Similarly, EV-C109, first reported in Nicaragua in 2008, has been associated with acute respiratory illness in children and has since been detected in Europe and the United States [3,10]. EV-C104, first detected in Switzerland in 2005, has since been infrequently reported [8,11,12,13,14]. Despite their potential clinical significance, the global incidence of these EV-C types appears to be low, likely due to underreporting and the infrequency of EV genomic surveillance [4,5].
In this study, we report EV-C105 detection from samples collected between 2023 and 2024 and tested at the Johns Hopkins Health System. Additionally, we describe prolonged infections with EV-C104 (2018) and EV-C109 (2024) in two immunocompromised patients. By employing next-generation sequencing and phylogenetic analysis, we attempted to elucidate the genetic diversity, evolutionary relationships, and potential clinical significance of these emerging viruses.
2. Methods
2.1. Ethics and Data Availability
The research was performed with a waiver of consent (the Johns Hopkins IRB protocol IRB00221396, initial approval on 29 October 2019). Genomes are available in GenBank: PV642522-PV642532.
2.2. Sample Collection
Remnant respiratory samples including nasopharyngeal swabs and bronchoalveolar lavage (BAL) that tested positive for rhinovirus/enterovirus (RV/EV) using the ePlex respiratory pathogen panels (Roche Diagnostics, Indianapolis, IN, USA) were used for genomic surveillance. Samples had collection dates between 2018 and 2024. For genotyping, nucleic acid was extracted from RV/EV-positive samples, and the VP4-VP2 region was amplified and sequenced as described previously [15,16].
Clinical and demographic data were collected from the electronic medical record system with a bulk query as previously described [17].
2.3. EV-C Whole-Genome Sequencing and Sequences Analysis
The Chemagic™ 360 system (Revvity, Walthan, MA, USA) was used for nucleic acid extraction. The LunaScript® RT SuperMix Kit (New England Biolabs (NEB), Ipswish, MA, USA) was used for reverse transcription.
For primary PCR amplification, Q5® High-Fidelity 2× Master Mix was used with primer pairs EV-ABC_80_F and EV-ABC4650_R for the P1 region and EVC-4220-F and C005-R [17,18] for the P2 region (Table S1). The thermal cycling protocol consisted of an initial denaturation at 98 °C for 30 s, followed by 35 cycles of 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 3 min, with a final extension at 72 °C for 10 min. Semi-nested and nested PCR were subsequently performed using primer pairs EV-ABC180_F and EV-ABC4450_R for the P1 region and EVC-4390-F and EVC-7405-R for the P2 region, respectively, while maintaining the same cycling conditions.
PCR products were pooled and purified using NEBNext Sample Purification Beads (NEB) before sequencing. The Native Barcoding (EXP-NBD196) and the NEBNext® ARTIC Library Prep Kits were used for library preparation. Sequencing was performed using the GridION (Oxford Nanopore Technologies, Oxford, England) and R10.4.1 flow cells. The resulting FASTQ files were analyzed using a previously described in-house pipeline (minimum depth cutoff was set at 100×) [19].
For phylogenetic analysis, complete genome sequences were used when available, while VP1 sequences were analyzed when only partial genomes were accessible. These sequences were aligned with all available GenBank references using MAFFT (version 7.450). Maximum likelihood trees were constructed with IQ-TREE3 (version 3.0.0) using 1000 bootstrap replicates, and the optimal nucleotide substitution model was determined with ModelFinder implemented in IQ-TREE3. An in-house script developed in R programming language (https://www.R-project.org/ accessed on 2 May 2024) was used for tree visualization.
To identify novel amino acid and nucleotide substitutions, we used a previously described Python (version 3.11.4). script [19].
3. Results
3.1. Demographic and Clinical Characteristics of Patients Positive for Uncommon EV-C
Of 3715 samples collected between January 2018 to November 2024 from a total of 3299 unique patients, EV-C105 was identified in 5 samples from 5 patients, EV-C104 in 4 samples from 1 patient, and EV-C109 in 4 samples from 1 patient. The five patients who tested positive for EV-C105 included two males and three females, aged 5 to 31 years and identified as either White (four patients) or Hispanic/Latino (one patient). Clinical presentations were heterogeneous but predominantly involved respiratory and gastrointestinal symptoms. Three patients presented with fever, abdominal pain, vomiting, and cough. One patient developed neurological manifestations including cerebellitis, nystagmus, dizziness, and migraine-like headaches and required intensive care. One patient presented with fever and gastrointestinal symptoms and had a history of asthma, immunosuppression, and hypertension. In total, three of the five patients were hospitalized, and one required ICU-level care. Interestingly, EV-C105 was found in patients from different geographic areas across Maryland, suggesting its widespread regional presence or multiple introductions despite the limited number of cases identified.
Of our screened cohort, we identified a case of a prolonged EV-C104 infection in an adult immunocompromised patient with uncontrolled HIV infection (CD4 count: 4 cells/mm3). The patient’s medical history was complicated with chronic hepatitis C, hypertension, hyperlipidemia, chronic pain, and history of substance use. The patient presented with respiratory symptoms, including shortness of breath, productive cough, and fever, along with systemic complaints such as chills, fatigue, and body ache. Gastrointestinal manifestations included diarrhea, nausea, and diffuse abdominal pain. Additional symptoms and signs included oropharyngeal thrush, anorexia, and mild intermittent headaches. Respiratory symptom onset occurred approximately two to three weeks prior to presentation. Despite the initiation of antiretroviral therapy, the patient experienced a persistent respiratory illness, with laboratory testing confirming EV-C104 positivity over a five-month period.
EV-C109 was identified in an adult patient with a history of Epstein–Barr virus (EBV)-positive NK/T-cell lymphoma who had undergone a myeloablative matched unrelated donor allogeneic stem cell transplantation (allo-SCT). The post-transplant course was complicated by chronic graft-versus-host disease (GVHD) affecting the lungs and eyes, resulting in bronchiolitis obliterans syndrome and progressive respiratory failure. The patient developed upper respiratory symptoms and tested positive for RV/EV, which was genotyped as EV-C109. Despite receiving two doses of intravenous immunoglobulin (IVIG), the patient remained persistently PCR-positive for RV/EV.
3.2. Phylogenetic Analysis and Viral Evolution
Complete genomes were successfully obtained for the four EV-C105 samples collected in 2024, while the 2023 sample yielded a partial genome with 66.1% coverage. Phylogenetic analysis indicated that three of the five sequences belonged to clade C1, clustering with strains identified from Italy in 2023, supported by a bootstrap value of 100% and nucleotide similarity exceeding 98.43% (Figure 1). The remaining two sequences, including the 2023 sample, formed a distinct cluster within clade C3 and were closely related to sequences from Massachusetts (USA) identified in 2023 and 2024, with a bootstrap value of 100% and nucleotide similarity above 97.74%. Notably, clades C1, C2, and C3 exhibit more than 5% sequence divergence from one another, consistent with established thresholds for the classification of well-characterized enteroviruses [20].
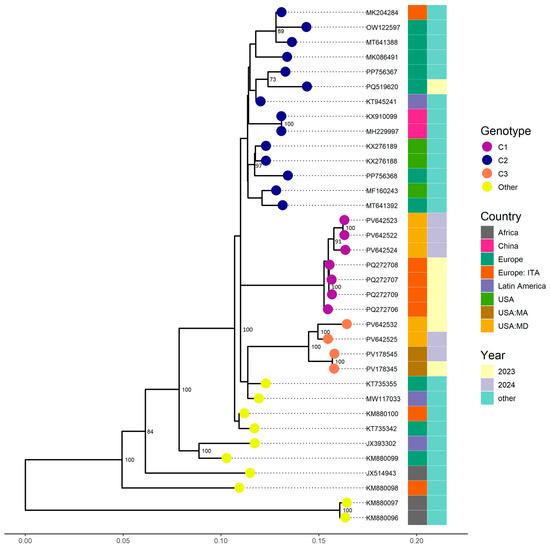
Figure 1.
Phylogenetic relationships of EV-C105 strains identified from the Johns Hopkins Health System in 2023 and 2024. Complete and partial genome sequences (VP1) from other countries available from GenBank were included. The phylogenetic tree was constructed using the Maximum Likelihood method in IQ-TREE2 with 1000 bootstrap replicates. Only bootstrap values above 70 were represented on corresponding branches.
For EV-C104, four samples collected at different time points from the same patient were sequenced. Sample 1 (S1) was the initial positive, followed by Sample 2 (S2), collected 1 day later; Sample 3 (S3), collected 108 days after S1; and Sample 4 (S4), collected 160 days after S1. Whole-genome sequences were successfully obtained for three of the samples, while a partial genome with 59.8% coverage was recovered from S2. No additional samples were collected from this patient after S4.
Phylogenetic analysis revealed that all strains belonged to genotype B, with a bootstrap value of 100 and were most closely related to a strain detected in Los Angeles, USA (MF160250, VP4-VP2 region). This strain exhibited a nucleotide similarity exceeding 97.3% with a complete genome sequence from France (PP756376), which had a similarity above 96.6%, both identified in 2014 (Figure 2).
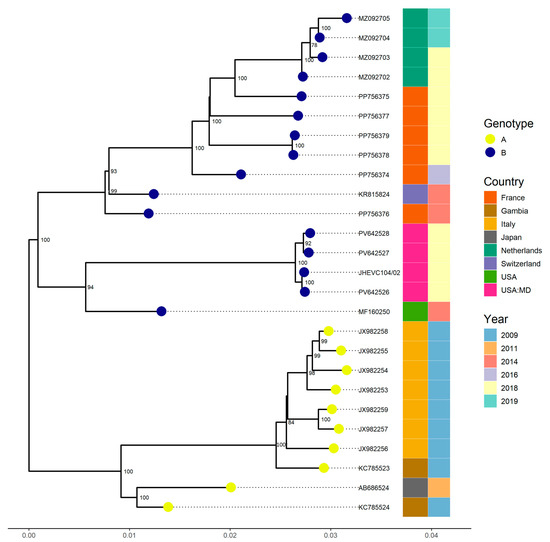
Figure 2.
Phylogenetic relationships of four EV-C104 strains identified from samples collected at different time points from the same patient at the Johns Hopkins Health System in 2018. Complete genome sequences from other countries available in GenBank, as well as one partial genome closely related to our strains based on BLAST (version 2.16.0) results were included. The phylogenetic tree was constructed using the Maximum Likelihood method implemented in IQ-TREE2 with 1000 bootstrap replicates. Only bootstrap values greater than 70% are shown on the corresponding branches.
To explore potential intra-host viral evolution, we analyzed single-nucleotide variants (SNVs) over time (Table 1). A total of 23 single-nucleotide variants (SNVs) were identified across the viral genome, 14 of which were shared by both samples (S3 and S4). These included five in the 5′ untranslated region (5′UTR), two each in VP2, VP3, and VP1, one in 2A, two each in 2B and 2C, four in 3D, and one in the 3′ UTR. Of these, six were non-synonymous substitutions, resulting in amino acid changes in both structural and non-structural proteins in each sample. These included E434G in VP3, D675G in VP1, I1618V and I1630T in the 3C protease, and V2133I and N2179D in the 3D polymerase. Notably, the VP1-D675G (D94G) mutation is located within the BC loop.

Table 1.
Intra-host variants among EV-C104-positive samples from a single patient. Samples S3 (collected 108 days after S1) and S4 (collected 160 days after S1) were compared to the earliest sample, S1, to assess viral variation over time.
Regarding EV-C109, four respiratory samples, including one BAL, collected from the same patient at different time points were sequenced. S1 was the initial specimen, followed by S2 which was collected 20 days later; S3 was collected 75 days after S1; and S4, a BAL sample, was collected 82 days after S1. Additionally, five other samples tested positive for RV/EV, including one that was collected 116 days after the initial sample; however, these were not available for sequencing. A subsequent negative RV/EV result was obtained 209 days after the first positive sample.
Complete genomes were successfully recovered from the three swab samples. Phylogenetic analysis revealed that our EV-C109 strains clustered with sequences (5′UTR region) detected in the Netherlands in 2014 and 2015, showing a nucleotide similarity of approximately 97.92% (Figure 3). However, a comparison with full genomes available in GenBank indicated a nucleotide similarity above 95.6% with strains from Florida (USA) detected in 2018.
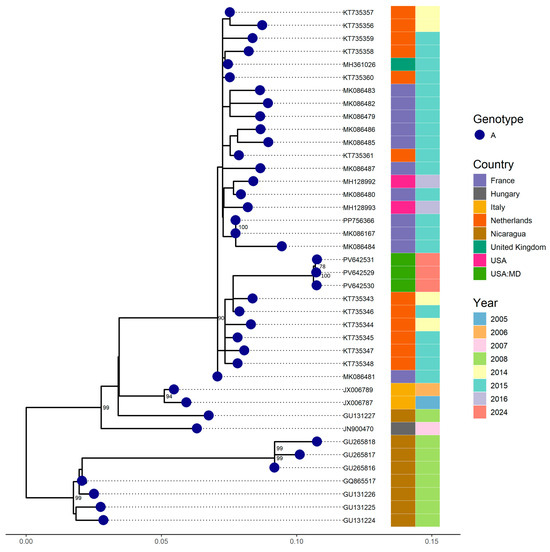
Figure 3.
Phylogenetic relationships among four EV-C109 strains identified from samples collected at different time points from the same patient at the Johns Hopkins Health System in 2024. Complete and partial genome (VP1) sequences from other countries retrieved from GenBank were included. A phylogenetic tree was constructed using the Maximum Likelihood method implemented in IQ-TREE2, with 1000 bootstrap replicates. Only bootstrap values above 70% are displayed on the corresponding branches.
Over time, several nucleotide mutations were detected between the initial EV-C109 S1 and the two subsequent samples (S2 and S3) (Table 2). A total of 21 SNVs were identified across the viral genome, distributed among both structural and non-structural regions. These included two SNVs in the 5′UTR, two in VP4, four in VP2, five in VP1, two in 2B, three in 2C, one in 3B, one in 3C, and one in 3D. Among the nucleotide changes, seven resulted in non-synonymous substitutions, leading to amino acid changes across both structural and non-structural proteins. Within the structural proteins, two amino acid changes were identified in VP2 (A190T and A205S), and three in VP1 (S597L, A607S, and S687T). For the non-structural proteins, one mutation was detected in 2C (I1235V) and one in 3D (T1764A). Notably, none of the non-synonymous substitutions were shared between samples.
Table 2.
Intra-host variants among EV-C109-positive samples from a single patient. Samples S2 (collected 20 days after S1) and S3 (collected at 75 days after S1) were compared to the earliest sample, S1, to assess viral variation over time.
4. Discussion
Of the sporadically reported enterovirus C types, we identified five EV-C105 strains from distinct geographic locations across Maryland between 2023 and 2024, which suggests local viral circulation. This observation aligns with recent EV-C105 detections in several countries, including Spain (5 cases) [4], Italy (6 cases) [5], and Russia (1 case) [21]. Globally, EV-C105 had previously been reported only sporadically between 2010 and 2022 [9,13,22,23,24,25]. The relatively increased recent reporting may be driven by increased global mobility, ongoing viral evolution, increased association with clinical symptomatic disease, or advances in sequencing technologies and surveillance efforts. Notably, the historical low detection rates of EV-C types might have been influenced by the limitations of commonly used cell lines for EV isolations. RD cells, which are widely used, are not optimal for EV-C types, whereas HEp-2C cells have demonstrated improved isolation efficiency [10,26]. Although our study utilized molecular surveillance methods, cell culture limitations may have contributed to the perceived rarity of EV-C105 in earlier studies.
Four of the five EV-C105 infections occurred in children, a trend consistent with prior reports [4,5]. This predominance may reflect immunological naivety, frequent exposure in communal settings, and possibly surveillance biases that prioritize testing in pediatric populations.
One of the EV-C105 cases was associated with cerebellitis, extending the known clinical spectrum of this virus beyond respiratory symptoms. While earlier studies primarily linked EV-C105 to mild respiratory illness [1,9], our observation aligns with rare reports of neurological involvement, including acute flaccid paralysis [4,24]. These findings indicate that further investigation into the neurotropic potential of EV-C105, including viral receptor use, cellular tropism, and host immune interactions is needed.
Phylogenetic analysis revealed the identification of two EV-C105 clades in Maryland: clade C1, identified in 2024 (3/5 sequences), and clade C3, identified in 2023 and 2024 (2/5). Clade C1 sequences were closely related to contemporary strains from Italy and Spain, suggesting recent transcontinental dissemination. Clade C3, newly defined in this study, showed > 5% nucleotide divergence from other clades, consistent with established thresholds for enterovirus classification [4,20]. This finding suggests the emergence of a novel EV-C105 clade circulating in the United States. The observed shift from clade C3 in 2023 to clade C1 in 2024 may reflect a clade replacement dynamic like patterns seen in Spain [4].
We also describe a case of prolonged EV-C104 infection in an immunocompromised patient, characterized by persistent viral detection over five months and evidence of intra-host genomic evolution. Previous studies have identified EV-C104 in both immunocompetent and immunocompromised individuals [6,8,22,27], but data on its evolution during chronic infection is limited.
Phylogenetic analysis confirmed that our EV-C104 strain belonged to genotype B, with >97.3% nucleotide identity to strains from Los Angeles (MF160250) and France (PP756376) identified in 2014, consistent with the global persistence of this lineage [11,13,28]. Intra-host analysis revealed 22 SNVs, including 14 shared across time points and 6 non-synonymous substitutions affecting both structural (e.g., VP1-D675G, VP3-E434G) and non-structural proteins (3C, 3D). These mutations persisted despite the anticipated lack of competent immune pressure, suggesting relaxed purifying selection, a pattern similarly observed in chronic EV infections [8]. Of particular interest, VP1-D675G is located in the BC loop, a region implicated in receptor binding and immune evasion, mirroring other conserved mutations seen in long-term infections [8].
Lastly, we present a case of prolonged EV-C109 infection in an immunocompromised patient. EV-C109 RNA was detected over four months, despite two rounds of IVIG therapy. This finding supports prior reports suggesting EV-C109 can cause persistent infections in immunocompromised individuals [6,8,22,27]. In our EV-C109 case, IVIG was administered, yet viral detection was prolonged. Recent case studies that reported prolonged enterovirus infections showed similar IVIG limitations. Chronic poliovirus infection in an immunodeficient patient and coxsackievirus A1 chronic diarrhea did not resolve after IVIG therapy [29,30]. This highlights the challenges of managing chronic infections caused by EV-C types in immunocompromised hosts.
Phylogenetic analysis revealed close relatedness to strains from the Netherlands (2014–2015) and Florida (2018), with >95.6% nucleotide identity, suggesting global but underrecognized circulation. Whole-genome sequencing of four sequential samples detected 21 nucleotide changes, including seven non-synonymous mutations in VP1, VP2, 2C, and 3D. Notably, VP1 substitutions (S597L, A607S, and S687T) may influence antigenicity or receptor interaction, while a 3D polymerase mutation (T1764A) may affect replication fidelity [31]. These amino acid changes are in both structural and non-structural regions of the genome, which may have functional consequences. Changes in VP1, a key capsid protein, could alter the surface charge or conformation, potentially affecting viral tropism, immune recognition, or receptor binding, even if synonymous. This aligns with previous work showing that conservative or synonymous substitutions in capsid regions can affect the overall protein structure and its interactions [32,33,34,35]. The observed genetic heterogeneity across samples, with limited shared non-synonymous variants, supports a quasispecies model and the presence of intra-host evolution under reduced immune pressure [36].
5. Limitations
This study has several limitations. The number of cases per EV-C type, particularly EV-C104 and EV-C109, was small, limiting generalizability. Incomplete genome coverage for some samples prevented comprehensive variant analysis. While several amino acid substitutions were identified, their functional relevance remains speculative in the absence of experimental validation. Samples were collected based on availability and are biased toward the population of patients tested clinically for RV/EV. Despite these constraints, the study contributes valuable insights into the genetic diversity and potential clinical significance of uncommon EV-C types in the USA.
6. Conclusions
Our study reports the detection of the rare enterovirus C types EV-C104, EV-C105, and EV-C109 in respiratory samples from Maryland. The identification of a novel EV-C105 clade and the documentation of prolonged EV-C104 and EV-C109 infections in immunocompromised individuals emphasize the need for increased clinical vigilance and ongoing genomic surveillance. Evidence of intra-host viral evolution during chronic infection emphasizes the potential of these viruses for persistence. Importantly, the lack of complete genome sequences for these rare EV-C types in public databases highlights a significant gap in global surveillance efforts and reinforces the need to prioritize full-genome sequencing to better inform diagnostics, treatment strategies, and epidemiological tracking.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/v17091183/s1, Table S1: Primers used for whole-genome amplification of the EV-C.
Author Contributions
Conceptualization, A.F. and H.H.M.; methodology, A.F., T.X.Z., D.V., A.D., J.M.N., O.A. and H.H.M.; software, A.F., E.K. and H.H.M.; validation, A.F., A.P., E.K. and H.H.M.; formal analysis, A.F.; investigation, A.F. and H.H.M.; resources, H.H.M.; data curation, A.F., E.K. and H.H.M.; writing—original draft preparation, A.F.; writing—review and editing, A.F., T.X.Z., A.D., J.M.N., A.P., E.K. and H.H.M.; visualization, A.F., A.P., E.K. and H.H.M.; supervision, H.H.M.; project administration, H.H.M.; funding acquisition, H.H.M. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Johns Hopkins Center of Excellence in Influenza Research and Response (HHS 75N93021C000045).
Data Availability Statement
Data is contained within the article.
Acknowledgments
The authors would like to acknowledge the staff of JHHS medical microbiology laboratories for their help with this study.
Conflicts of Interest
H.H.M. collaborates for research with Hologic, Qiagen, and Diasorin. H.H.M. received honoraria from BioMérieux, Roche Diagnostics, Qiagen, Diasorin, and BD Diagnostics and serves on the advisory board of Seegene. All other authors report no potential conflicts.
Abbreviations
The following abbreviations are used in this manuscript:
| EV | Enteroviruses |
| EV-C | Enterovirus C |
| RV/EV | Rhinovirus/enterovirus |
| BAL | Bronchoalveolar lavage |
| EBV | Epstein–Barr virus |
| allo-SCT | Allogeneic stem cell transplantation |
| GVHD | Graft-versus-host disease |
| IVIG | Intravenous immunoglobulin |
| SNVs | Single-nucleotide variants |
References
- Pallansch, M.A.; Oberste, M.S. Enterovirus 71 encephalitis: A new vaccine on the horizon? Lancet 2013, 381, 976–977. [Google Scholar] [CrossRef]
- Holm-Hansen, C.C.; Midgley, S.E.; Fischer, T.K. Global emergence of enterovirus D68: A systematic review. Lancet Infect. Dis. 2016, 16, e64–e75. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Yglesias, J.A.; Stevenson-Yuen, T.A.; Wolfe, C.M.; Cone, M.R.; Heberlein-Larson, L.A.; Maher, K.; Rogers, S.; Chern, S.W.; Montmayeur, A.; et al. Characterization of the Genome Sequences of Enterovirus C109 from Two Respiratory Disease Cases in Florida, 2016. Microbiol. Resour. Announc. 2018, 7, e00803-18. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Camacho, J.; Diez-Fuertes, F.; Ruiz de Pedro, E.; Garcia-Ibanez, N.; Navascues, A.; Berengua, C.; Antequera-Rodriguez, P.; Ruiz-Garcia, M.; Pastor-Fajardo, M.T.; et al. Detections of rare enterovirus C105 linked to an emerging novel clade, Spain, 2019 to 2024. Eurosurveillance 2025, 30, 2500073. [Google Scholar] [CrossRef]
- Pomari, E.; Malago, S.; Ferrari, G.; Romano, G.; Mori, A.; Matucci, A.; Feletti, R.; Bonetti, P.; Baldanti, F.; Castilletti, C.; et al. Unexpected Pediatric Cluster of Enterovirus C105, Verona, Italy. Viruses 2025, 17, 255. [Google Scholar] [CrossRef]
- van Leer-Buter, C.C.; Poelman, R.; Borger, R.; Niesters, H.G. Newly Identified Enterovirus C Genotypes, Identified in the Netherlands through Routine Sequencing of All Enteroviruses Detected in Clinical Materials from 2008 to 2015. J. Clin. Microbiol. 2016, 54, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; van der Sanden, S.M.G.; Calis, J.C.J.; Bruning, A.H.L.; Wang, S.; Wildenbeest, J.G.; Rebers, S.P.H.; Phiri, K.S.; Westerhuis, B.M.; van Hensbroek, M.B.; et al. High frequency of Polio-like Enterovirus C strains with differential clustering of CVA-13 and EV-C99 subgenotypes in a cohort of Malawian children. Arch. Virol. 2018, 163, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, H.; Schuele, L.; Lizarazo-Forero, E.; Couto, N.; Rossen, J.W.A.; Friedrich, A.W.; van Leer-Buter, C.; Niesters, H.G.M. Exploring a prolonged enterovirus C104 infection in a severely ill patient using nanopore sequencing. Virus Evol. 2022, 8, veab109. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Drexler, J.F.; Kotova, V.O.; Amjaga, E.N.; Reznik, V.I.; Gmyl, A.P.; Grard, G.; Taty Taty, R.; Trotsenko, O.E.; Leroy, E.M.; et al. Novel serotypes 105 and 116 are members of distinct subgroups of human enterovirus C. J. Gen. Virol. 2012, 93, 2357–2362. [Google Scholar] [CrossRef] [PubMed]
- Yozwiak, N.L.; Skewes-Cox, P.; Gordon, A.; Saborio, S.; Kuan, G.; Balmaseda, A.; Ganem, D.; Harris, E.; DeRisi, J.L. Human enterovirus 109: A novel interspecies recombinant enterovirus isolated from a case of acute pediatric respiratory illness in Nicaragua. J. Virol. 2010, 84, 9047–9058. [Google Scholar] [CrossRef]
- Kaida, A.; Kubo, H.; Sekiguchi, J.; Hase, A.; Iritani, N. Enterovirus 104 infection in adult, Japan, 2011. Emerg. Infect. Dis. 2012, 18, 882–883. [Google Scholar] [CrossRef]
- Piralla, A.; Fiorina, L.; Daleno, C.; Esposito, S.; Baldanti, F. Complete genome characterization of enterovirus 104 circulating in Northern Italy shows recombinant origin of the P3 region. Infect. Genet. Evol. 2013, 20, 111–117. [Google Scholar] [CrossRef]
- Tokarz, R.; Hirschberg, D.L.; Sameroff, S.; Haq, S.; Luna, G.; Bennett, A.J.; Silva, M.; Leguia, M.; Kasper, M.; Bausch, D.G.; et al. Genomic analysis of two novel human enterovirus C genotypes found in respiratory samples from Peru. J. Gen. Virol. 2013, 94, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Wollants, E.; Maes, P.; Merino, M.; Bloemen, M.; Van Ranst, M.; Vanmechelen, B. First genomic characterization of a Belgian Enterovirus C104 using sequence-independent Nanopore sequencing. Infect. Genet. Evol. 2020, 81, 104267. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.H.; Fall, A.; Norton, J.M.; Sachithanandham, J.; Yunker, M.; Abdullah, O.; Hanlon, A.; Gluck, L.; Morris, C.P.; Pekosz, A.; et al. Respiratory virus disease and outcomes at a large academic medical center in the United States: A retrospective observational study of the early 2023/2024 respiratory viral season. Microbiol. Spectr. 2024, 12, e0111624. [Google Scholar] [CrossRef]
- Fall, A.; Norton, J.M.; Abdullah, O.; Pekosz, A.; Klein, E.; Mostafa, H.H. Enhanced genomic surveillance of enteroviruses reveals a surge in enterovirus D68 cases, the Johns Hopkins health system, Maryland, 2024. J. Clin. Microbiol. 2025, 63, e0046925. [Google Scholar] [CrossRef] [PubMed]
- Fall, A.; Gallagher, N.; Morris, C.P.; Norton, J.M.; Pekosz, A.; Klein, E.; Mostafa, H.H. Circulation of Enterovirus D68 during Period of Increased Influenza-Like Illness, Maryland, USA, 2021. Emerg. Infect. Dis. 2022, 28, 1525–1527. [Google Scholar] [CrossRef] [PubMed]
- Joffret, M.L.; Polston, P.M.; Razafindratsimandresy, R.; Bessaud, M.; Heraud, J.M.; Delpeyroux, F. Whole Genome Sequencing of Enteroviruses Species A to D by High-Throughput Sequencing: Application for Viral Mixtures. Front. Microbiol. 2018, 9, 2339. [Google Scholar] [CrossRef]
- Fall, A.; Abdullah, O.; Han, L.; Norton, J.M.; Gallagher, N.; Forman, M.; Morris, C.P.; Klein, E.; Mostafa, H.H. Enterovirus D68: Genomic and Clinical Comparison of 2 Seasons of Increased Viral Circulation and Discrepant Incidence of Acute Flaccid Myelitis-Maryland, USA. Open Forum Infect. Dis. 2024, 11, ofae656. [Google Scholar] [CrossRef]
- Benschop, K.S.M.; Broberg, E.K.; Hodcroft, E.; Schmitz, D.; Albert, J.; Baicus, A.; Bailly, J.L.; Baldvinsdottir, G.; Berginc, N.; Blomqvist, S.; et al. Molecular Epidemiology and Evolutionary Trajectory of Emerging Echovirus 30, Europe. Emerg. Infect. Dis. 2021, 27, 1616–1626. [Google Scholar] [CrossRef]
- Nokhova, A.R.; Saroyan, T.A.; Solomatina, M.V.; Gutova, T.A.; Derko, A.A.; Dubovitskiy, N.A.; Murashkina, T.A.; Sharshov, K.A.; Shestopalov, A.M.; Kurskaya, O.G. Genetic Diversity and Epidemiology of Enteroviruses and Rhinoviruses in Children Hospitalized with Acute Respiratory Infections in Novosibirsk, Russia (2023–2024). Viruses 2024, 16, 1924. [Google Scholar] [CrossRef]
- Barnadas, C.; Midgley, S.E.; Skov, M.N.; Jensen, L.; Poulsen, M.W.; Fischer, T.K. An enhanced Enterovirus surveillance system allows identification and characterization of rare and emerging respiratory enteroviruses in Denmark, 2015–2016. J. Clin. Virol. 2017, 93, 40–44. [Google Scholar] [CrossRef]
- Piralla, A.; Daleno, C.; Girello, A.; Esposito, S.; Baldanti, F. Circulation of two Enterovirus C105 (EV-C105) lineages in Europe and Africa. J. Gen. Virol. 2015, 96, 1374–1379. [Google Scholar] [CrossRef]
- Horner, L.M.; Poulter, M.D.; Brenton, J.N.; Turner, R.B. Acute Flaccid Paralysis Associated with Novel Enterovirus C105. Emerg. Infect. Dis. 2015, 21, 1858–1860. [Google Scholar] [CrossRef]
- Li, M.; Zhang, T.; Gong, C.; Li, A.; Luo, M.; Dong, M.; Huang, F. Human Enterovirus C105, China, 2017. Emerg. Infect. Dis. 2019, 25, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Pillet, S.; Ibrahim, W.; Joffret, M.L.; Pozzetto, B.; Delpeyroux, F.; Gouandjika-Vasilache, I. Molecular characterization of human enteroviruses in the Central African Republic: Uncovering wide diversity and identification of a new human enterovirus A71 genogroup. J. Clin. Microbiol. 2012, 50, 1650–1658. [Google Scholar] [CrossRef]
- Piralla, A.; Rovida, F.; Baldanti, F.; Gerna, G. Enterovirus genotype EV-104 in humans, Italy, 2008-2009. Emerg. Infect. Dis. 2010, 16, 1018–1021. [Google Scholar] [CrossRef]
- Xiang, Z.; Xie, Z.; Wang, Z.; Ren, L.; Xiao, Y.; Li, L.; Vernet, G.; Paranhos-Baccala, G.; Shen, K.; Wang, J. Human enterovirus genotype C104, China. Emerg. Infect. Dis. 2013, 19, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, W.H.; Canning, B.; Wilton, T.; Kidd, M.; Klapsa, D.; Majumdar, M.; Sooriyakumar, K.; Martin, J.; Huissoon, A.P. Case report: Clearance of longstanding, immune-deficiency-associated, vaccine-derived polio virus infection following remdesivir therapy for chronic SARS-CoV-2 infection. Front. Immunol. 2023, 14, 1135834. [Google Scholar] [CrossRef] [PubMed]
- Moreni, G.; Calitz, C.; Koen, G.; van Eijk, H.; Johannesson, N.; De Ruijter, J.; Benschop, K.S.M.; Cremer, J.; Pajkrt, D.; Sridhar, A.; et al. Toward Personalized Medicine: The Effect of Treatment of Chronic Enterovirus Diarrhea in an Immunocompromised Patient and the Correlation With In Vitro Models. Open Forum Infect. Dis. 2025, 12, ofaf212. [Google Scholar] [CrossRef]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef]
- Pechmann, S.; Frydman, J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 2013, 20, 237–243. [Google Scholar] [CrossRef]
- Machado, R.S.; Gomes-Neto, F.; Aguiar-Oliveira, M.L.; Burlandy, F.M.; Tavares, F.N.; da Silva, E.E.; Sousa, I.P., Jr. Analysis of Coxsackievirus B5 Infections in the Central Nervous System in Brazil: Insights into Molecular Epidemiology and Genetic Diversity. Viruses 2022, 14, 899. [Google Scholar] [CrossRef]
- Leser, J.S.; Frost, J.L.; Wilson, C.J.; Rudy, M.J.; Clarke, P.; Tyler, K.L. VP1 is the primary determinant of neuropathogenesis in a mouse model of enterovirus D68 acute flaccid myelitis. J. Virol. 2024, 98, e0039724. [Google Scholar] [CrossRef] [PubMed]
- Dhindsa, R.S.; Copeland, B.R.; Mustoe, A.M.; Goldstein, D.B. Natural Selection Shapes Codon Usage in the Human Genome. Am. J. Hum. Genet. 2022, 109, 1559. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).