
In the Absence of Type-1 IFN, HSV-1 LAT Increases γ34.5 Expression and Enhances Mortality in Infected Mice

Abstract
1. Importance
2. Introduction
3. Results
3.1. LAT Protects Against Neurovirulence in the Absence of Type-1 Interferons
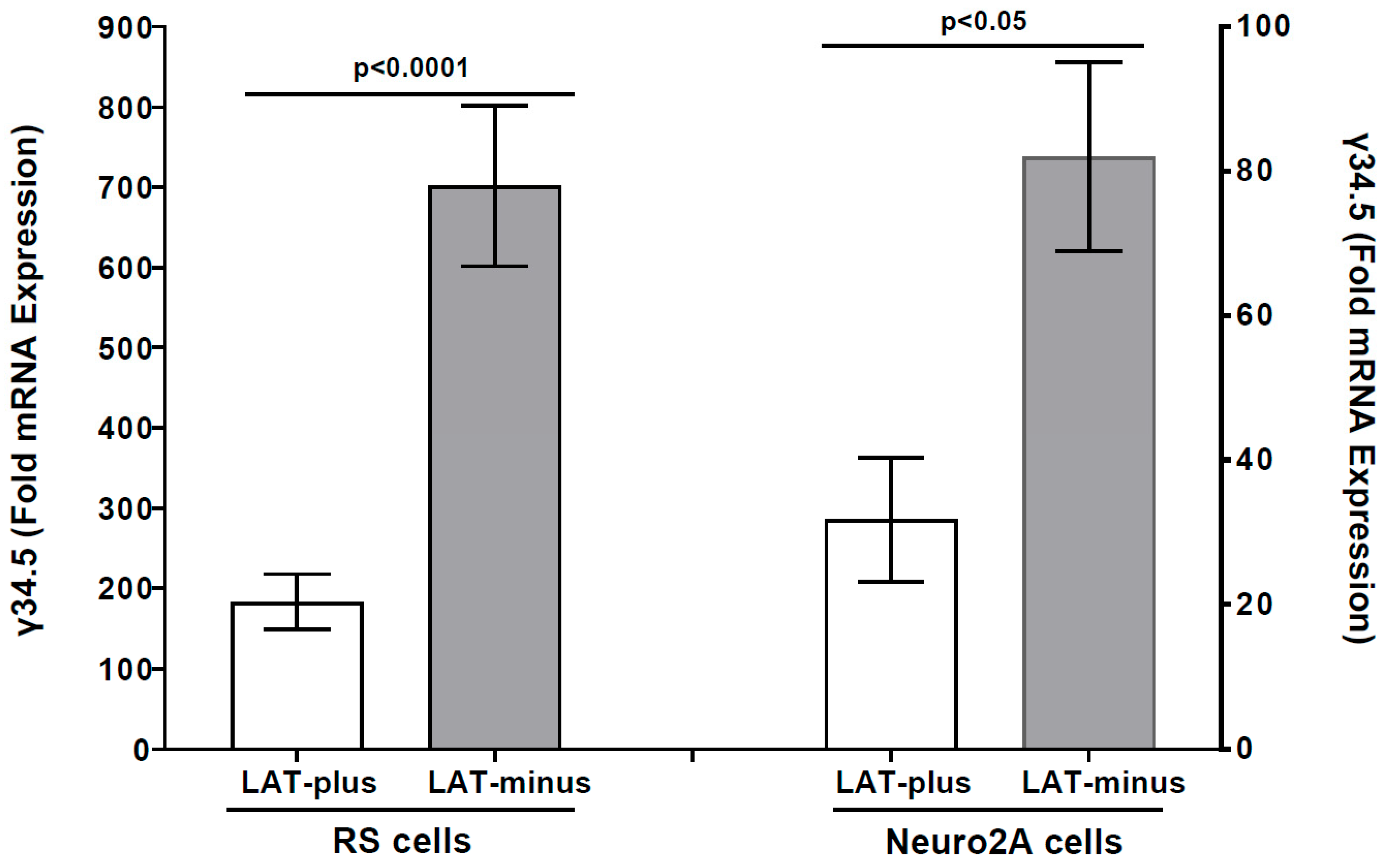
3.2. The γ34.5 Gene Is Upregulated in the Absence of LAT in LAT-Minus-Infected RS and Neuro2A Cells in Vitro
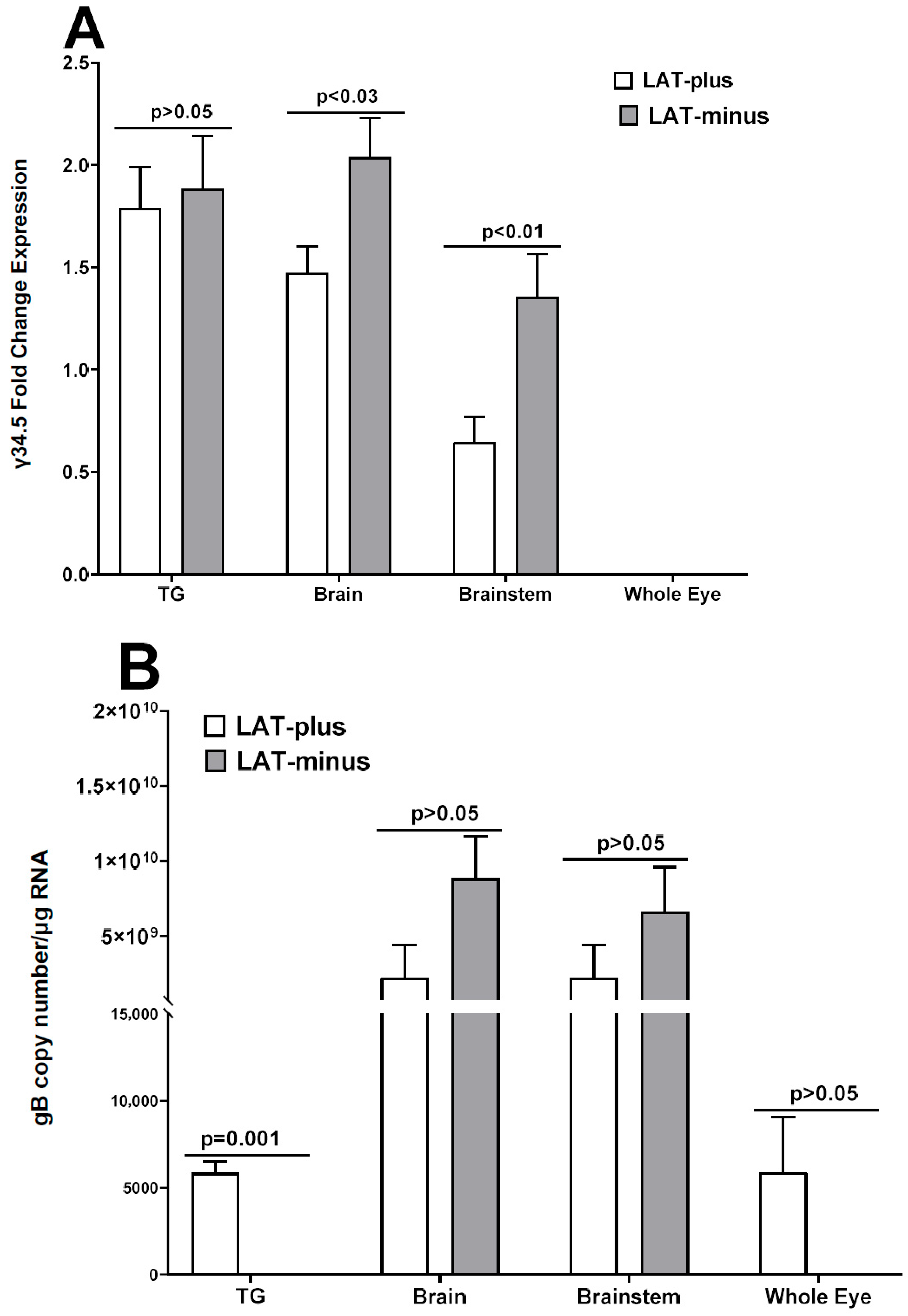
3.3. Upregulation of γ34.5, but Not gB, in the Absence of LAT in the CNS of Infected Mice
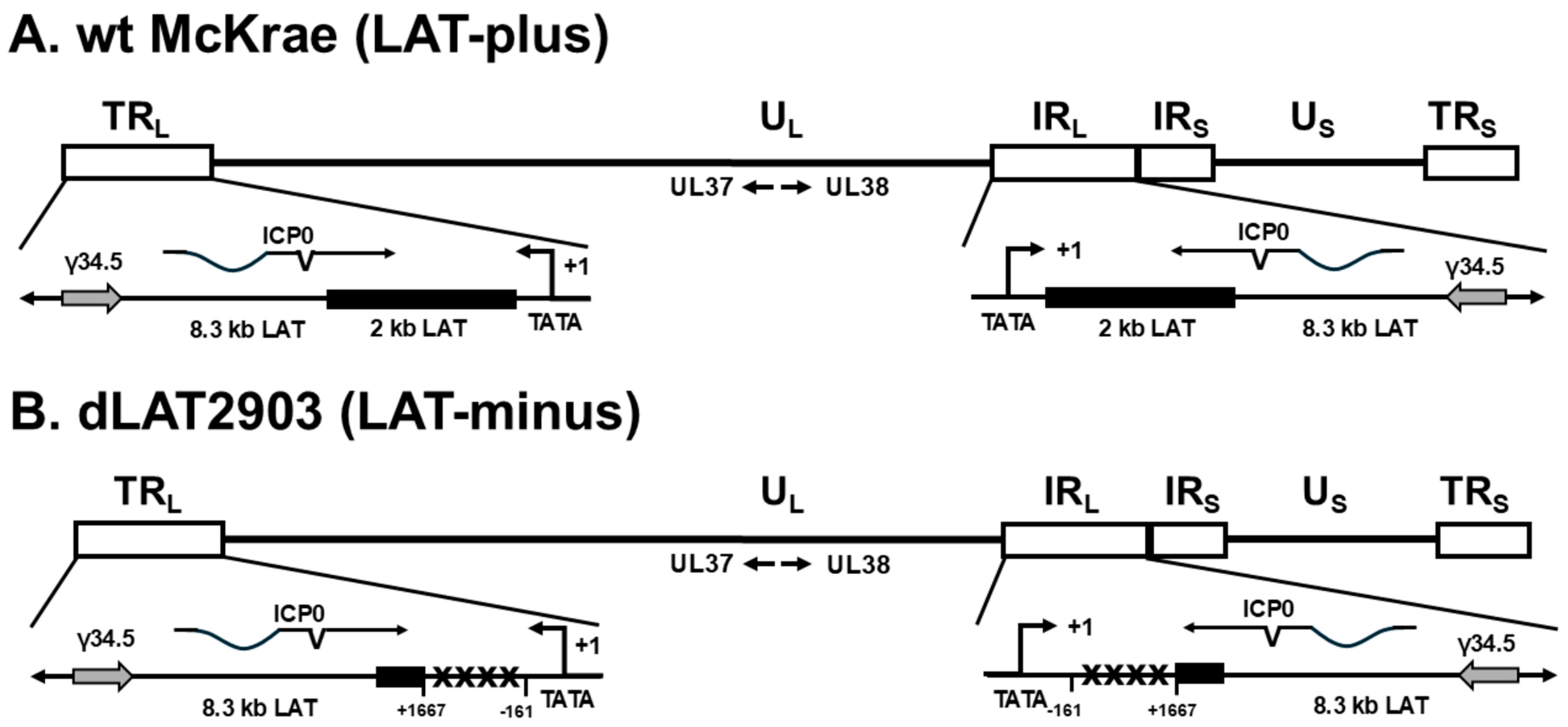
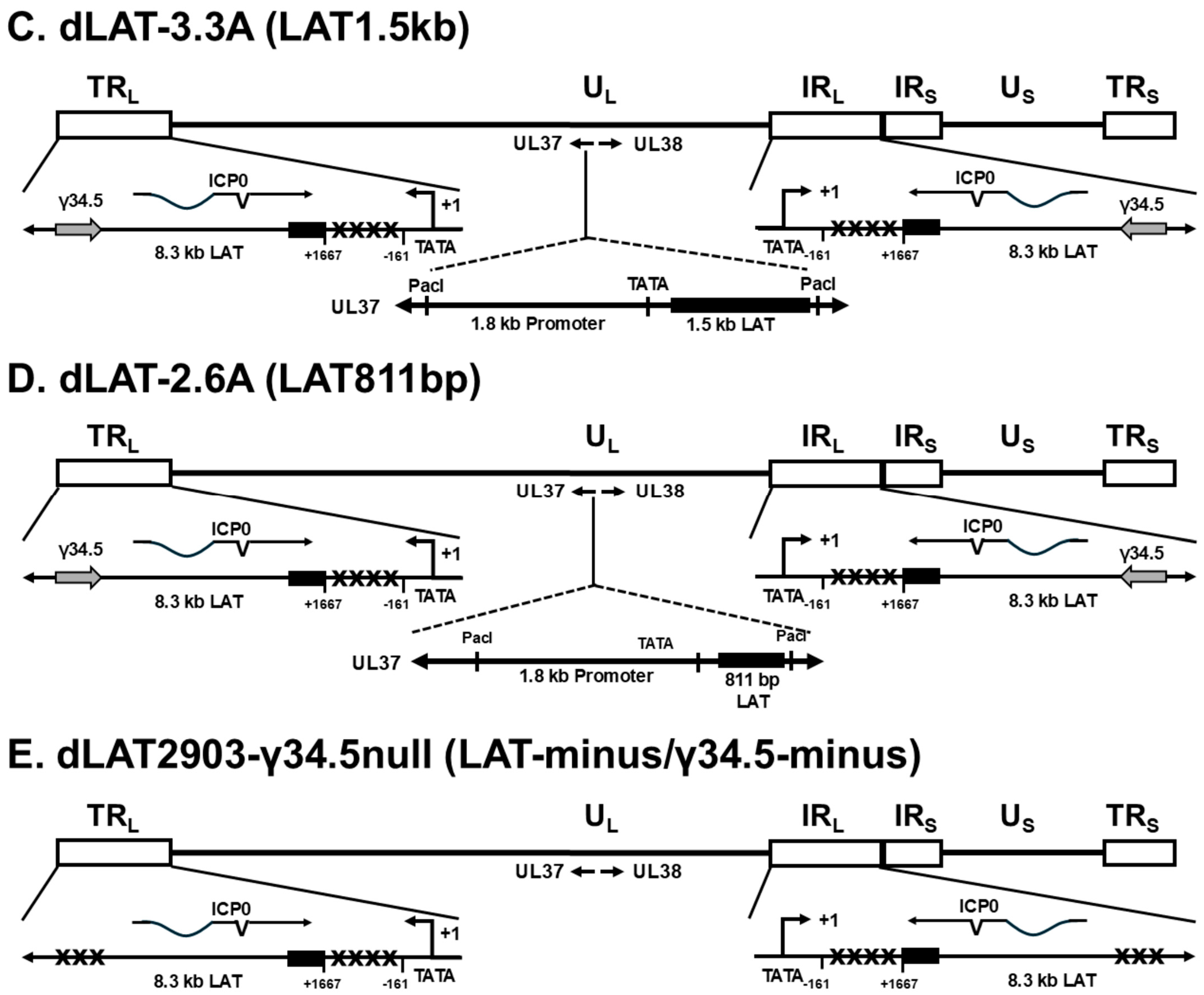
3.4. Mapping the Neurovirulence Region of LAT That Contributes to Increased Mortality and γ34.5 Upregulation in Infected IFNαβR−/− Mice
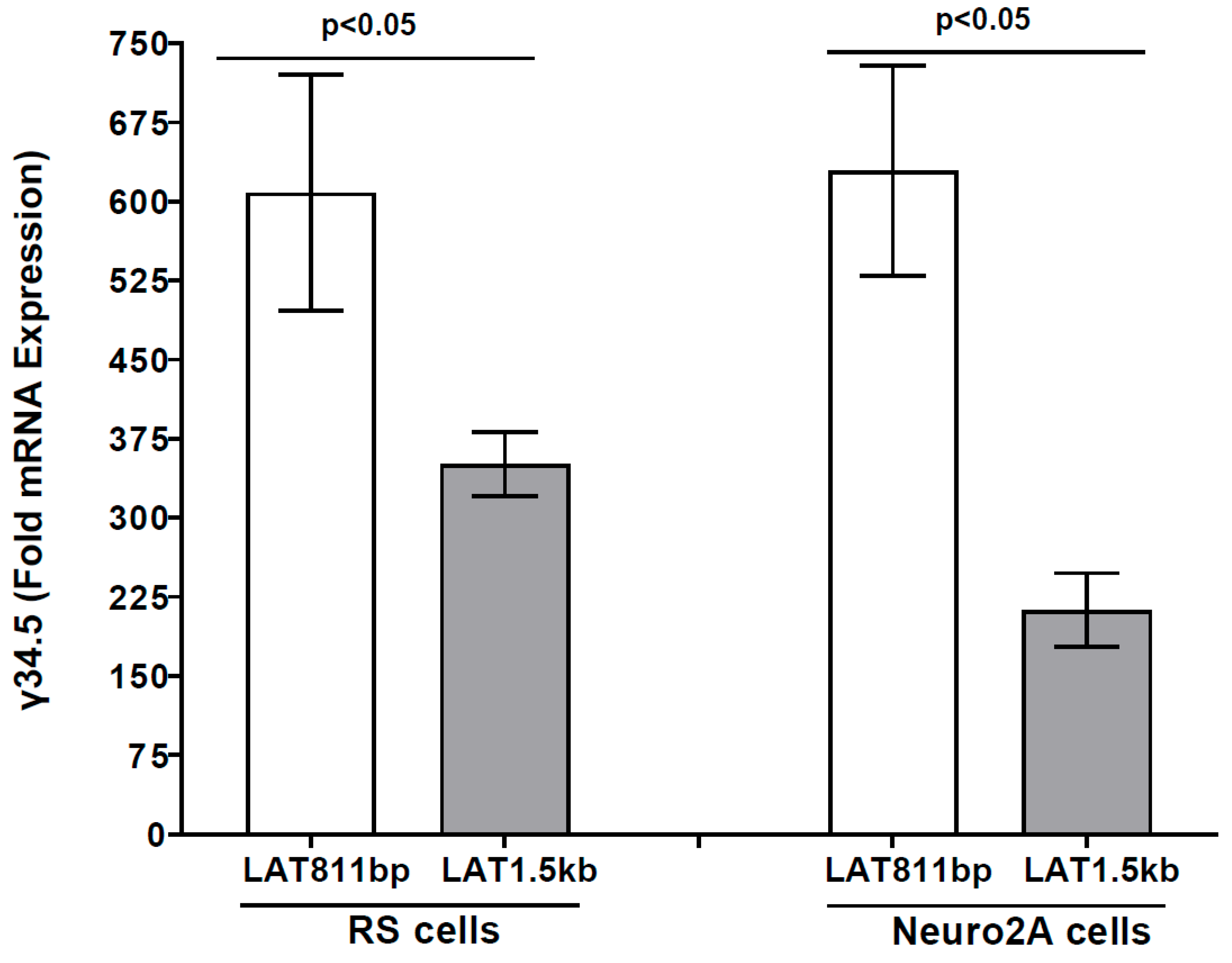
3.5. γ34.5 Gene Is Upregulated in the Absence of the Second Half of LAT in LAT811bp-Infected RS and Neuro2A Cells In Vitro
3.6. Upregulation of γ34.5 in the Absence of LAT Affects Survival in Infected Mice
4. Discussion
5. Materials and Methods
5.1. Ethics Statement
5.2. Structure of Recombinant Viruses
5.3. Cell Lines and Mice
5.4. Ocular Infection and Survival of Infected Mice
5.5. In Vitro Infection of RS and Neuro2A Cells
5.6. RNA Extraction, cDNA Synthesis, and TaqMan RT-PCR
5.7. Isolation of RNA from TG, Brainstem, Brain, and the Whole Eye of Infected Mice
5.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dawson, C.R. Ocular herpes simplex virus infections. Clin. Dermatol. 1984, 2, 56–66. [Google Scholar] [CrossRef]
- Wilhelmus, K.R.; Dawson, C.R.; Barron, B.A.; Bacchetti, P.; Gee, L.; Jones, D.B.; Kaufman, H.E.; Sugar, J.; Hyndiuk, R.A.; Laibson, P.R.; et al. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br. J. Ophthalmol. 1996, 80, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.G. Human herpesviruses: A consideration of the latent state. Microbiol. Rev. 1989, 53, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Rock, D.L.; Nesburn, A.B.; Ghiasi, H.; Ong, J.; Lewis, T.L.; Lokensgard, J.R.; Wechsler, S.L. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol. 1987, 61, 3820–3826. [Google Scholar] [CrossRef]
- Fraser, N.W.; Valyi-Nagy, T. Viral, neuronal and immune factors which may influence herpes simplex virus (HSV) latency and reactivation. Microb. Pathog. 1993, 15, 83–91. [Google Scholar] [CrossRef]
- Hill, J.M.; Sedarati, F.; Javier, R.T.; Wagner, E.K.; Stevens, J.G. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology 1990, 174, 117–125. [Google Scholar] [CrossRef]
- Wechsler, S.L.; Nesburn, A.B.; Watson, R.; Slanina, S.; Ghiasi, H. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J. Gen. Virol. 1988, 69, 3101–3106. [Google Scholar] [CrossRef]
- Wechsler, S.L.; Nesburn, A.B.; Watson, R.; Slanina, S.M.; Ghiasi, H. Fine mapping of the latency-related gene of herpes simplex virus type 1: Alternative splicing produces distinct latency-related RNAs containing open reading frames. J. Virol. 1988, 62, 4051–4058. [Google Scholar] [CrossRef]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef]
- Deatly, A.M.; Spivack, J.G.; Lavi, E.H.; O’Boyle 2nd, D.R.; Fraser, N.W. Latent herpes simplex virus type 1 transcripts in peripheral and central nervous system tissues of mice map to similar regions of the viral genome. J. Virol. 1988, 62, 749–756. [Google Scholar] [CrossRef]
- Perng, G.C.; Dunkel, E.C.; Geary, P.A.; Slanina, S.M.; Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol. 1994, 68, 8045–8055. [Google Scholar] [CrossRef] [PubMed]
- Perng, G.C.; Ghiasi, H.; Slanina, S.M.; Nesburn, A.B.; Wechsler, S.L. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3- kilobase primary transcript. J. Virol. 1996, 70, 976–984. [Google Scholar] [CrossRef]
- Perng, G.C.; Slanina, S.M.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J. Virol. 1996, 70, 2014–2018. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef]
- Henderson, G.; Peng, W.; Jin, L.; Perng, G.C.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J. Neurovirol. 2002, 8 (Suppl. S2), 103–111. [Google Scholar] [CrossRef]
- Perng, G.C.; Maguen, B.; Jin, L.; Mott, K.R.; Osorio, N.; Slanina, S.M.; Yukht, A.; Ghiasi, H.; Nesburn, A.B.; Inman, M.; et al. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J. Virol. 2002, 76, 1224–1235. [Google Scholar] [CrossRef]
- Jin, L.; Perng, G.C.; Mott, K.R.; Osorio, N.; Naito, J.; Brick, D.J.; Carpenter, D.; Jones, C.; Wechsler, S.L. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J. Virol. 2005, 79, 12286–12295. [Google Scholar] [CrossRef]
- Peng, W.; Henderson, G.; Inman, M.; BenMohamed, L.; Perng, G.C.; Wechsler, S.L.; Jones, C. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal Ganglia of acutely infected mice. J. Virol. 2005, 79, 6162–6171. [Google Scholar] [CrossRef]
- Jin, L.; Perng, G.C.; Carpenter, D.; Mott, K.R.; Osorio, N.; Naito, J.; Brick, D.J.; Jones, C.; Wechsler, S.L. Reactivation phenotype in rabbits of a herpes simplex virus type 1 mutant containing an unrelated antiapoptosis gene in place of latency-associated transcript. J. Neurovirol. 2007, 13, 78–84. [Google Scholar] [CrossRef]
- Branco, F.J.; Fraser, N.W. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J. Virol. 2005, 79, 9019–9025. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 262415. [Google Scholar] [CrossRef]
- Allen, S.J.; Rhode-Kurnow, A.; Mott, K.R.; Jiang, X.; Carpenter, D.; Rodriguez-Barbosa, J.I.; Jones, C.; Wechsler, S.L.; Ware, C.F.; Ghiasi, H. Interactions between herpesvirus entry mediator (tnfrsf14) and latency-associated transcript during herpes simplex virus 1 latency. J. Virol. 2014, 88, 1961–1971. [Google Scholar] [CrossRef]
- Tormanen, K.; Allen, S.; Mott, K.R.; Ghiasi, H. The Latency-Associated Transcript Inhibits Apoptosis via Downregulation of Components of the Type I Interferon Pathway during Latent Herpes Simplex Virus 1 Ocular Infection. J. Virol. 2019, 93, e00103–e00119. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A.; Biron, C.A. Type 1 interferons and the virus-host relationship: A lesson in detente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Navratil, V.; de Chassey, B.; Meyniel, L.; Pradezynski, F.; Andre, P.; Rabourdin-Combe, C.; Lotteau, V. System-level comparison of protein-protein interactions between viruses and the human type I interferon system network. J. Proteome. Res. 2010, 9, 3527–3536. [Google Scholar] [CrossRef]
- Lin, R.J.; Liao, C.L.; Lin, E.; Lin, Y.L. Blocking of the alpha interferon-induced Jak-Stat signaling pathway by Japanese encephalitis virus infection. J. Virol. 2004, 78, 9285–9294. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Pestka, S. The human interferon alpha species and receptors. Biopolymers 2000, 55, 254–287. [Google Scholar] [CrossRef]
- Hardy, M.P.; Owczarek, C.M.; Trajanovska, S.; Liu, X.; Kola, I.; Hertzog, P.J. The soluble murine type I interferon receptor Ifnar-2 is present in serum, is independently regulated, and has both agonistic and antagonistic properties. Blood 2001, 97, 473–482. [Google Scholar] [CrossRef]
- Hardy, M.P.; Sanij, E.P.; Hertzog, P.J.; Owczarek, C.M. Characterization and transcriptional analysis of the mouse Chromosome 16 cytokine receptor gene cluster. Mamm. Genome 2003, 14, 105–118. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Samarajiwa, S.A.; Hertzog, P.J. Type I interferon receptors: Biochemistry and biological functions. J. Biol. Chem. 2007, 282, 20053–20057. [Google Scholar] [CrossRef] [PubMed]
- Brierley, M.M.; Fish, E.N. Review: IFN-alpha/beta receptor interactions to biologic outcomes: Understanding the circuitry. J. Interferon. Cytokine Res. 2002, 22, 835–845. [Google Scholar] [CrossRef]
- Thomas, C.; Moraga, I.; Levin, D.; Krutzik, P.O.; Podoplelova, Y.; Trejo, A.; Lee, C.; Yarden, G.; Vleck, S.E.; Glenn, J.S.; et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 2011, 146, 621–632. [Google Scholar] [CrossRef]
- Mittnacht, S.; Straub, P.; Kirchner, H.; Jacobsen, H. Interferon treatment inhibits onset of herpes simplex virus immediate- early transcription. Virology 1988, 164, 201–210. [Google Scholar] [CrossRef]
- Mikloska, Z.; Cunningham, A.L. Alpha and gamma interferons inhibit herpes simplex virus type 1 infection and spread in epidermal cells after axonal transmission. J. Virol. 2001, 75, 11821–11826. [Google Scholar] [CrossRef]
- Leib, D.A.; Harrison, T.E.; Laslo, K.M.; Machalek, M.A.; Moorman, N.J.; Virgin, H.W. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 1999, 189, 663–672. [Google Scholar] [CrossRef]
- De Regge, N.; Van Opdenbosch, N.; Nauwynck, H.J.; Efstathiou, S.; Favoreel, H.W. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PLoS ONE 2010, 5, e13076. [Google Scholar] [CrossRef]
- Mossman, K.L.; Macgregor, P.F.; Rozmus, J.J.; Goryachev, A.B.; Edwards, A.M.; Smiley, J.R. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 2001, 75, 750–758. [Google Scholar] [CrossRef]
- Leib, D.A. Counteraction of interferon-induced antiviral responses by herpes simplex viruses. Curr. Top Microbiol. Immunol. 2002, 269, 171–185. [Google Scholar]
- Hwang, S.Y.; Hertzog, P.J.; Holland, K.A.; Sumarsono, S.H.; Tymms, M.J.; Hamilton, J.A.; Whitty, G.; Bertoncello, I.; Kola, I. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc. Natl. Acad. Sci. USA 1995, 92, 11284–11288. [Google Scholar] [CrossRef]
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Platanias, L.C.; Uddin, S.; Colamonici, O.R. Tyrosine phosphorylation of the alpha and beta subunits of the type I interferon receptor. Interferon-beta selectively induces tyrosine phosphorylation of an alpha subunit-associated protein. J. Biol. Chem. 1994, 269, 17761–17764. [Google Scholar] [CrossRef] [PubMed]
- Conrady, C.D.; Halford, W.P.; Carr, D.J. Loss of the type I interferon pathway increases vulnerability of mice to genital herpes simplex virus 2 infection. J. Virol. 2011, 85, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Conrady, C.D.; Thapa, M.; Wuest, T.; Carr, D.J. Loss of mandibular lymph node integrity is associated with an increase in sensitivity to HSV-1 infection in CD118-deficient mice. J. Immunol. 2009, 182, 3678–3687. [Google Scholar] [CrossRef]
- Luker, G.D.; Prior, J.L.; Song, J.; Pica, C.M.; Leib, D.A. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J. Virol. 2003, 77, 11082–11093. [Google Scholar] [CrossRef]
- Wang, S.; Jaggi, U.; Katsumata, M.; Ghiasi, H. The importance of IFNalpha2A (Roferon-A) in HSV-1 latency and T cell exhaustion in ocularly infected mice. PLoS Pathog. 2024, 20, e1012612. [Google Scholar] [CrossRef]
- Wang, S.; Jaggi, U.; Oh, J.J.; Ghiasi, H. IFNβ absence compensates for LAT functions in latency-reactivation and T cell exhaustion. J. Virol. 2001, 99, e0201023. [Google Scholar] [CrossRef]
- Bourdon, M.; Manet, C.; Montagutelli, X. 2020. Host genetic susceptibility to viral infections: The role of type I interferon induction. Genes Immun. 2025, 21, 365–379. [Google Scholar] [CrossRef]
- Duncan, C.J.A.; Randall, R.E.; Hambleton, S. Genetic Lesions of Type I Interferon Signalling in Human Antiviral Immunity. Trends Genet. 2021, 37, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bucciol, G.; Effort, C.H.G.; Meyts, I. Inherited and acquired errors of type I interferon immunity govern susceptibility to COVID-19 and multisystem inflammatory syndrome in children. J. Allergy Clin. Immunol. 2023, 151, 832–840. [Google Scholar] [CrossRef]
- Sancho-Shimizu, V.; Perez de Diego, R.; Jouanguy, E.; Zhang, S.Y.; Casanova, J.L. Inborn errors of anti-viral interferon immunity in humans. Curr. Opin. Virol. 2011, 1, 487–496. [Google Scholar] [CrossRef]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [CrossRef]
- Garber, D.A.; Schaffer, P.A.; Knipe, D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 1997, 71, 5885–5893. [Google Scholar] [CrossRef]
- Ahmed, M.; Lock, M.; Miller, C.G.; Fraser, N.W. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J. Virol. 2002, 76, 717–729. [Google Scholar] [CrossRef]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef]
- Mossman, K.L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 2000, 74, 2052–2056. [Google Scholar] [CrossRef]
- Kew, C.; Lui, P.Y.; Chan, C.P.; Liu, X.; Au, S.W.; Mohr, I.; Jin, D.Y.; Kok, K.H. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J. Virol. 2013, 87, 13141–13149. [Google Scholar] [CrossRef]
- Cotter, C.R.; Nguyen, M.L.; Yount, J.S.; Lopez, C.B.; Blaho, J.A.; Moran, T.M. The virion host shut-off (vhs) protein blocks a TLR-independent pathway of herpes simplex virus type 1 recognition in human and mouse dendritic cells. PLoS ONE 2010, 5, e8684. [Google Scholar] [CrossRef]
- Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J. Virol. 2013, 87, 12814–12827. [Google Scholar] [CrossRef]
- Paladino, P.; Mossman, K.L. Mechanisms employed by herpes simplex virus 1 to inhibit the interferon response. J. Interferon. Cytokine Res. 2009, 29, 599–607. [Google Scholar] [CrossRef]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3266–3270. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc. Natl. Acad. Sci. USA 1994, 91, 5247–5251. [Google Scholar] [CrossRef]
- Wilcox, D.R.; Longnecker, R. The Herpes Simplex Virus Neurovirulence Factor gamma34.5: Revealing Virus-Host Interactions. PLoS Pathog. 2016, 12, e1005449. [Google Scholar] [CrossRef]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Drolet, B.S.; Perng, G.C.; Villosis, R.J.; Slanina, S.M.; Nesburn, A.B.; Wechsler, S.L. Expression of the first 811 nucleotides of the herpes simplex virus type 1 latency-associated transcript (LAT) partially restores wild-type spontaneous reactivation to a LAT-null mutant. Virology 1999, 253, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Samoto, K.; Perng, G.C.; Ehtesham, M.; Liu, Y.; Wechsler, S.L.; Nesburn, A.B.; Black, K.L.; Yu, J.S. A herpes simplex virus type 1 mutant deleted for gamma34.5 and LAT kills glioma cells in vitro and is inhibited for in vivo reactivation. Cancer Gene Ther. 2001, 8, 269–277. [Google Scholar] [CrossRef]
- Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res. 1992, 22, 25–39. [Google Scholar] [CrossRef]
- Mador, N.; Goldenberg, D.; Cohen, O.; Panet, A.; Steiner, I. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. J. Virol. 1998, 72, 5067–5075. [Google Scholar] [CrossRef]
- Peng, W.; Vitvitskaia, O.; Carpenter, D.; Wechsler, S.L.; Jones, C. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J. Neurovirol. 2008, 14, 41–52. [Google Scholar] [CrossRef]
- Tormanen, K.; Matundan, H.H.; Wang, S.; Jaggi, U.; Mott, K.R.; Ghiasi, H. Small Noncoding RNA (sncRNA1) within the Latency-Associated Transcript Modulates Herpes Simplex Virus 1 Virulence and the Host Immune Response during Acute but Not Latent Infection. J. Virol. 2022, 96, e0005422. [Google Scholar] [CrossRef]
- Tormanen, K.; Wang, S.; Matundan, H.H.; Yu, J.; Jaggi, U.; Ghiasi, H. Herpes Simplex Virus 1 Small Noncoding RNAs 1 and 2 Activate the Herpesvirus Entry Mediator Promoter. J. Virol. 2022, 96, e0198521. [Google Scholar] [CrossRef]
- Oh, J.J.; Jaggi, U.; Tormanen, K.; Wang, S.; Hirose, S.; Ghiasi, H. The anti-apoptotic function of HSV-1 LAT in neuronal cell cultures but not its function during reactivation correlates with expression of two small non-coding RNAs, sncRNA1&2. PLoS Pathog. 2024, 20, e1012307. [Google Scholar]
- Jurak, I.; Kramer, M.F.; Mellor, J.C.; van Lint, A.L.; Roth, F.P.; Knipe, D.M.; Coen, D.M. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J. Virol. 2010, 84, 4659–4672. [Google Scholar] [CrossRef]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Griffiths, A.; Li, G.; Silva, L.M.; Kramer, M.F.; Gaasterland, T.; Wang, X.J.; Coen, D.M. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J. Virol. 2006, 80, 5499–5508. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.F.; Jurak, I.; Pesola, J.M.; Boissel, S.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 2011, 417, 239–247. [Google Scholar] [CrossRef]
- Tang, S.; Bertke, A.S.; Patel, A.; Wang, K.; Cohen, J.I.; Krause, P.R. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc. Natl. Acad. Sci. USA 2008, 105, 10931–10936. [Google Scholar] [CrossRef]
- Farrell, M.J.; Dobson, A.T.; Feldman, L.T. Herpes simplex virus latency-associated transcript is a stable intron. Proc. Natl. Acad. Sci. USA 1991, 88, 790–794. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 latent transcription and non-coding RNA: A critical retrospective. J. Neuroimmunol. 2017, 308, 65–101. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, H.; Cai, S.; Perng, G.C.; Nesburn, A.B.; Wechsler, S.L. Both CD4+ and CD8+ T cells are involved in protection against HSV-1 induced corneal scarring. Br. J. Ophthalmol. 2000, 84, 408–412. [Google Scholar] [CrossRef]
- Erlich, K.S.; Wofsy, D.; Dix, R.D.; Mills, J. Effects of selective depletion of L3T4+ T-lymphocytes on herpes simplex virus encephalitis. Clin. Immunol. Immunopathol. 1989, 52, 190–201. [Google Scholar] [CrossRef]
- Newell, C.K.; Martin, S.; Sendele, D.; Mercadal, C.M.; Rouse, B.T. Herpes simplex virus-induced stromal keratitis: Role of T-lymphocyte subsets in immunopathology. J. Virol. 1989, 63, 769–775. [Google Scholar] [CrossRef]
- Newell, C.K.; Sendele, D.; Rouse, B.T. Effects of CD4+ and CD8+ T-lymphocyte depletion on the induction and expression of herpes simplex stromal keratitis. Reg. Immunol. 1989, 2, 366–369. [Google Scholar]
- Wang, S.; Ljubimov, A.V.; Jin, L.; Pfeffer, K.; Kronenberg, M.; Ghiasi, H. Herpes Simplex Virus 1 Latency and the Kinetics of Reactivation Are Regulated by a Complex Network of Interactions between the Herpesvirus Entry Mediator, Its Ligands (gD, BTLA, LIGHT, and CD160), and the Latency-Associated Transcript. J. Virol. 2018, 92, e01451-18. [Google Scholar] [CrossRef]
- Mott, K.; Brick, D.J.; van Rooijen, N.; Ghiasi, H. Macrophages Are Important Determinants of Acute Ocular HSV-1 Infection in Immunized Mice. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5605–5615. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Hamrah, P.; Gate, D.M.; Mott, K.R.; Mantopoulos, D.; Zheng, L.; Town, T.; Jones, C.; von Andrian, U.H.; Freeman, G.J.; et al. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J. Virol. 2011, 85, 4184–4197. [Google Scholar] [CrossRef]
- Osorio, Y.; Ghiasi, H. Comparison of adjuvant efficacy of herpes simplex virus type 1 recombinant viruses expressing TH1 and TH2 cytokine genes. J. Virol. 2003, 77, 5774–5783. [Google Scholar] [CrossRef]
- Mott, K.R.; Perng, G.C.; Osorio, Y.; Kousoulas, K.G.; Ghiasi, H. A Recombinant Herpes Simplex Virus Type 1 Expressing Two Additional Copies of gK Is More Pathogenic than Wild-Type Virus in Two Different Strains of Mice. J. Virol. 2007, 81, 12962–12972. [Google Scholar] [CrossRef]
- Jaggi, U.; Matundan, H.H.; Tormanen, K.; Wang, S.; Yu, J.; Mott, K.R.; Ghiasi, H. Expression of Murine CD80 by Herpes Simplex Virus 1 in Place of Latency-Associated Transcript (LAT) Can Compensate for Latency Reactivation and Anti-apoptotic Functions of LAT. J. Virol. 2020, 94, e01798-19. [Google Scholar] [CrossRef]
- Ghiasi, H.; Osorio, Y.; Perng, G.C.; Nesburn, A.B.; Wechsler, S.L. Overexpression of interleukin-2 by a recombinant herpes simplex virus type 1 attenuates pathogenicity and enhances antiviral immunity. J. Virol. 2002, 76, 9069–9078. [Google Scholar] [CrossRef]
- Mott, K.R.; Osorio, Y.; Brown, D.J.; Morishige, N.; Wahlert, A.; Jester, J.V.; Ghiasi, H. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol. Vis. 2007, 13, 1802–1812. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Survival/Total | ||||
|---|---|---|---|---|
| PFU/Eye | ||||
| IFN-αβR−/− | WT | |||
| Virus strain | 1 × 104 | 1 × 103 b | 1 × 102 b | 1 × 104 b |
| McKrae (LAT-plus) | 0/5 (0%) | 9/16 (56%) b | 10/10 (100%) b | 10/10 (100%) |
| dLAT2903 (LAT-minus) | 0/5 (0%) | 1/16 (6%) b | 0/10 (0%) b | 10/10 (100%) |
| Survival/Total | ||||
|---|---|---|---|---|
| PFU/Eye | ||||
| IFN-αβR−/− | WT | |||
| Virus strain | 1 × 104 | 1 × 103 | 1 × 102 | 1 × 104 |
| dLAT3.3A (LAT1.5kb) | 0/5 (0%) | 3/9 (33%) b | 9/9 (100%) b | 5/5 (100%) |
| dLAT2.6A (LAT811bp) | 0/5 (0%) | 0/8 (0%) b | 0/10 (0%) b | 5/5 (100%) |
| dLAT2903-γ34.5null (LAT-γ34.5 minus) | 0/5 (0%) | 2/5 (40%) | 5/5 (100%) | 5/5 (100%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.J.; Jaggi, U.; Arya, D.; Wang, S.; Ghiasi, H. In the Absence of Type-1 IFN, HSV-1 LAT Increases γ34.5 Expression and Enhances Mortality in Infected Mice. Viruses 2025, 17, 1061. https://doi.org/10.3390/v17081061
Oh JJ, Jaggi U, Arya D, Wang S, Ghiasi H. In the Absence of Type-1 IFN, HSV-1 LAT Increases γ34.5 Expression and Enhances Mortality in Infected Mice. Viruses. 2025; 17(8):1061. https://doi.org/10.3390/v17081061
Chicago/Turabian StyleOh, Jay J., Ujjaldeep Jaggi, Deepak Arya, Shaohui Wang, and Homayon Ghiasi. 2025. "In the Absence of Type-1 IFN, HSV-1 LAT Increases γ34.5 Expression and Enhances Mortality in Infected Mice" Viruses 17, no. 8: 1061. https://doi.org/10.3390/v17081061
APA StyleOh, J. J., Jaggi, U., Arya, D., Wang, S., & Ghiasi, H. (2025). In the Absence of Type-1 IFN, HSV-1 LAT Increases γ34.5 Expression and Enhances Mortality in Infected Mice. Viruses, 17(8), 1061. https://doi.org/10.3390/v17081061