
SARS-CoV-2 Remdesivir Exposure Leads to Different Evolutionary Pathways That Converge in Moderate Levels of Drug Resistance

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Cell Culture
2.2. Generation of the RDV Escape Virus
2.3. Reverse Genetics Experiments
2.4. Determination of Viral Infectivity Titers
2.5. Antiviral Activity Assays
2.6. Growth Kinetics and Competition Assays for the Determination of Viral Fitness
2.7. In Vivo Experiments
2.8. Lung Tissue Processing
2.9. Determination of Viral RNA Titers
2.10. Neutralization Assay
2.11. Next-Generation Sequencing Analysis
2.12. In Silico Structural Analysis of Remdesivir Resistance-Associated Substitutions
2.13. Statistical Analysis
3. Results
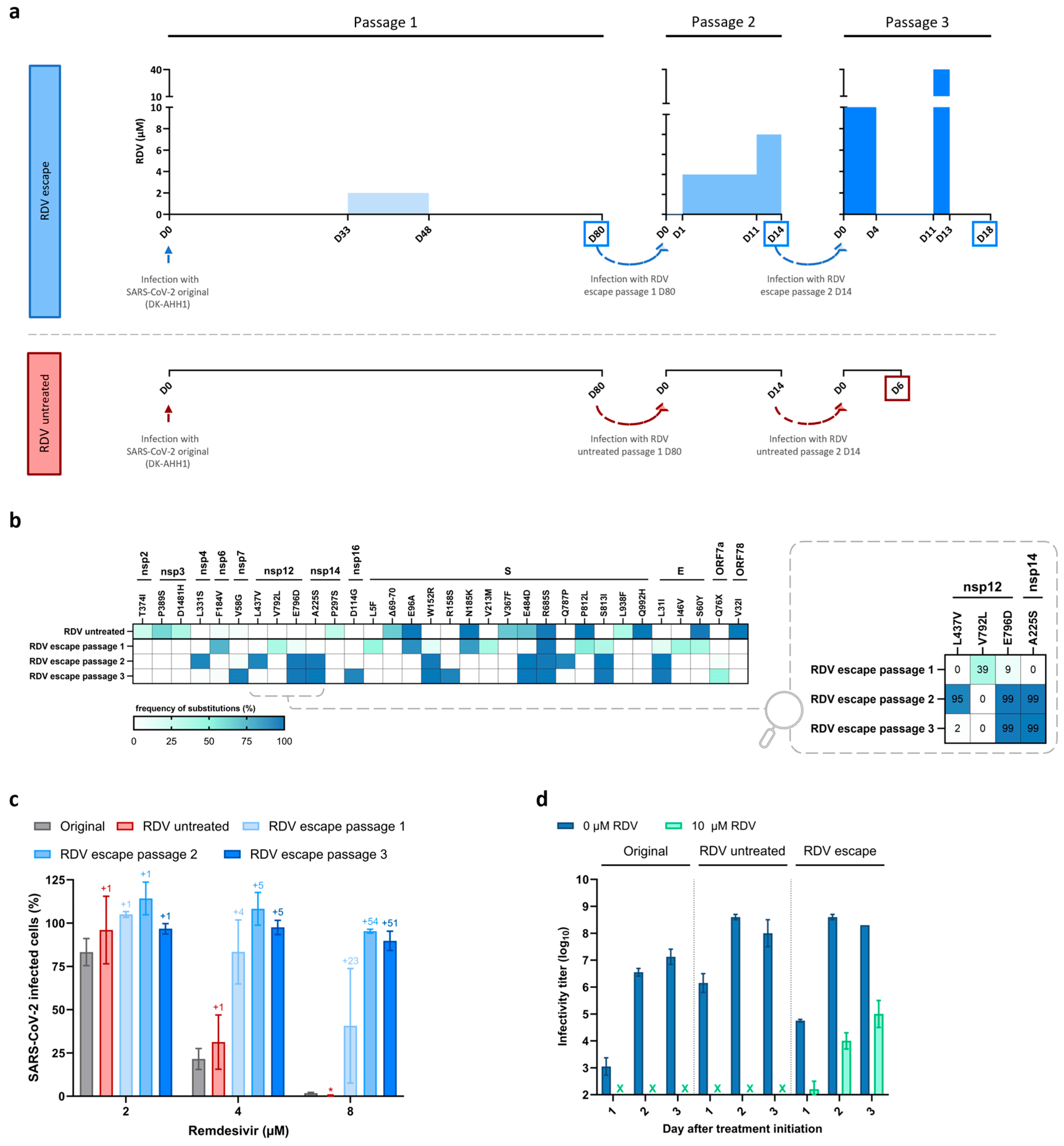
3.1. Emergence of SARS-CoV-2 Resistance to Remdesivir Following Serial Passage in Vero E6 Cells
3.2. Substitution nsp12-E796D Reduces Remdesivir Susceptibility
3.3. Viral Fitness Might Contribute to Remdesivir Resistance in Vero E6 Cells
3.4. Substitution nsp12-E796D Does Not Alter SARS-CoV-2 Viability In Vivo
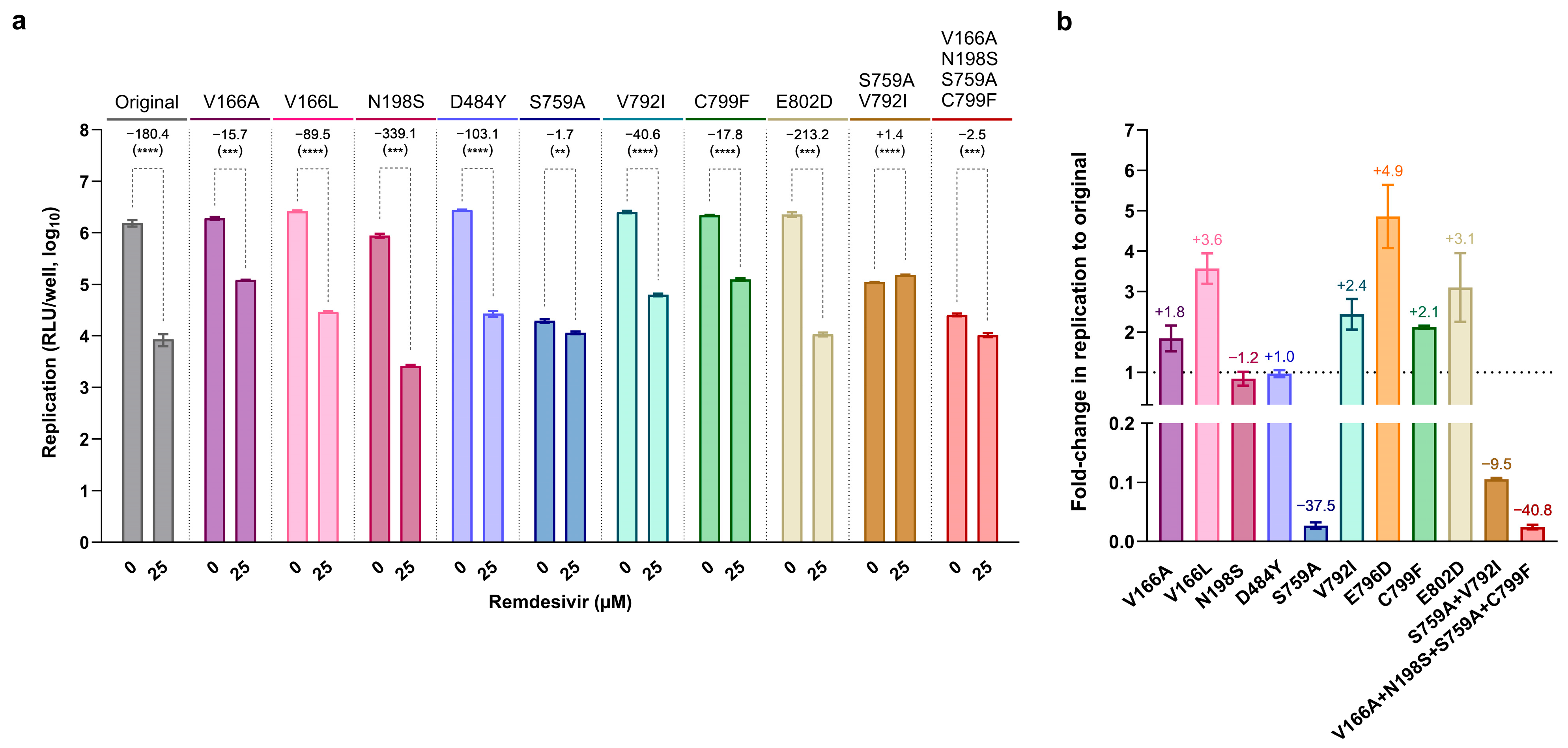
3.5. Most nsp12-RAS Confer Moderate Resistance to Remdesivir
3.6. In Silico Structural Analysis Correlates the Location of nsp12-RAS with Levels of Remdesivir Resistance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cihlar, T.; Mackman, R.L. Journey of remdesivir from the inhibition of hepatitis C virus to the treatment of COVID-19. Antivir. Ther. 2022, 27, 135965352210827. [Google Scholar] [CrossRef]
- Cho, A.; Saunders, O.L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J.E.; Feng, J.Y.; Ray, A.S.; Kim, C.U. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorganic Med. Chem. Lett. 2012, 22, 2705–2707. [Google Scholar] [CrossRef]
- Lo, M.K.; Jordan, R.; Arvey, A.; Sudhamsu, J.; Shrivastava-Ranjan, P.; Hotard, A.L.; Flint, M.; McMullan, L.K.; Siegel, D.; Clarke, M.O.; et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017, 7, 43395. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L.; Hui, H.C.; Perron, M.; Murakami, E.; Palmiotti, C.; Lee, G.; Stray, K.; Zhang, L.; Goyal, B.; Chun, K.; et al. Prodrugs of a 1′-CN-4-Aza-7,9-dideazaadenosine C-Nucleoside Leading to the Discovery of Remdesivir (GS-5734) as a Potent Inhibitor of Respiratory Syncytial Virus with Efficacy in the African Green Monkey Model of RSV. J. Med. Chem. 2021, 64, 5001–5017. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Binderup, A.; Galli, A.; Fossat, N.; Fernandez-Antunez, C.; Mikkelsen, L.S.; Rivera-Rangel, L.R.; Scheel, T.K.; Fahnøe, U.; Bukh, J.; Ramirez, S. Differential activity of nucleotide analogs against tick-borne encephalitis and yellow fever viruses in human cell lines. Virology 2023, 585, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H.; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Pérez-Pérez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- Ramirez, S.; Fernandez-Antunez, C.; Galli, A.; Underwood, A.; Pham, L.V.; Ryberg, L.A.; Feng, S.; Pedersen, M.S.; Mikkelsen, L.S.; Belouzard, S.; et al. Overcoming Culture Restriction for SARS-CoV-2 in Human Cells Facilitates the Screening of Compounds Inhibiting Viral Replication. Antimicrob. Agents Chemother. 2021, 65, e0009721. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium. Remdesivir and three other drugs for hospitalised patients with COVID-19: Final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet 2022, 399, 1941–1953. [Google Scholar] [CrossRef]
- Godwin, P.O.; Polsonetti, B.; Caron, M.F.; Oppelt, T.F. Remdesivir for the Treatment of COVID-19: A Narrative Review. Infect. Dis. Ther. 2024, 13, 1–19. [Google Scholar] [CrossRef]
- EMA (European Medicines Agency). Authorised COVID-19 Treatments n.d. Available online: https://www.ema.europa.eu/en/human-regulatory-overview/public-health-threats/coronavirus-disease-covid-19/covid-19-medicines (accessed on 3 July 2025).
- FDA (US Food and Drug Administration). Coronavirus (COVID-19)|Drugs. n.d. Available online: https://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs (accessed on 3 July 2025).
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M. Retreatment of Hepatitis C Virus-Infected Patients with Direct-Acting Antiviral Failures. Semin. Liver Dis. 2019, 39, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.A.; Monit, C.; Gupta, R.K. The Impact of HIV-1 Drug Escape on the Global Treatment Landscape. Cell Host Microbe 2019, 26, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Smyk, J.M.; Szydłowska, N.; Szulc, W.; Majewska, A. Evolution of Influenza Viruses—Drug Resistance, Treatment Options, and Prospects. Int. J. Mol. Sci. 2022, 23, 12244. [Google Scholar] [CrossRef]
- Zhou, Y.; Gammeltoft, K.A.; Ryberg, L.A.; Pham, L.V.; Tjørnelund, H.D.; Binderup, A.; Hernandez, C.R.; Fernandez-Antunez, C.; Offersgaard, A.; Fahnøe, U.; et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 2022, 8, eadd7197. [Google Scholar] [CrossRef]
- Hedskog, C.; Rodriguez, L.; Roychoudhury, P.; Huang, M.-L.; Jerome, K.R.; Hao, L.; Ireton, R.C.; Li, J.; Perry, J.K.; Han, D.; et al. Viral Resistance Analyses from the Remdesivir Phase 3 Adaptive COVID-19 Treatment Trial-1 (ACTT-1). J. Infect. Dis. 2023, 228, 1263–1273. [Google Scholar] [CrossRef]
- Hedskog, C.; Spinner, C.D.; Protzer, U.; Hoffmann, D.; Ko, C.; Gottlieb, R.L.; Askar, M.; Roestenberg, M.; de Vries, J.J.; Carbo, E.C.; et al. No Remdesivir Resistance Observed in the Phase 3 Severe and Moderate COVID-19 SIMPLE Trials. Viruses 2024, 16, 546. [Google Scholar] [CrossRef]
- Rodriguez, L.; Lee, H.W.; Li, J.; Martin, R.; Han, D.; Xu, S.; Moshiri, J.; Peinovich, N.; Camus, G.; Perry, J.K.; et al. SARS-CoV-2 resistance analyses from the Phase 3 PINETREE study of remdesivir treatment in nonhospitalized participants. Antimicrob. Agents Chemother. 2024, 69, e01238-24. [Google Scholar] [CrossRef]
- Yang, S.; Multani, A.; Garrigues, J.M.; Oh, M.S.; Hemarajata, P.; Burleson, T.; Green, N.M.; Oliai, C.; Gaynor, P.T.; Beaird, O.E.; et al. Transient SARS-CoV-2 RNA-Dependent RNA Polymerase Mutations after Remdesivir Treatment for Chronic COVID-19 in Two Transplant Recipients: Case Report and Intra-Host Viral Genomic Investigation. Microorganisms 2023, 11, 2096. [Google Scholar] [CrossRef] [PubMed]
- Heyer, A.; Günther, T.; Robitaille, A.; Lütgehetmann, M.; Addo, M.M.; Jarczak, D.; Kluge, S.; Aepfelbacher, M.; Wiesch, J.S.; Fischer, N.; et al. Remdesivir-induced emergence of SARS-CoV2 variants in patients with prolonged infection. Cell Rep. Med. 2022, 3, 100735. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Kobayashi, H.; Kakizaki, Y.; Saito, A.; Tsutsui, T.; Kawaguchi, M.; Shimamura, S.; Hata, K.; Hanawa, S.; Toyama, J.; et al. Multidrug-resistant mutations to antiviral and antibody therapy in an immunocompromised patient infected with SARS-CoV-2. Med 2023, 4, 813–824.e4. [Google Scholar] [CrossRef]
- Martinot, M.; Jary, A.; Fafi-Kremer, S.; Leducq, V.; Delagreverie, H.; Garnier, M.; Pacanowski, J.; Mékinian, A.; Pirenne, F.; Tiberghien, P.; et al. Emerging RNA-Dependent RNA Polymerase Mutation in a Remdesivir-Treated B-cell Immunodeficient Patient with Protracted Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 73, e1762–e1765. [Google Scholar] [CrossRef]
- Tanino, Y.; Nishioka, K.; Yamamoto, C.; Watanabe, Y.; Daidoji, T.; Kawamoto, M.; Uda, S.; Kirito, S.; Nakagawa, Y.; Kasamatsu, Y.; et al. Emergence of SARS-CoV-2 with Dual-Drug Resistant Mutations During a Long-Term Infection in a Kidney Transplant Recipient. Infect. Drug Resist. 2024, 17, 531–541. [Google Scholar] [CrossRef]
- Hogan, J.I.; Duerr, R.; Dimartino, D.; Marier, C.; Hochman, S.E.; Mehta, S.; Wang, G.; Heguy, A. Remdesivir Resistance in Transplant Recipients with Persistent Coronavirus Disease 2019. Clin. Infect. Dis. 2023, 76, 342–345. [Google Scholar] [CrossRef]
- Nooruzzaman, M.; Johnson, K.E.E.; Rani, R.; Finkelsztein, E.J.; Caserta, L.C.; Kodiyanplakkal, R.P.; Wang, W.; Hsu, J.; Salpietro, M.T.; Banakis, S.; et al. Emergence of transmissible SARS-CoV-2 variants with decreased sensitivity to antivirals in immunocompromised patients with persistent infections. Nat. Commun. 2024, 15, 7999. [Google Scholar] [CrossRef]
- Igari, H.; Sakao, S.; Ishige, T.; Saito, K.; Murata, S.; Yahaba, M.; Taniguchi, T.; Suganami, A.; Matsushita, K.; Tamura, Y.; et al. Dynamic diversity of SARS-CoV-2 genetic mutations in a lung transplantation patient with persistent COVID-19. Nat. Commun. 2024, 15, 3604. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.J.; Peña-Hernández, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Bakhash, S.A.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. Nat. Commun. 2022, 13, 1547. [Google Scholar] [CrossRef]
- Tamura, T.J.; Choudhary, M.C.; Deo, R.; Yousuf, F.; Gomez, A.N.; Edelstein, G.E.; Boucau, J.; Glover, O.T.; Barry, M.; Gilbert, R.F.; et al. Emerging SARS-CoV-2 Resistance After Antiviral Treatment. JAMA Netw. Open 2024, 7, e2435431. [Google Scholar] [CrossRef] [PubMed]
- Checkmahomed, L.; Carbonneau, J.; Du Pont, V.; Riola, N.C.; Perry, J.K.; Li, J.; Paré, B.; Simpson, S.M.; Smith, M.A.; Porter, D.P.; et al. In Vitro Selection of Remdesivir-Resistant SARS-CoV-2 Demonstrates High Barrier to Resistance. Antimicrob. Agents Chemother. 2022, 66, e00198-22. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Kim, K.S.; Koseki, J.; Suzuki, R.; Iwanami, S.; Fujita, Y.; Jeong, Y.D.; Ito, J.; Asakura, H.; Nagashima, M.; et al. Increased flexibility of the SARS-CoV-2 RNA-binding site causes resistance to remdesivir. PLoS Pathog. 2023, 19, e1011231. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef]
- Sama, B.; Selisko, B.; Falcou, C.; Fattorini, V.; Piorkowski, G.; Touret, F.; Donckers, K.; Neyts, J.; Jochmans, D.; Shannon, A.; et al. The effects of Remdesivir’s functional groups on its antiviral potency and resistance against the SARS-CoV-2 polymerase. Antivir. Res. 2024, 232, 106034. [Google Scholar] [CrossRef]
- Schreiber, A.; Rodner, F.; Oberberg, N.; Anhlan, D.; Bletz, S.; Mellmann, A.; Planz, O.; Ludwig, S. The host-targeted antiviral drug Zapnometinib exhibits a high barrier to the development of SARS-CoV-2 resistance. Antivir. Res. 2024, 225, 105840. [Google Scholar] [CrossRef]
- Gammeltoft, K.A.; Zhou, Y.; Ryberg, L.A.; Pham, L.V.; Binderup, A.; Hernandez, C.R.D.; Offersgaard, A.; Fahnøe, U.; Peters, G.H.; Ramirez, S.; et al. Substitutions in SARS-CoV-2 Mpro Selected by Protease Inhibitor Boceprevir Confer Resistance to Nirmatrelvir. Viruses 2023, 15, 1970. [Google Scholar] [CrossRef]
- Zhou, Y.; Gilmore, K.; Ramirez, S.; Settels, E.; Gammeltoft, K.A.; Pham, L.V.; Fahnøe, U.; Feng, S.; Offersgaard, A.; Trimpert, J.; et al. In vitro efficacy of artemisinin-based treatments against SARS-CoV-2. Sci. Rep. 2021, 11, 14571. [Google Scholar] [CrossRef]
- Fahnøe, U.; Pham, L.V.; Fernandez-Antunez, C.; Costa, R.; Rivera-Rangel, L.R.; Galli, A.; Feng, S.; Mikkelsen, L.S.; Gottwein, J.M.; Scheel, T.K.; et al. Versatile SARS-CoV-2 Reverse-Genetics Systems for the Study of Antiviral Resistance and Replication. Viruses 2022, 14, 172. [Google Scholar] [CrossRef]
- Pham, L.V.; Underwood, A.P.; Binderup, A.; Fahnøe, U.; Fernandez-Antunez, C.; Lopez-Mendez, B.; Ryberg, L.A.; Galli, A.; Sølund, C.; Weis, N.; et al. Neutralisation resistance of SARS-CoV-2 spike-variants is primarily mediated by synergistic receptor binding domain substitutions. Emerg. Microbes Infect. 2024, 13, 2412643. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Gammeltoft, K.A.; Zhou, Y.; Duarte Hernandez, C.R.; Galli, A.; Offersgaard, A.; Costa, R.; Pham, L.V.; Fahnøe, U.; Feng, S.; Scheel, T.K.; et al. Hepatitis C Virus Protease Inhibitors Show Differential Efficacy and Interactions with Remdesivir for Treatment of SARS-CoV-2 In Vitro. Antimicrob. Agents Chemother. 2021, 65, e02680-20. [Google Scholar] [CrossRef]
- Offersgaard, A.; Duarte Hernandez, C.R.; Feng, S.; Marichal-Gallardo, P.; Holmbeck, K.; Pihl, A.F.; Fernandez-Antunez, C.; Alzua, G.P.; Hartmann, K.T.; Pham, L.V.; et al. An inactivated SARS-CoV-2 vaccine induced cross-neutralizing persisting antibodies and protected against challenge in small animals. iScience 2023, 26, 105949. [Google Scholar] [CrossRef] [PubMed]
- Underwood, A.P.; Sølund, C.; Fernandez-Antunez, C.; Villadsen, S.L.; Winckelmann, A.A.; Bollerup, S.; Mikkelsen, L.S.; Sørensen, A.-L.; Feng, S.; Fahnøe, U.; et al. Neutralisation titres against SARS-CoV-2 are sustained 6 months after onset of symptoms in individuals with mild COVID-19. EBioMedicine 2021, 71, 103519. [Google Scholar] [CrossRef]
- Mejer, N.; Fahnøe, U.; Galli, A.; Ramirez, S.; Weiland, O.; Benfield, T.; Bukh, J. Mutations Identified in the Hepatitis C Virus (HCV) Polymerase of Patients with Chronic HCV Treated with Ribavirin Cause Resistance and Affect Viral Replication Fidelity. Antimicrob. Agents Chemother. 2020, 64, e01417-20. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.5.0; Schrödinger, LLC.: New York, NY, USA, 2021.
- Malone, B.F.; Perry, J.K.; Olinares, P.D.B.; Lee, H.W.; Chen, J.; Appleby, T.C.; Feng, J.Y.; Bilello, J.P.; Ng, H.; Sotiris, J.; et al. Structural basis for substrate selection by the SARS-CoV-2 replicase. Nature 2023, 614, 781–787. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- D.E. Shaw Research. Molecular Dynamics Simulations Related to SARS-CoV-2. D.E. Shaw Research Technical Data. 2020. Available online: https://www.deshawresearch.com/downloads/download_trajectory_sarscov2.cgi/ (accessed on 22 January 2021).
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Li, Y.; Cao, L.; Li, G.; Cong, F.; Li, Y.; Sun, J.; Luo, Y.; Chen, G.; Li, G.; Wang, P.; et al. Remdesivir Metabolite GS-441524 Effectively Inhibits SARS-CoV-2 Infection in Mouse Models. J. Med. Chem. 2022, 65, 2785–2793. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L.; Kalla, R.V.; Babusis, D.; Pitts, J.; Barrett, K.T.; Chun, K.; Du Pont, V.; Rodriguez, L.; Moshiri, J.; Xu, Y.; et al. Discovery of GS-5245 (Obeldesivir), an Oral Prodrug of Nucleoside GS-441524 That Exhibits Antiviral Efficacy in SARS-CoV-2-Infected African Green Monkeys. J. Med. Chem. 2023, 66, 11701–11717. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Peng, R.; Yuan, B.; Wang, M.; Zhao, J.; Fu, L.; Qi, J.; Shi, Y. Structural Basis of SARS-CoV-2 Polymerase Inhibition by Favipiravir. Innovation 2021, 2, 100080. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, S.; Itoh, S.G.; Okumura, H. “Bucket brigade” using lysine residues in RNA-dependent RNA polymerase of SARS-CoV-2. Biophys. J. 2021, 120, 3615–3627. [Google Scholar] [CrossRef]
- Truong, T.T.; Ryutov, A.; Pandey, U.; Yee, R.; Goldberg, L.; Bhojwani, D.; Aguayo-Hiraldo, P.; Pinsky, B.A.; Pekosz, A.; Shen, L.; et al. Increased viral variants in children and young adults with impaired humoral immunity and persistent SARS-CoV-2 infection: A consecutive case series. EBioMedicine 2021, 67, 103355. [Google Scholar] [CrossRef]
- Gallego, I.; Sheldon, J.; Moreno, E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Barrier-Independent, Fitness-Associated Differences in Sofosbuvir Efficacy against Hepatitis C Virus. Antimicrob. Agents Chemother. 2016, 60, 3786–3793. [Google Scholar] [CrossRef]
- Fernandez-Antunez, C.; Wang, K.; Fahnøe, U.; Mikkelsen, L.S.; Gottwein, J.M.; Bukh, J.; Ramirez, S. Characterization of multi-DAA resistance using a novel hepatitis C virus genotype 3a infectious culture system. Hepatology 2023, 78, 621–636. [Google Scholar] [CrossRef]
- Graepel, K.W.; Lu, X.; Case, J.B.; Sexton, N.R.; Smith, E.C.; Denison, M.R.; Subbarao, K. Proofreading-Deficient Coronaviruses Adapt for Increased Fitness over Long-Term Passage without Reversion of Exoribonuclease-Inactivating Mutations. mBio 2017, 8, e01503-17. [Google Scholar] [CrossRef]
- Sun, C.; Xie, C.; Bu, G.-L.; Zhong, L.-Y.; Zeng, M.-S. Molecular characteristics, immune evasion, and impact of SARS-CoV-2 variants. Signal Transduct. Target. Ther. 2022, 7, 202. [Google Scholar] [CrossRef]
- Ling-Hu, T.; Simons, L.M.; Rios-Guzman, E.; Carvalho, A.M.; Agnes, M.F.R.; Alisoltanidehkordi, A.; Ozer, E.A.; Lorenzo-Redondo, R.; Hultquist, J.F. The impact of remdesivir on SARS-CoV-2 evolution in vivo. JCI Insight 2025, 10, e182376. [Google Scholar] [CrossRef]
- Lo, C.-W.; Kariv, O.; Hao, C.; Gammeltoft, K.A.; Bukh, J.; Gottwein, J.; Westberg, M.; Lin, M.Z.; Einav, S. Replication capacity and susceptibility of nirmatrelvir-resistant mutants to next-generation Mpro inhibitors in a SARS-CoV-2 replicon system. Antivir. Res. 2024, 231, 106022. [Google Scholar] [CrossRef]
- Shannon, A.; Canard, B. Kill or corrupt: Mechanisms of action and drug-resistance of nucleotide analogues against SARS-CoV-2. Antivir. Res. 2023, 210, 105501. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Wu, L.J.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.N.; Lou, Z.Y.; Yan, L.M.; Zhang, R.G.; Rao, Z.H. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Antunez, C.; Ryberg, L.A.; Wang, K.; Pham, L.V.; Mikkelsen, L.S.; Fahnøe, U.; Hartmann, K.T.; Jensen, H.E.; Holmbeck, K.; Bukh, J.; et al. SARS-CoV-2 Remdesivir Exposure Leads to Different Evolutionary Pathways That Converge in Moderate Levels of Drug Resistance. Viruses 2025, 17, 1055. https://doi.org/10.3390/v17081055
Fernandez-Antunez C, Ryberg LA, Wang K, Pham LV, Mikkelsen LS, Fahnøe U, Hartmann KT, Jensen HE, Holmbeck K, Bukh J, et al. SARS-CoV-2 Remdesivir Exposure Leads to Different Evolutionary Pathways That Converge in Moderate Levels of Drug Resistance. Viruses. 2025; 17(8):1055. https://doi.org/10.3390/v17081055
Chicago/Turabian StyleFernandez-Antunez, Carlota, Line A. Ryberg, Kuan Wang, Long V. Pham, Lotte S. Mikkelsen, Ulrik Fahnøe, Katrine T. Hartmann, Henrik E. Jensen, Kenn Holmbeck, Jens Bukh, and et al. 2025. "SARS-CoV-2 Remdesivir Exposure Leads to Different Evolutionary Pathways That Converge in Moderate Levels of Drug Resistance" Viruses 17, no. 8: 1055. https://doi.org/10.3390/v17081055
APA StyleFernandez-Antunez, C., Ryberg, L. A., Wang, K., Pham, L. V., Mikkelsen, L. S., Fahnøe, U., Hartmann, K. T., Jensen, H. E., Holmbeck, K., Bukh, J., & Ramirez, S. (2025). SARS-CoV-2 Remdesivir Exposure Leads to Different Evolutionary Pathways That Converge in Moderate Levels of Drug Resistance. Viruses, 17(8), 1055. https://doi.org/10.3390/v17081055