
The Role of SARS-CoV-2 Nucleocapsid Protein in Host Inflammation




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. N Protein
2.1. N Protein Structure
2.2. N Protein and Viral Life Cycle
3. N Protein and Inflammation
3.1. N Protein and Innate Immunity
3.1.1. N Protein and Intracellular Inflammatory Signaling
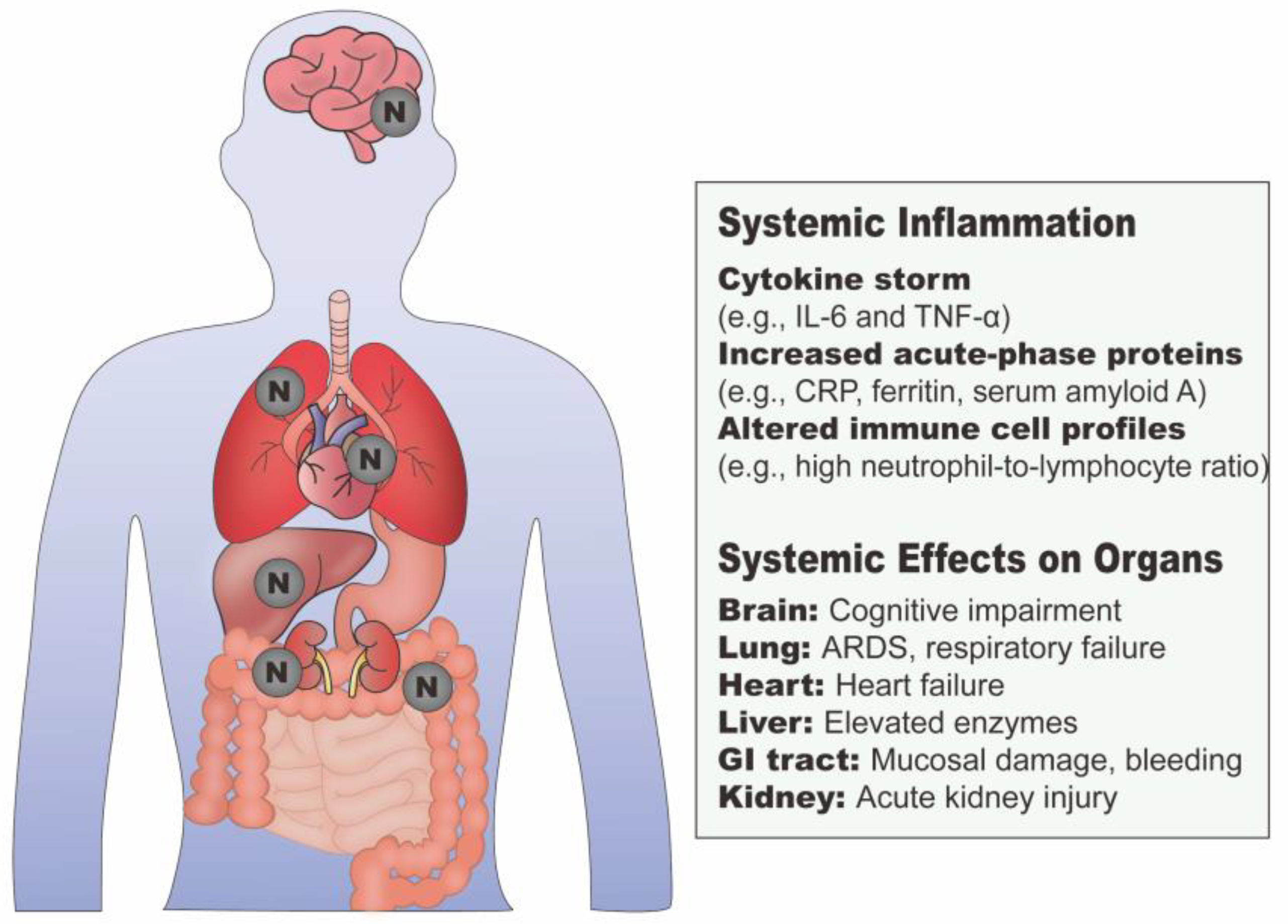
3.1.2. N Protein and Systemic Inflammation
3.2. N Protein and Adaptive Immunity
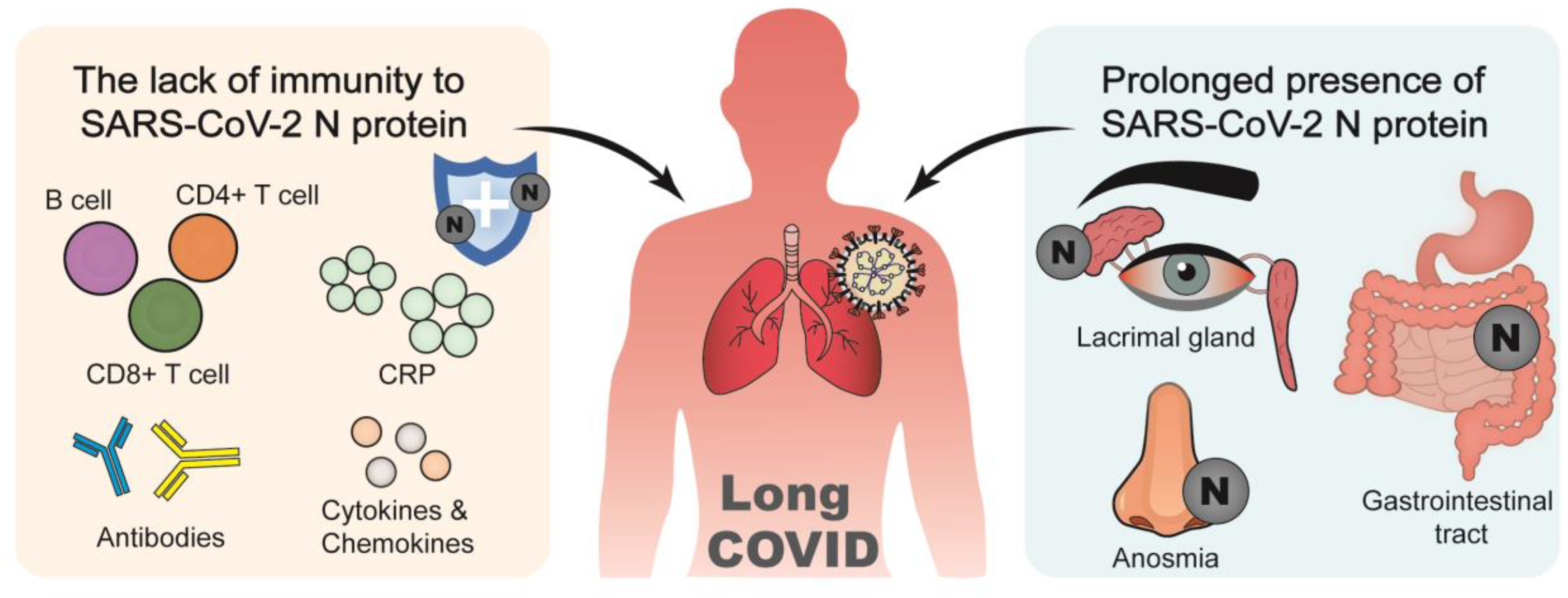
3.3. N Protein and Long COVID
3.4. N Protein and Pre-Existing Inflammatory Diseases
4. Conclusions and Future Work
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP3B1 | Adaptor-related protein complex 3 subunit beta 1 |
| ASC | Apoptosis-associated speck-like protein containing CARD |
| ASCC3 | Activating signal co-integrator 1 complex subunit 3 |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| COVID-19 | Coronavirus disease 2019 |
| CoVs | Coronaviruses |
| CRP | C-reactive protein |
| CTD | C-terminal domain |
| dsRNA | Double-stranded RNA |
| E | Envelope protein |
| ERGIC | Endoplasmic reticulum–Golgi intermediate compartment |
| ERK1/2 | Extracellular signal-regulated kinases 1 and 2 |
| G3BP1 | GTPase-activating protein-binding protein 1 |
| GADD34 | Growth arrest and DNA damage-inducible 34 |
| GSDMD | Gasdermin D |
| GSK-3 | Glycogen synthase kinase- |
| HCoV | Human coronavirus |
| HDAC6 | Histone deacetylases 6 |
| HERC5 | HECT and RLD domain containing E3 ubiquitin protein ligase 5 |
| HLA | Human leukocyte antigen |
| ICU | Intensive care unit |
| IDRs | Intrinsically disordered regions |
| IFN-1 | Type I interferon |
| IgG | Immunoglobulin G |
| IgM | Immunoglobulin M |
| IKKα | IκB kinase-α |
| IL | Interleukin |
| IMPDH2 | Inosine monophosphate dehydrogenase 2 |
| IP-10 | IFN-gamma-inducible protein 10 kD |
| IRF | Interferon regulatory factor |
| ISG15 | IFN-stimulated gene 15 |
| JAK-STAT | Janus kinase-signal transducer and activator of transcription |
| KPNA | Karyopherin alpha |
| KPNB1 | Karyopherin beta 1 |
| LKR | Linking region |
| LLPS | Liquid–liquid phase separation |
| M | Membrane protein |
| MAPK | Mitogen-activated protein kinase |
| MASP-2 | Mannan-binding lectin serine protease 2 |
| MAVS | Mitochondrial antiviral signaling |
| MDA5 | Melanoma differentiation-associated gene 5 |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| MHV | Mouse Hepatitis Virus |
| MIS-C | Multisystem inflammatory syndrome |
| N | Nucleocapsid |
| NF-κB | Nuclear factor kappa B |
| NFKB1 | NF-κB subunit 1 |
| NFKBIA | NF-κB inhibitor alpha |
| NLRP3 | NLR family pyrin domain containing-3 |
| NLRs | NOD like receptors |
| NMD | Nonsense-mediated mRNA decay |
| NOD | Nucleotide-binding oligomerization domain proteins |
| NSPs | Nonstructural proteins |
| NTD | N-terminal domain |
| ORFs | Open reading frames |
| PAMPs | Pathogen-associated molecular patterns |
| PASC | Post-acute sequelae of SARS-CoV-2 infection |
| PCT | Procalcitonin |
| PDE4 | Phosphodiesterase 4 |
| PKM | Pyruvate kinase M |
| PLpro | Papain-like protease |
| poly(I:C) | Polyinosinic: polycytidylic acid |
| PRRs | Pattern recognition receptors |
| PTX3 | Pentraxin 3 |
| RAGE | Glycation end products |
| RAN | Ras-related nuclear protein |
| RdRp | RNA polymerase |
| RIG-I | Retinoic acid-inducible gene I |
| RISC | RNA-induced silencing complex |
| RLRs | RIG-I-like receptors |
| RNAi | RNA interference |
| RNP | Ribonucleoprotein |
| S | Spike protein |
| SAA | Serum amyloid A |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SARSr-CoV | SARS-related coronavirus |
| SCAP | SREBPs cleavage-activating protein |
| SGK1 | Serum glucocorticoid regulated kinase 1 |
| SGs | Stress granules |
| siRNA | Small interfering RNAs |
| SL | Stem-loop |
| SNX8 | Sorting nexin 8 |
| SR | Ser/Arg |
| SREBPs | Sterol regulatory element-binding proteins |
| SUMO | Small ubiquitin-like modifiers |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TBK1 | Tank-binding kinase 1 |
| TCR | T cell receptor |
| TDP-43 | Transactive response-binding protein 43 kDa |
| Th1 | T helper type 1 |
| TLR2 | Toll-like receptor 2 |
| TMAO | Trimethylamine N-oxide |
| TNF | Tumor necrosis factor |
| TNFR2 | Tumor necrosis factor receptor 2 |
| TRIM25 | Tripartite motif protein 25 |
| TRIM6 | Tripartite motif protein 6 |
| UBC9 | Ubiquitin-conjugating enzyme 9 |
| UPF1 | UP-frameshift-1 |
| VOCs | Variants of concern |
| YBX1 | Y-box binding protein 1 |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Zmasek, C.M.; Lefkowitz, E.J.; Niewiadomska, A.; Scheuermann, R.H. Genomic evolution of the Coronaviridae family. Virology 2022, 570, 123–133. [Google Scholar] [CrossRef]
- Zandi, M.; Shafaati, M.; Kalantar-Neyestanaki, D.; Pourghadamyari, H.; Fani, M.; Soltani, S.; Kaleji, H.; Abbasi, S. The role of SARS-CoV-2 accessory proteins in immune evasion. Biomed. Pharmacother. 2022, 156, 113889. [Google Scholar] [CrossRef]
- Wu, W.; Cheng, Y.; Zhou, H.; Sun, C.; Zhang, S. The SARS-CoV-2 nucleocapsid protein: Its role in the viral life cycle, structure and functions, and use as a potential target in the development of vaccines and diagnostics. Virol. J. 2023, 20, 6. [Google Scholar] [CrossRef]
- Schiavina, M.; Pontoriero, L.; Uversky, V.N.; Felli, I.C.; Pierattelli, R. The highly flexible disordered regions of the SARS-CoV-2 nucleocapsid N protein within the 1–248 residue construct: Sequence-specific resonance assignments through NMR. Biomol. NMR Assign. 2021, 15, 219–227. [Google Scholar] [CrossRef]
- Whittaker, G.R.; Daniel, S.; Millet, J.K. Coronavirus entry: How we arrived at SARS-CoV-2. Curr. Opin. Virol. 2021, 47, 113–120. [Google Scholar] [CrossRef]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular host factors for SARS-CoV-2 infection. Nat. Microbiol. 2021, 6, 1219–1232. [Google Scholar] [CrossRef]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and function of major SARS-CoV-2 and SARS-CoV proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef]
- Zeng, W.; Liu, G.; Ma, H.; Zhao, D.; Yang, Y.; Liu, M.; Mohammed, A.; Zhao, C.; Yang, Y.; Xie, J. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochem. Biophys. Res. Commun. 2020, 527, 618–623. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, R.; Li, Z.; Ma, J. Liquid-liquid phase separation in viral function. J. Mol. Biol. 2023, 435, 167955. [Google Scholar] [CrossRef]
- Chen, S.-C.; Xu, C.-T.; Chang, C.-F.; Yang, C.-S.; Lin, P.-H.; Liu, W.-M.; Chen, Y.; Yu, C.-H. Characterization of the binding features between SARS-CoV-2 5’-proximal transcripts of genomic RNA and nucleocapsid proteins. RNA Biol. 2025, 22, 1–16. [Google Scholar] [CrossRef]
- Zhao, D.; Xu, W.; Zhang, X.; Wang, X.; Ge, Y.; Yuan, E.; Xiong, Y.; Wu, S.; Li, S.; Wu, N. Understanding the phase separation characteristics of nucleocapsid protein provides a new therapeutic opportunity against SARS-CoV-2. Protein Cell 2021, 12, 734–740. [Google Scholar] [CrossRef]
- Zhao, M.; Yu, Y.; Sun, L.-M.; Xing, J.-Q.; Li, T.; Zhu, Y.; Wang, M.; Yu, Y.; Xue, W.; Xia, T. GCG inhibits SARS-CoV-2 replication by disrupting the liquid phase condensation of its nucleocapsid protein. Nat. Commun. 2021, 12, 2114. [Google Scholar] [CrossRef]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D. Nucleocapsid mutations in SARS-CoV-2 augment replication and pathogenesis. PLoS Pathog. 2022, 18, e1010627. [Google Scholar] [CrossRef]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef]
- Yaron, T.M.; Heaton, B.E.; Levy, T.M.; Johnson, J.L.; Jordan, T.X.; Cohen, B.M.; Kerelsky, A.; Lin, T.-Y.; Liberatore, K.M.; Bulaon, D.K. Host protein kinases required for SARS-CoV-2 nucleocapsid phosphorylation and viral replication. Sci. Signal. 2022, 15, eabm0808. [Google Scholar] [CrossRef]
- Wu, C.-H.; Chen, P.-J.; Yeh, S.-H. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host Microbe 2014, 16, 462–472. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, H.; Liu, C.; Wang, W.; Qin, Y.; Chen, M. SARS-CoV-2 N protein coordinates viral particle assembly through multiple domains. J. Virol. 2024, 98, e01036-24. [Google Scholar] [CrossRef]
- Dutta, M.; Su, Y.; Plescia, C.B.; Voth, G.A.; Stahelin, R.V. The SARS-CoV-2 nucleoprotein associates with anionic lipid membranes. J. Biol. Chem. 2024, 300, 107456. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, Y.; Wei, X.F.; Fan, L.; Gao, X.; Li, Y.; Wu, Y.; Feng, W.; Shen, X.; Liu, L. TRIM6 facilitates SARS-CoV-2 proliferation by catalyzing the K29-typed ubiquitination of NP to enhance the ability to bind viral genomes. J. Med. Virol. 2024, 96, e29531. [Google Scholar] [CrossRef]
- Dang, M.; Song, J. CTD of SARS-CoV-2 N protein is a cryptic domain for binding ATP and nucleic acid that interplay in modulating phase separation. Protein Sci. 2022, 31, 345–356. [Google Scholar] [CrossRef]
- Brennan, K.; Bowie, A.G. Activation of host pattern recognition receptors by viruses. Curr. Opin. Microbiol. 2010, 13, 503–507. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, Y.; Liu, J.; Li, T. SARS-CoV-2 modulation of RIG-I-MAVS signaling: Potential mechanisms of impairment on host antiviral immunity and therapeutic approaches. MedComm–Future Med. 2022, 1, e29. [Google Scholar] [CrossRef]
- Aoshi, T.; Koyama, S.; Kobiyama, K.; Akira, S.; Ishii, K.J. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011, 1, 226–232. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 nucleocapsid protein targets RIG-I-like receptor pathways to inhibit the induction of interferon response. Cells 2021, 10, 530. [Google Scholar] [CrossRef]
- Chen, K.; Xiao, F.; Hu, D.; Ge, W.; Tian, M.; Wang, W.; Pan, P.; Wu, K.; Wu, J. SARS-CoV-2 nucleocapsid protein interacts with RIG-I and represses RIG-mediated IFN-β production. Viruses 2020, 13, 47. [Google Scholar] [CrossRef]
- Li, Y.; Li, M.; Xiao, H.; Liao, F.; Shen, M.; Ge, W.; Ou, J.; Liu, Y.; Chen, L.; Zhao, Y. The R203M and D377Y mutations of the nucleocapsid protein promote SARS-CoV-2 infectivity by impairing RIG-I-mediated antiviral signaling. PLoS Pathog. 2025, 21, e1012886. [Google Scholar] [CrossRef]
- Zheng, Z.-Q.; Wang, S.-Y.; Xu, Z.-S.; Fu, Y.-Z.; Wang, Y.-Y. SARS-CoV-2 nucleocapsid protein impairs stress granule formation to promote viral replication. Cell Discov. 2021, 7, 38. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.-W.; Xu, Y.; Zhang, J.; Nan, M.-L.; Xiao, Y.; Zhan, P.; Liu, X. SARS-CoV-2 NSP5 and N protein counteract the RIG-I signaling pathway by suppressing the formation of stress granules. Signal Transduct. Target. Ther. 2022, 7, 22. [Google Scholar] [CrossRef]
- Kim, S.S.-Y.; Sze, L.; Lam, K.-P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-β response. J. Biol. Chem. 2019, 294, 6430–6438. [Google Scholar] [CrossRef]
- Somasekharan, S.P.; Gleave, M. SARS-CoV-2 nucleocapsid protein interacts with immunoregulators and stress granules and phase separates to form liquid droplets. FEBS Lett. 2021, 595, 2872–2896. [Google Scholar] [CrossRef]
- Mukherjee, A.; Lo, M.; Chandra, P.; Datta Chaudhuri, R.; De, P.; Dutta, S.; Chawla-Sarkar, M. SARS-CoV-2 nucleocapsid protein promotes self-deacetylation by inducing HDAC6 to facilitate viral replication. Virol. J. 2024, 21, 186. [Google Scholar] [CrossRef]
- Mulloy, R.P.; Evseev, D.; Bui-Marinos, M.P.; Sharlin, N.; Corcoran, J.A. A truncated SARS-CoV-2 nucleocapsid protein enhances virus fitness by evading antiviral responses. bioRxiv 2025. [Google Scholar] [CrossRef]
- Liu, J.; Guan, G.; Wu, C.; Wang, B.; Chu, K.; Zhang, X.; He, S.; Zhang, N.; Yang, G.; Jin, Z. SARS-CoV-2 Nucleocapsid Protein Antagonizes GADD34-Mediated Innate Immune Pathway through Atypical Foci. Molecules 2024, 29, 4792. [Google Scholar] [CrossRef]
- Wang, S.; Dai, T.; Qin, Z.; Pan, T.; Chu, F.; Lou, L.; Zhang, L.; Yang, B.; Huang, H.; Lu, H. Targeting liquid–liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 2021, 23, 718–732. [Google Scholar] [CrossRef]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef]
- Zotta, A.; Hooftman, A.; O’Neill, L.A. SARS-CoV-2 targets MAVS for immune evasion. Nat. Cell Biol. 2021, 23, 682–683. [Google Scholar] [CrossRef]
- Choi, G.W.; Lee, Y.; Yun, M.; Kang, J.; Lee, S.-B. Formation of SUMO3-conjugated chains of MAVS induced by poly (dA:dT), a ligand of RIG-I, enhances the aggregation of MAVS that drives the secretion of interferon-β in human keratinocytes. Biochem. Biophys. Res. Commun. 2020, 522, 939–944. [Google Scholar] [CrossRef]
- Huang, C.; Yin, Y.; Pan, P.; Huang, Y.; Chen, S.; Chen, J.; Wang, J.; Xu, G.; Tao, X.; Xiao, X. The Interaction between SARS-CoV-2 Nucleocapsid Protein and UBC9 Inhibits MAVS Ubiquitination by Enhancing Its SUMOylation. Viruses 2023, 15, 2304. [Google Scholar] [CrossRef]
- Ren, J.; Wang, S.; Zong, Z.; Pan, T.; Liu, S.; Mao, W.; Huang, H.; Yan, X.; Yang, B.; He, X. TRIM28-mediated nucleocapsid protein SUMOylation enhances SARS-CoV-2 virulence. Nat. Commun. 2024, 15, 244. [Google Scholar] [CrossRef]
- Mao, S.; Cai, X.; Niu, S.; Wei, J.; Jiang, N.; Deng, H.; Wang, W.; Zhang, J.; Shen, S.; Ma, Y. TRIM21 promotes ubiquitination of SARS-CoV-2 nucleocapsid protein to regulate innate immunity. J. Med. Virol. 2023, 95, e28719. [Google Scholar] [CrossRef]
- Shajahan, A.; Pepi, L.E.; Rouhani, D.S.; Heiss, C.; Azadi, P. Glycosylation of SARS-CoV-2: Structural and functional insights. Anal. Bioanal. Chem. 2021, 413, 7179–7193. [Google Scholar] [CrossRef]
- Rump, A.; Risti, R.; Kristal, M.-L.; Reut, J.; Syritski, V.; Lookene, A.; Boudinot, S.R. Dual ELISA using SARS-CoV-2 nucleocapsid protein produced in E. coli and CHO cells reveals epitope masking by N-glycosylation. Biochem. Biophys. Res. Commun. 2021, 534, 457–460. [Google Scholar] [CrossRef]
- Nilsson-Payant, B.E.; Uhl, S.; Grimont, A.; Doane, A.S.; Cohen, P.; Patel, R.S.; Higgins, C.A.; Acklin, J.A.; Bram, Y.; Chandar, V. The NF-κB transcriptional footprint is essential for SARS-CoV-2 replication. J. Virol. 2021, 95, e0125721. [Google Scholar] [CrossRef]
- Qian, Y.; Lei, T.; Patel, P.S.; Lee, C.H.; Monaghan-Nichols, P.; Xin, H.-B.; Qiu, J.; Fu, M. Direct activation of endothelial cells by SARS-CoV-2 nucleocapsid protein is blocked by Simvastatin. J. Virol. 2021, 95, e0139621. [Google Scholar] [CrossRef]
- Wang, M.; Valadez-Ingersoll, M.; Gilmore, T.D. Control of nuclear localization of the nucleocapsid protein of SARS-CoV-2. Virology 2024, 600, 110232. [Google Scholar] [CrossRef]
- Lu, Y.; Ye, Z.; Liu, X.; Zhou, L.; Ding, X.; Hou, Y. Role of SARS-CoV-2 nucleocapsid protein in affecting immune cells and insights on its molecular mechanisms. Exp. Ther. Med. 2023, 26, 504. [Google Scholar] [CrossRef]
- Wang, Y.C.; Tsai, C.-H.; Wang, Y.-C.; Yen, L.-C.; Chang, Y.-W.; Sun, J.-R.; Lin, T.-Y.; Chiu, C.-H.; Chao, Y.-C.; Chang, F.-Y. SARS-CoV-2 nucleocapsid protein, rather than spike protein, triggers a cytokine storm originating from lung epithelial cells in patients with COVID-19. Infection 2024, 52, 955–983. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Xia, J.; Tang, W.; Wang, J.; Lai, D.; Xu, Q.; Huang, R.; Hu, Y.; Gong, X.; Fan, J.; Shu, Q. SARS-CoV-2 N protein induces acute lung injury in mice via NF-ĸB activation. Front. Immunol. 2021, 12, 791753. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, L.; Cai, S.; Zhuang, Z.; Zhao, Z.; Jin, S.; Xie, W.; Zhou, L.; Zhang, L.; Zhao, J. RNA-induced liquid phase separation of SARS-CoV-2 nucleocapsid protein facilitates NF-κB hyper-activation and inflammation. Signal Transduct. Target. Ther. 2021, 6, 167. [Google Scholar] [CrossRef]
- Xia, J.; Wang, J.; Ying, L.; Huang, R.; Zhang, K.; Zhang, R.; Tang, W.; Xu, Q.; Lai, D.; Zhang, Y. RAGE is a receptor for SARS-CoV-2 N protein and mediates N protein–induced acute lung injury. Am. J. Respir. Cell Mol. Biol. 2023, 69, 508–520. [Google Scholar] [CrossRef]
- Yue, Z.; Zhang, X.; Gu, Y.; Liu, Y.; Lan, L.-M.; Liu, Y.; Li, Y.; Yang, G.; Wan, P.; Chen, X. Regulation and functions of the NLRP3 inflammasome in RNA virus infection. Front. Cell. Infect. Microbiol. 2024, 13, 1309128. [Google Scholar] [CrossRef]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef]
- Ma, J.; Zhu, F.; Zhao, M.; Shao, F.; Yu, D.; Ma, J.; Zhang, X.; Li, W.; Qian, Y.; Zhang, Y. SARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking Gasdermin D cleavage. EMBO J. 2021, 40, e108249. [Google Scholar] [CrossRef]
- Tan, F.L.; Yin, J.Q. RNAi, a new therapeutic strategy against viral infection. Cell Res. 2004, 14, 460–466. [Google Scholar] [CrossRef]
- van Rij, R.P.; Andino, R. The silent treatment: RNAi as a defense against virus infection in mammals. Trends Biotechnol. 2006, 24, 186–193. [Google Scholar] [CrossRef]
- Mu, J.; Xu, J.; Zhang, L.; Shu, T.; Wu, D.; Huang, M.; Ren, Y.; Li, X.; Geng, Q.; Xu, Y. SARS-CoV-2-encoded nucleocapsid protein acts as a viral suppressor of RNA interference in cells. Sci. China Life Sci. 2020, 63, 1413–1416. [Google Scholar] [CrossRef]
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X. The nucleocapsid protein of coronaviruses acts as a viral suppressor of RNA silencing in mammalian cells. J. Virol. 2015, 89, 9029–9043. [Google Scholar] [CrossRef]
- Nuccetelli, V.; Mghezzi-Habellah, M.; Deymier, S.; Roisin, A.; Gérard-Baraggia, F.; Rocchi, C.; Coureux, P.-D.; Gouet, P.; Cimarelli, A.; Mocquet, V. The SARS-CoV-2 nucleocapsid protein interferes with the full enzymatic activation of UPF1 and its interaction with UPF2. Nucleic Acids Res. 2025, 53, gkaf010. [Google Scholar] [CrossRef]
- Jiang, P.; Dai, Z.; Yang, C.; Ding, L.; Li, S.; Xu, X.; Cheng, C.; Wang, J.; Liu, S. CFTR Inhibitors Display Antiviral Activity against Herpes Simplex Virus. Viruses 2024, 16, 1308. [Google Scholar] [CrossRef]
- Chen, L.; Guan, W.-J.; Qiu, Z.-E.; Xu, J.-B.; Bai, X.; Hou, X.-C.; Sun, J.; Qu, S.; Huang, Z.-X.; Lei, T.-L. SARS-CoV-2 nucleocapsid protein triggers hyperinflammation via protein-protein interaction-mediated intracellular Cl− accumulation in respiratory epithelium. Signal Transduct. Target. Ther. 2022, 7, 255. [Google Scholar] [CrossRef]
- Dzimianski, J.V.; Scholte, F.E.; Bergeron, É.; Pegan, S.D. ISG15: It’s complicated. J. Mol. Biol. 2019, 431, 4203–4216. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, G.; Sayyad, Z.; Goins, C.M.; Stauffer, S.R.; Gack, M.U. ISGylation of the SARS-CoV-2 N protein by HERC5 impedes N oligomerization and thereby viral RNA synthesis. J. Virol. 2024, 98, e00869-24. [Google Scholar] [CrossRef]
- Rhamadianti, A.F.; Abe, T.; Tanaka, T.; Ono, C.; Katayama, H.; Makino, Y.; Deng, L.; Matsui, C.; Moriishi, K.; Shima, F. SARS-CoV-2 papain-like protease inhibits ISGylation of the viral nucleocapsid protein to evade host anti-viral immunity. J. Virol. 2024, 98, e00855-24. [Google Scholar] [CrossRef]
- Bang, W.; Kim, J.; Seo, K.; Lee, J.; Han, J.H.; Park, D.; Cho, J.H.; Shin, D.; Kim, K.-H.; Song, M.J. Suppression of SARS-CoV-2 nucleocapsid protein dimerization by ISGylation and its counteraction by viral PLpro. Front. Microbiol. 2024, 15, 1490944. [Google Scholar] [CrossRef]
- Fujita, T. Evolution of the lectin–complement pathway and its role in innate immunity. Nat. Rev. Immunol. 2002, 2, 346–353. [Google Scholar] [CrossRef]
- Gao, T.; Zhu, L.; Liu, H.; Zhang, X.; Wang, T.; Fu, Y.; Li, H.; Dong, Q.; Hu, Y.; Zhang, Z. Highly pathogenic coronavirus N protein aggravates inflammation by MASP-2-mediated lectin complement pathway overactivation. Signal Transduct. Target. Ther. 2022, 7, 318. [Google Scholar] [CrossRef]
- Kocsis, A.; Bartus, D.; Hirsch, E.; Józsi, M.; Hajdú, I.; Dobó, J.; Balczer, J.; Pál, G.; Gál, P. SARS-CoV-2 Nucleocapsid Protein Is Not Responsible for Over-Activation of Complement Lectin Pathway. Int. J. Mol. Sci. 2024, 25, 7343. [Google Scholar] [CrossRef]
- Bally, I.; Drumont, G.; Rossi, V.; Guseva, S.; Botova, M.; Reiser, J.-B.; Thépaut, M.; Dergan Dylon, S.; Dumestre-Pérard, C.; Gaboriaud, C. Revisiting the interaction between complement lectin pathway protease MASP-2 and SARS-CoV-2 nucleoprotein. Front. Immunol. 2024, 15, 1419165. [Google Scholar] [CrossRef]
- Zhao, Y.; Sui, L.; Wu, P.; Wang, W.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q.; Wang, G. A dual-role of SARS-CoV-2 nucleocapsid protein in regulating innate immune response. Signal Transduct. Target. Ther. 2021, 6, 331. [Google Scholar] [CrossRef]
- Reuschl, A.-K.; Thorne, L.G.; Whelan, M.V.; Ragazzini, R.; Furnon, W.; Cowton, V.M.; De Lorenzo, G.; Mesner, D.; Turner, J.L.; Dowgier, G. Evolution of enhanced innate immune suppression by SARS-CoV-2 Omicron subvariants. Nat. Microbiol. 2024, 9, 451–463. [Google Scholar] [CrossRef]
- Shekhawat, J.; Gauba, K.; Gupta, S.; Purohit, P.; Mitra, P.; Garg, M.; Misra, S.; Sharma, P.; Banerjee, M. Interleukin-6 perpetrator of the COVID-19 cytokine storm. Indian J. Clin. Biochem. 2021, 36, 440–450. [Google Scholar] [CrossRef]
- Karwaciak, I.; Sałkowska, A.; Karaś, K.; Dastych, J.; Ratajewski, M. Nucleocapsid and spike proteins of the coronavirus SARS-CoV-2 induce il6 in monocytes and macrophages—Potential implications for cytokine storm syndrome. Vaccines 2021, 9, 54. [Google Scholar] [CrossRef]
- Vora, S.M.; Lieberman, J.; Wu, H. Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 2021, 21, 694–703. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Chen, L.-D.; Zhang, Z.-Y.; Wei, X.-J.; Cai, Y.-Q.; Yao, W.-Z.; Wang, M.-H.; Huang, Q.-F.; Zhang, X.-B. Association between cytokine profiles and lung injury in COVID-19 pneumonia. Respir. Res. 2020, 21, 201. [Google Scholar] [CrossRef]
- McConnell, M.J.; Kawaguchi, N.; Kondo, R.; Sonzogni, A.; Licini, L.; Valle, C.; Bonaffini, P.A.; Sironi, S.; Alessio, M.G.; Previtali, G. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J. Hepatol. 2021, 75, 647–658. [Google Scholar] [CrossRef]
- Wick, K.D.; Leligdowicz, A.; Willmore, A.; Carrillo, S.A.; Ghale, R.; Jauregui, A.; Chak, S.S.; Nguyen, V.; Lee, D.; Jones, C. Plasma SARS-CoV-2 nucleocapsid antigen levels are associated with progression to severe disease in hospitalized COVID-19. Crit. Care 2022, 26, 278. [Google Scholar] [CrossRef]
- ACTIV-3/TICO Bamlanivimab Study Group; Lundgren, J.D.; Grund, B.; Barkauskas, C.E.; Holland, T.L.; Gottlieb, R.L.; Sandkovsky, U.; Brown, S.M.; Knowlton, K.U.; Self, W.H. Responses to a neutralizing monoclonal antibody for hospitalized patients with COVID-19 according to baseline antibody and antigen levels: A randomized controlled trial. Ann. Intern. Med. 2022, 175, 234–243. [Google Scholar] [CrossRef]
- Thudium, R.F.; Stoico, M.P.; Høgdall, E.; Høgh, J.; Krarup, H.B.; Larsen, M.A.; Madsen, P.H.; Nielsen, S.D.; Ostrowski, S.R.; Palombini, A. Early laboratory diagnosis of COVID-19 by antigen detection in blood samples of the SARS-CoV-2 nucleocapsid protein. J. Clin. Microbiol. 2021, 59, e01001-21. [Google Scholar] [CrossRef]
- Favresse, J.; Bayart, J.-L.; David, C.; Dogné, J.-M.; Douxfils, J. Nucleocapsid serum antigen determination in SARS-CoV-2 infected patients using the single molecule array technology and prediction of disease severity. J. Infect. 2022, 84, e4. [Google Scholar] [CrossRef]
- Yuan, C.; Ma, Z.; Xie, J.; Li, W.; Su, L.; Zhang, G.; Xu, J.; Wu, Y.; Zhang, M.; Liu, W. The role of cell death in SARS-CoV-2 infection. Signal Transduct. Target. Ther. 2023, 8, 357. [Google Scholar] [CrossRef]
- Machhi, J.; Shahjin, F.; Das, S.; Patel, M.; Abdelmoaty, M.M.; Cohen, J.D.; Singh, P.A.; Baldi, A.; Bajwa, N.; Kumar, R. A role for extracellular vesicles in SARS-CoV-2 therapeutics and prevention. J. Neuroimmune Pharmacol. 2021, 16, 270–288. [Google Scholar] [CrossRef]
- Perna, F.; Bruzzaniti, S.; Piemonte, E.; Maddaloni, V.; Atripaldi, L.; Sale, S.; Sanduzzi, A.; Nicastro, C.; Pepe, N.; Bifulco, M. Serum levels of SARS-CoV-2 nucleocapsid antigen associate with inflammatory status and disease severity in COVID-19 patients. Clin. Immunol. 2021, 226, 108720. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, S.; Yan, H.; Huang, Y.; Wei, C.; Liu, J.; Sun, J. Plasma SARS-CoV-2 nucleocapsid antigen levels are associated with lung infection and tissue-damage biomarkers. Virus Res. 2025, 356, 199580. [Google Scholar] [CrossRef]
- Wang, L. C-reactive protein levels in the early stage of COVID-19. Med. Mal. Infect. 2020, 50, 332–334. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, K.I.; Liu, S.; Yan, Z.; Xu, C.; Qiao, Z. Plasma CRP level is positively associated with the severity of COVID-19. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 18. [Google Scholar] [CrossRef]
- Stravalaci, M.; Pagani, I.; Paraboschi, E.M.; Pedotti, M.; Doni, A.; Scavello, F.; Mapelli, S.N.; Sironi, M.; Perucchini, C.; Varani, L. Recognition and inhibition of SARS-CoV-2 by humoral innate immunity pattern recognition molecules. Nat. Immunol. 2022, 23, 275–286. [Google Scholar] [CrossRef]
- Feitosa, T.A.; de Souza Sá, M.V.; Pereira, V.C.; de Andrade Cavalcante, M.K.; Pereira, V.R.A.; da Costa Armstrong, A.; do Carmo, R.F. Association of polymorphisms in long pentraxin 3 and its plasma levels with COVID-19 severity. Clin. Exp. Med. 2023, 23, 1225–1233. [Google Scholar] [CrossRef]
- Oprinca, G.C.; Mohor, C.-I.; Bereanu, A.-S.; Oprinca-Muja, L.-A.; Bogdan-Duică, I.; Fleacă, S.R.; Hașegan, A.; Diter, A.; Boeraș, I.; Cristian, A.N. Detection of SARS-CoV-2 Viral Genome and Viral Nucleocapsid in Various Organs and Systems. Int. J. Mol. Sci. 2024, 25, 5755. [Google Scholar] [CrossRef]
- Orsini, F.; Bosica, M.; Martucci, A.; De Paola, M.; Comolli, D.; Pascente, R.; Forloni, G.; Fraser, P.E.; Arancio, O.; Fioriti, L. SARS-CoV-2 Nucleocapsid Protein Induces Tau Pathological Changes That Can Be Counteracted by SUMO2. Int. J. Mol. Sci. 2024, 25, 7169. [Google Scholar] [CrossRef]
- Low, Z.Y.; Zabidi, N.Z.; Yip, A.J.W.; Puniyamurti, A.; Chow, V.T.; Lal, S.K. SARS-CoV-2 non-structural proteins and their roles in host immune evasion. Viruses 2022, 14, 1991. [Google Scholar] [CrossRef]
- Demmer, R.T.; Wu, C.; Kim, J.S.; Sun, Y.; Balte, P.; Cushman, M.; Boyle, R.; Tracy, R.P.; Styer, L.M.; Bell, T.D. Demographic and Clinical Factors Associated With SARS-CoV-2 Anti-Nucleocapsid Antibody Response Among Previously Infected US Adults: The C4R Study. Open Forum Infect. Dis. 2025, 12, ofaf123. [Google Scholar] [CrossRef]
- Sokal, A.; Chappert, P.; Barba-Spaeth, G.; Roeser, A.; Fourati, S.; Azzaoui, I.; Vandenberghe, A.; Fernandez, I.; Meola, A.; Bouvier-Alias, M. Maturation and persistence of the anti-SARS-CoV-2 memory B cell response. Cell 2021, 184, 1201–1213.E14. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Koerber, N.; Priller, A.; Yazici, S.; Bauer, T.; Cheng, C.-C.; Mijočević, H.; Wintersteller, H.; Jeske, S.; Vogel, E.; Feuerherd, M. Dynamics of spike-and nucleocapsid specific immunity during long-term follow-up and vaccination of SARS-CoV-2 convalescents. Nat. Commun. 2022, 13, 153. [Google Scholar] [CrossRef]
- Chansaenroj, J.; Yorsaeng, R.; Posuwan, N.; Puenpa, J.; Wanlapakorn, N.; Sudhinaraset, N.; Sripramote, M.; Chalongviriyalert, P.; Jirajariyavej, S.; Kiatpanabhikul, P. Long-term specific IgG response to SARS-CoV-2 nucleocapsid protein in recovered COVID-19 patients. Sci. Rep. 2021, 11, 23216. [Google Scholar] [CrossRef]
- Chia, W.N.; Zhu, F.; Ong, S.W.X.; Young, B.E.; Fong, S.-W.; Le Bert, N.; Tan, C.W.; Tiu, C.; Zhang, J.; Tan, S.Y. Dynamics of SARS-CoV-2 neutralising antibody responses and duration of immunity: A longitudinal study. Lancet Microbe 2021, 2, e240–e249. [Google Scholar] [CrossRef]
- Eser, T.M.; Baranov, O.; Huth, M.; Ahmed, M.I.; Deák, F.; Held, K.; Lin, L.; Pekayvaz, K.; Leunig, A.; Nicolai, L. Nucleocapsid-specific T cell responses associate with control of SARS-CoV-2 in the upper airways before seroconversion. Nat. Commun. 2023, 14, 2952. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deitchman, A.N.; Torres, L.; Iyer, N.S.; Munter, S.E.; Nixon, C.C.; Donatelli, J.; Thanh, C.; Takahashi, S.; Hakim, J. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 2021, 36, 109518. [Google Scholar] [CrossRef]
- Taus, E.; Hofmann, C.; Ibarrondo, F.J.; Hausner, M.A.; Fulcher, J.A.; Krogstad, P.; Ferbas, K.G.; Tobin, N.H.; Rimoin, A.W.; Aldrovandi, G.M. Dominant CD8+ T cell nucleocapsid targeting in SARS-CoV-2 infection and broad spike targeting from vaccination. Front. Immunol. 2022, 13, 835830. [Google Scholar] [CrossRef]
- López-Muñoz, A.D.; Kosik, I.; Holly, J.; Yewdell, J.W. Cell surface SARS-CoV-2 nucleocapsid protein modulates innate and adaptive immunity. Sci. Adv. 2022, 8, eabp9770. [Google Scholar] [CrossRef]
- Lineburg, K.E.; Grant, E.J.; Swaminathan, S.; Chatzileontiadou, D.S.; Szeto, C.; Sloane, H.; Panikkar, A.; Raju, J.; Crooks, P.; Rehan, S. CD8+ T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope cross-react with selective seasonal coronaviruses. Immunity 2021, 54, 1055–1065.E5. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Rowntree, L.C.; Petersen, J.; Chua, B.Y.; Hensen, L.; Kedzierski, L.; van de Sandt, C.E.; Chaurasia, P.; Tan, H.-X.; Habel, J.R. CD8+ T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope display high naive precursor frequency and TCR promiscuity. Immunity 2021, 54, 1066–1082.E5. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Jia, X.; Cao, S.; Lee, A.S.; Manohar, M.; Sindher, S.B.; Ahuja, N.; Artandi, M.; Blish, C.A.; Blomkalns, A.L.; Chang, I. Anti-nucleocapsid antibody levels and pulmonary comorbid conditions are linked to post–COVID-19 syndrome. JCI Insight 2022, 7, e156713. [Google Scholar] [CrossRef]
- Altmann, D.M.; Whettlock, E.M.; Liu, S.; Arachchillage, D.J.; Boyton, R.J. The immunology of long COVID. Nat. Rev. Immunol. 2023, 23, 618–634. [Google Scholar] [CrossRef]
- Littlefield, K.M.; Watson, R.O.; Schneider, J.M.; Neff, C.P.; Yamada, E.; Zhang, M.; Campbell, T.B.; Falta, M.T.; Jolley, S.E.; Fontenot, A.P. SARS-CoV-2-specific T cells associate with inflammation and reduced lung function in pulmonary post-acute sequalae of SARS-CoV-2. PloS Pathog. 2022, 18, e1010359. [Google Scholar] [CrossRef]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.-S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef]
- Patrascu, R.; Dumitru, C.S. Advances in Understanding Inflammation and Tissue Damage: Markers of Persistent Sequelae in COVID-19 Patients. J. Clin. Med. 2025, 14, 1475. [Google Scholar] [CrossRef]
- Yin, J.-X.; Agbana, Y.L.; Sun, Z.-S.; Fei, S.-W.; Zhao, H.-Q.; Zhou, X.-N.; Chen, J.-H.; Kassegne, K. Increased interleukin-6 is associated with long COVID-19: A systematic review and meta-analysis. Infect. Dis. Poverty 2023, 12, 43. [Google Scholar] [CrossRef]
- Santa Cruz, A.; Mendes-Frias, A.; Azarias-da-Silva, M.; André, S.; Oliveira, A.I.; Pires, O.; Mendes, M.; Oliveira, B.; Braga, M.; Lopes, J.R. Post-acute sequelae of COVID-19 is characterized by diminished peripheral CD8+ β7 integrin+ T cells and anti-SARS-CoV-2 IgA response. Nat. Commun. 2023, 14, 1772. [Google Scholar] [CrossRef]
- Grote, K.; Schaefer, A.-C.; Soufi, M.; Ruppert, V.; Linne, U.; Mukund Bhagwat, A.; Szymanski, W.; Graumann, J.; Gercke, Y.; Aldudak, S. Targeting the high-density lipoprotein proteome for the treatment of post-acute sequelae of SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 4522. [Google Scholar] [CrossRef]
- Visvabharathy, L.; Hanson, B.A.; Orban, Z.S.; Lim, P.H.; Palacio, N.M.; Jimenez, M.; Clark, J.R.; Graham, E.L.; Liotta, E.M.; Tachas, G. Neuro-PASC is characterized by enhanced CD4+ and diminished CD8+ T cell responses to SARS-CoV-2 Nucleocapsid protein. Front. Immunol. 2023, 14, 1155770. [Google Scholar] [CrossRef]
- Bodansky, A.; Mettelman, R.C.; Sabatino, J.J., Jr.; Vazquez, S.E.; Chou, J.; Novak, T.; Moffitt, K.L.; Miller, H.S.; Kung, A.F.; Rackaityte, E. Molecular mimicry in multisystem inflammatory syndrome in children. Nature 2024, 632, 622–629. [Google Scholar] [CrossRef]
- Oprinca, G.-C.; Mohor, C.-I.; Oprinca-Muja, A.; Hașegan, A.; Cristian, A.-N.; Fleacă, S.-R.; Boeraș, I.; Cardoș, R.; Atasie, D.; Mihalache, M. Unveiling the Pathological Mechanisms of Death Induced by SARS-CoV-2 Viral Pneumonia. Microorganisms 2024, 12, 459. [Google Scholar] [CrossRef]
- Yan, Y.; Diao, B.; Liu, Y.; Zhang, W.; Wang, G.; Chen, X. Severe acute respiratory syndrome coronavirus 2 nucleocapsid protein in the ocular tissues of a patient previously infected with coronavirus disease 2019. JAMA Ophthalmol. 2020, 138, 1201–1204. [Google Scholar] [CrossRef]
- Yokoyama, K.; Sato, Y.; Suimon, Y.; Kase, S. Late onset paediatric dacryoadenitis associated with SARS-CoV-2 confirmed by histological analysis. BMJ Case Rep. CP 2024, 17, e257615. [Google Scholar] [CrossRef]
- de Melo, G.D.; Lazarini, F.; Levallois, S.; Hautefort, C.; Michel, V.; Larrous, F.; Verillaud, B.; Aparicio, C.; Wagner, S.; Gheusi, G. COVID-19–related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021, 13, eabf8396. [Google Scholar] [CrossRef]
- El-Baky, N.A.; Amara, A.A.; Uversky, V.N.; Redwan, E.M. Intrinsic factors behind long COVID: III. Persistence of SARS-CoV-2 and its components. J. Cell. Biochem. 2024, 125, e30514. [Google Scholar] [CrossRef]
- Eltayeb, A.; Al-Sarraj, F.; Alharbi, M.; Albiheyri, R.; Mattar, E.H.; Abu Zeid, I.M.; Bouback, T.A.; Bamagoos, A.; Uversky, V.N.; Rubio-Casillas, A. Intrinsic factors behind long COVID: IV. Hypothetical roles of the SARS-CoV-2 nucleocapsid protein and its liquid–liquid phase separation. J. Cell. Biochem. 2024, 125, e30530. [Google Scholar] [CrossRef]
- Rizzo, R.; Neri, L.M.; Simioni, C.; Bortolotti, D.; Occhionorelli, S.; Zauli, G.; Secchiero, P.; Semprini, C.M.; Laface, I.; Sanz, J.M. SARS-CoV-2 nucleocapsid protein and ultrastructural modifications in small bowel of a 4-week-negative COVID-19 patient. Clin. Microbiol. Infect. 2021, 27, 936. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kompoti, M. Obesity and infection. Lancet Infect. Dis. 2006, 6, 438–446. [Google Scholar] [CrossRef]
- Chakravarty, D.; Ratnani, P.; Sobotka, S.; Lundon, D.; Wiklund, P.; Nair, S.S.; Tewari, A.K. Increased hospitalization and mortality from COVID-19 in prostate cancer patients. Cancers 2021, 13, 1630. [Google Scholar] [CrossRef]
- Drucker, D.J. Diabetes, obesity, metabolism, and SARS-CoV-2 infection: The end of the beginning. Cell Metab. 2021, 33, 479–498. [Google Scholar] [CrossRef]
- Steinke, I.; Ghanei, N.; Govindarajulu, M.; Yoo, S.; Zhong, J.; Amin, R.H. Drug discovery and development of novel therapeutics for inhibiting TMAO in models of atherosclerosis and diabetes. Front. Physiol. 2020, 11, 567899. [Google Scholar] [CrossRef]
- Constantino-Jonapa, L.A.; Espinoza-Palacios, Y.; Escalona-Montaño, A.R.; Hernández-Ruiz, P.; Amezcua-Guerra, L.M.; Amedei, A.; Aguirre-García, M.M. Contribution of trimethylamine N-oxide (TMAO) to chronic inflammatory and degenerative diseases. Biomedicines 2023, 11, 431. [Google Scholar] [CrossRef]
- Liu, M.-H.; Lin, X.-L.; Xiao, L.-L. SARS-CoV-2 nucleocapsid protein promotes TMAO-induced NLRP3 inflammasome activation by SCAP–SREBP signaling pathway. Tissue Cell 2024, 86, 102276. [Google Scholar] [CrossRef]
- Liu, M.-H.; Lin, X.-L.; Xiao, L.-L. Excess phosphate promotes SARS-CoV-2 N protein-induced NLRP3 inflammasome activation via the SCAP-SREBP2 signaling pathway. Mol. Med. Rep. 2024, 29, 1–12. [Google Scholar] [CrossRef]
- Strong, M.J.; McLellan, C.; Kaplanis, B.; Droppelmann, C.A.; Junop, M. Phase Separation of SARS-CoV-2 Nucleocapsid Protein with TDP-43 Is Dependent on C-Terminus Domains. Int. J. Mol. Sci. 2024, 25, 8779. [Google Scholar] [CrossRef]
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, B.; Gu, Y.; Yue, Z.; Liu, Y.; Lei, Z.; Yang, G.; Deng, M.; Zhang, X.; Luo, Z. SARS-CoV-2 nucleocapsid protein interaction with YBX1 displays oncolytic properties through PKM mRNA destabilization. Mol. Cancer 2024, 23, 248. [Google Scholar] [CrossRef]
- El-Maradny, Y.A.; Badawy, M.A.; Mohamed, K.I.; Ragab, R.F.; Moharm, H.M.; Abdallah, N.A.; Elgammal, E.M.; Rubio-Casillas, A.; Uversky, V.N.; Redwan, E.M. Unraveling the role of the nucleocapsid protein in SARS-CoV-2 pathogenesis: From viral life cycle to vaccine development. Int. J. Biol. Macromol. 2024, 279, 135201. [Google Scholar] [CrossRef]
- Mendoza-Ramírez, N.J.; García-Cordero, J.; Shrivastava, G.; Cedillo-Barrón, L. The key to increase immunogenicity of next-generation COVID-19 vaccines lies in the inclusion of the SARS-CoV-2 nucleocapsid protein. J. Immunol. Res. 2024, 2024, 9313267. [Google Scholar] [CrossRef]
- Kang, S.; Yang, M.; He, S.; Wang, Y.; Chen, X.; Chen, Y.Q.; Hong, Z.S.; Liu, J.; Jiang, G.M.; Chen, Q.Y.; et al. A SARS-CoV-2 antibody curbs viral nucleocapsid protein-induced complement hyperactivation. Nat. Commun. 2021, 12, 2697. [Google Scholar] [CrossRef]
- Dangi, T.; Sanchez, S.; Class, J.; Richner, M.; Visvabharathy, L.; Chung, Y.R.; Bentley, K.; Staton, R.J.; Koralnik, I.J.; Richner, J.M.; et al. Improved control of SARS-CoV-2 by treatment with a nucleocapsid-specific monoclonal antibody. J. Clin. Investig. 2022, 132, e162282. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Wang, Y.; Huang, D.; Tan, Y.-J. The Role of SARS-CoV-2 Nucleocapsid Protein in Host Inflammation. Viruses 2025, 17, 1046. https://doi.org/10.3390/v17081046
Cao Y, Wang Y, Huang D, Tan Y-J. The Role of SARS-CoV-2 Nucleocapsid Protein in Host Inflammation. Viruses. 2025; 17(8):1046. https://doi.org/10.3390/v17081046
Chicago/Turabian StyleCao, Yujia, Yaju Wang, Dejian Huang, and Yee-Joo Tan. 2025. "The Role of SARS-CoV-2 Nucleocapsid Protein in Host Inflammation" Viruses 17, no. 8: 1046. https://doi.org/10.3390/v17081046
APA StyleCao, Y., Wang, Y., Huang, D., & Tan, Y.-J. (2025). The Role of SARS-CoV-2 Nucleocapsid Protein in Host Inflammation. Viruses, 17(8), 1046. https://doi.org/10.3390/v17081046