
Genome-Wide Screening Reveals the Oncolytic Mechanism of Newcastle Disease Virus in a Human Colonic Carcinoma Cell Line

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Plasmids, and Virus Strain
2.2. Virus Infection
2.3. Absolute Quantitative PCR (qPCR)
2.4. Flow Cytometric Analysis
2.5. RNA Sequencing
2.6. CRISPR-Cas9 Knockout Screening
2.7. High-Throughput Data Analysis
2.8. Statistical Analysis
3. Results
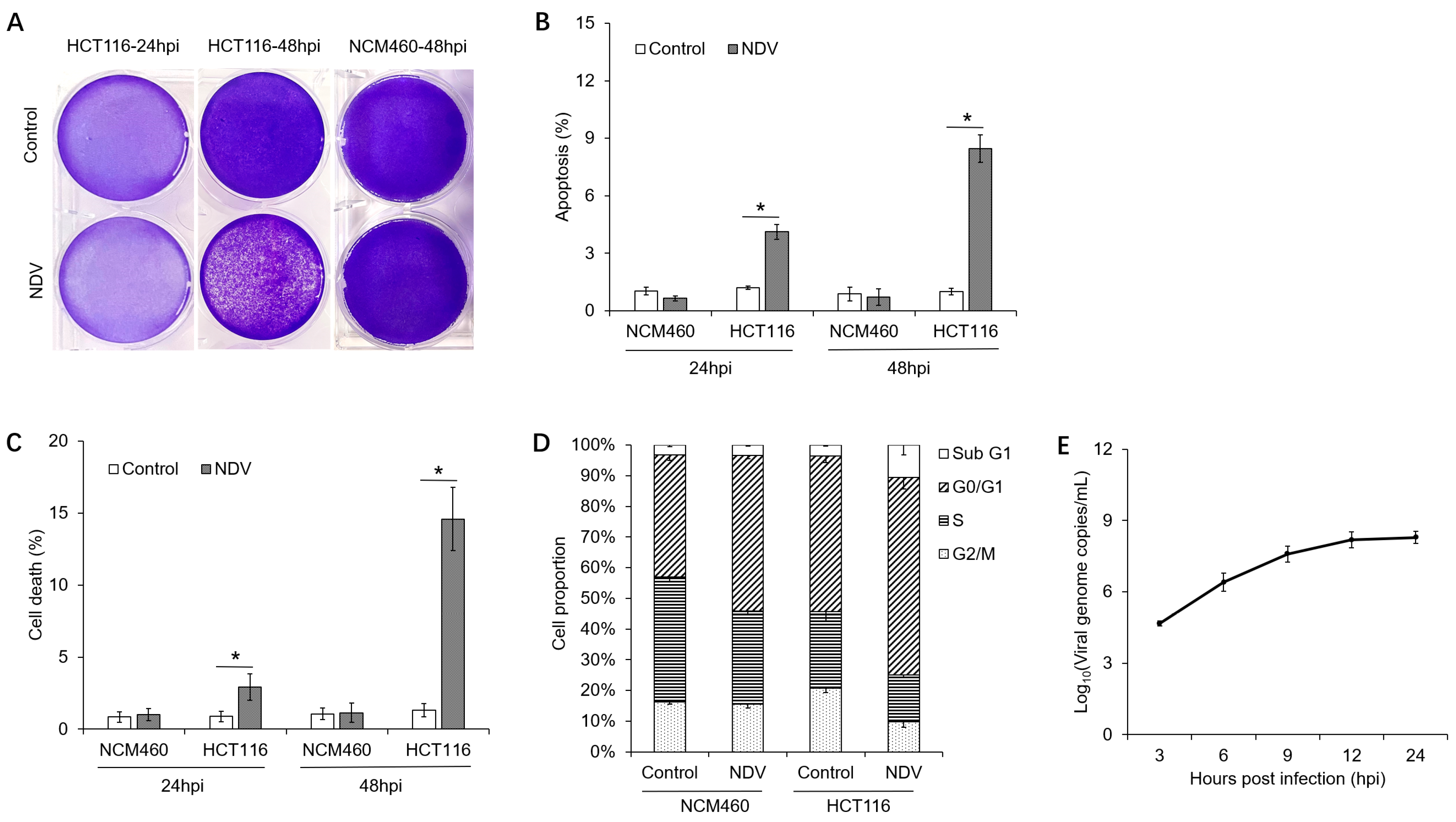
3.1. Oncolytic Effects of NDV on Different Cells
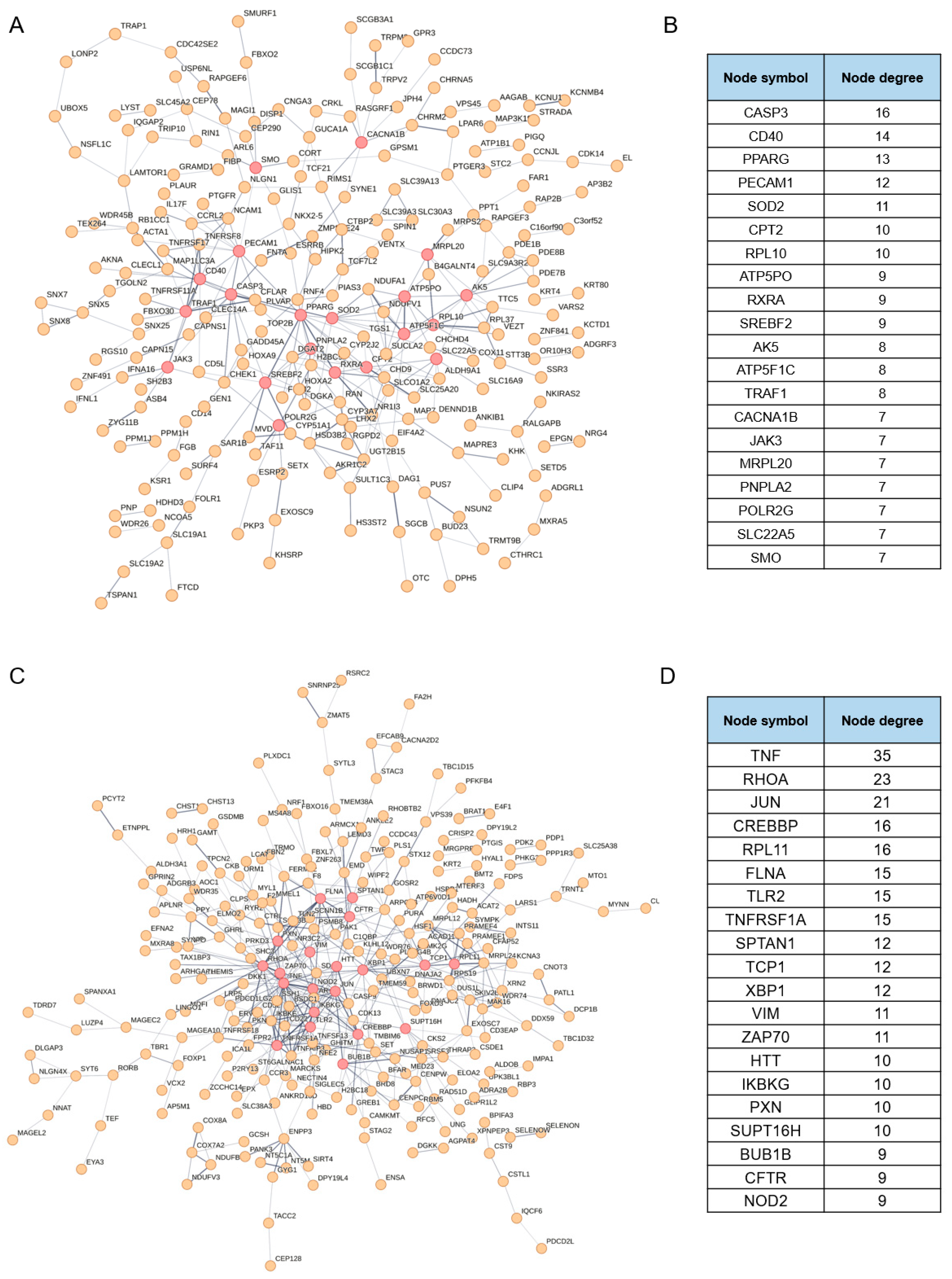
3.2. Identification of the Putative Regulatory Factors Involved in NDV Oncolysis
3.3. Identification of the Putative Effectors Involved in NDV Oncolysis
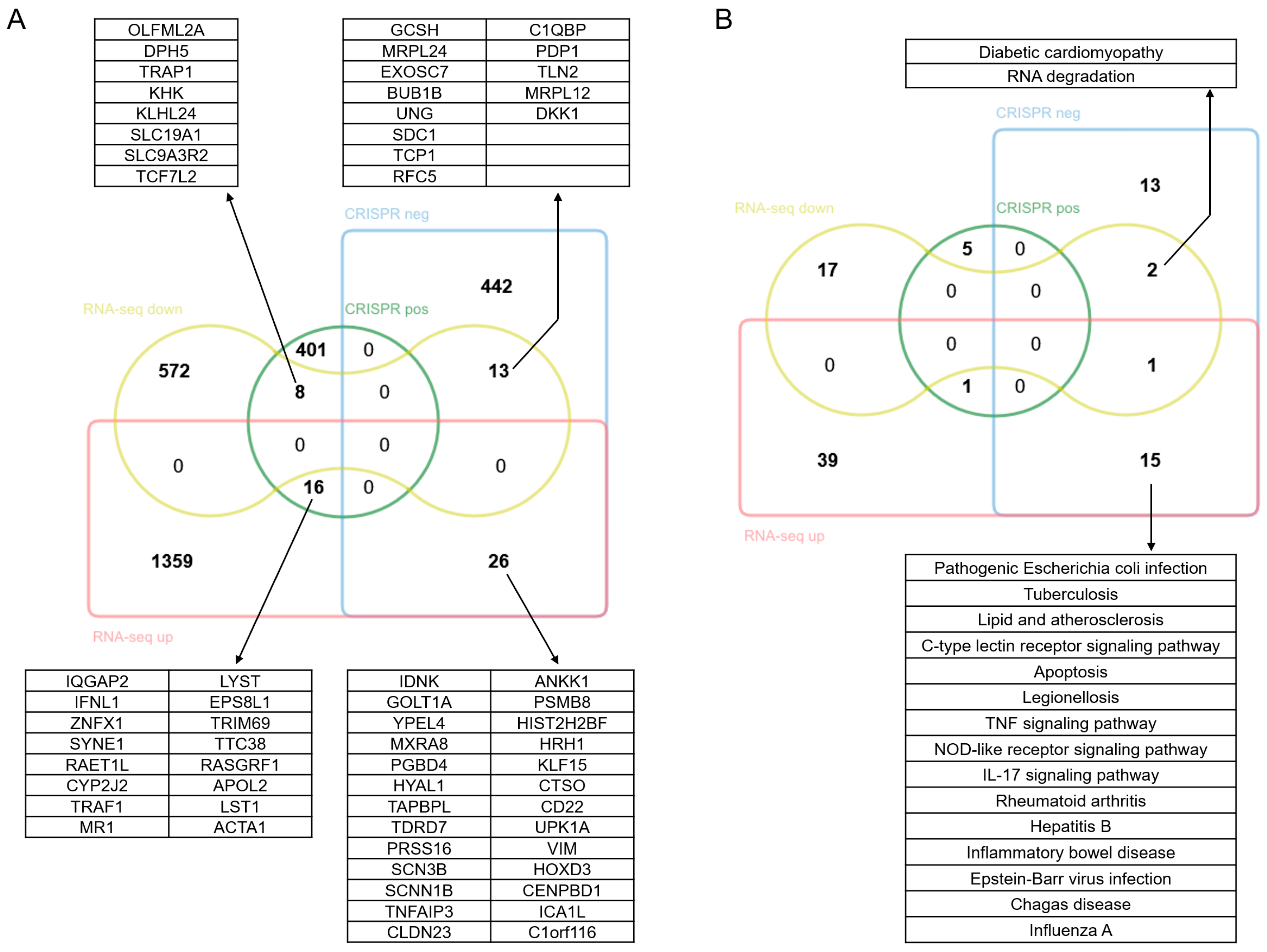
3.4. Comprehensive Analysis of the Regulatory Mechanisms Involved in NDV Oncolysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- De Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; van den Hoogen, B.G. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef]
- Zhong, L.; Gan, L.; Wang, B.; Wu, T.; Yao, F.; Gong, W.; Peng, H.; Deng, Z.; Xiao, G.; Liu, X.; et al. Hyperacute rejection-engineered oncolytic virus for interventional clinical trial in refractory cancer patients. Cell 2025, 188, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Coffin, R.S. From virotherapy to oncolytic immunotherapy: Where are we now? Curr. Opin. Virol. 2015, 13, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- De Munck, J.; Binks, A.; McNeish, I.A.; Aerts, J.L. Oncolytic virus-induced cell death and immunity: A match made in heaven? J. Leukoc. Biol. 2017, 102, 631–643. [Google Scholar] [CrossRef]
- Cassel, W.A.; Garrett, R.E. Newcastle Disease Virus as an Antineoplastic Agent. Cancer 1965, 18, 863–868. [Google Scholar] [CrossRef]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, J.L.; Goldberg, S.; Gross, P.; O’Neil, J.D.; Groene, W.S.; et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef]
- Hutcheson, J.M.; Susta, L.; Stice, S.L.; Afonso, C.L.; West, F.D. Delayed Newcastle disease virus replication using RNA interference to target the nucleoprotein. Biologicals 2015, 43, 274–280. [Google Scholar] [CrossRef]
- Ren, S.; Rehman, Z.U.; Shi, M.; Yang, B.; Qu, Y.; Yang, X.F.; Shao, Q.; Meng, C.; Yang, Z.; Gao, X.; et al. Syncytia generated by hemagglutinin-neuraminidase and fusion proteins of virulent Newcastle disease virus induce complete autophagy by activating AMPK-mTORC1-ULK1 signaling. Vet. Microbiol. 2019, 230, 283–290. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Z.; Wu, Y.; Zheng, D.; Sun, C.; Bi, D.; Zuo, Y.; Xu, T. Molecular epidemiological analysis of Newcastle disease virus isolated in China in 2005. J. Virol. Methods 2007, 140, 206–211. [Google Scholar] [CrossRef]
- Zhao, H.; Peeters, B.P.H. Recombinant Newcastle disease virus as a viral vector: Effect of genomic location of foreign gene on gene expression and virus replication. J. Gen. Virol. 2003, 84 Pt 4, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Bahreyni, A.; Mohamud, Y.; Luo, H. Oncolytic virus-based combination therapy in breast cancer. Cancer Lett. 2024, 585, 216634. [Google Scholar] [CrossRef] [PubMed]
- Wilden, H.; Fournier, P.; Zawatzky, R.; Schirrmacher, V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle Disease Virus. Int. J. Oncol. 2009, 34, 971–982. [Google Scholar]
- Bian, J.; Wang, K.; Kong, X.; Liu, H.; Chen, F.; Hu, M.; Zhang, X.; Jiao, X.; Ge, B.; Wu, Y.; et al. Caspase- and p38-MAPK-dependent induction of apoptosis in A549 lung cancer cells by Newcastle disease virus. Arch. Virol. 2011, 156, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Elankumaran, S.; Rockemann, D.; Samal, S.K. Newcastle disease virus exerts oncolysis by both intrinsic and extrinsic caspase-dependent pathways of cell death. J. Virol. 2006, 80, 7522–7534. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, H.X.; Mao, X.; Fang, H.; Wang, H.; Li, Y.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; et al. RIP1 is a central signaling protein in regulation of TNF-α/TRAIL mediated apoptosis and necroptosis during Newcastle disease virus infection. Oncotarget 2017, 8, 43201–43217. [Google Scholar] [CrossRef]
- Tayeb, S.; Zakay-Rones, Z.; Panet, A. Therapeutic potential of oncolytic Newcastle disease virus: A critical review. Oncolytic Virother. 2015, 4, 49–62. [Google Scholar]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Smyth, G.K. Camera: A competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012, 40, e133. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, D.; Von Kuster, G.; Coraor, N.; Ananda, G.; Lazarus, R.; Mangan, M.; Nekrutenko, A.; Taylor, J. Galaxy: A web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010, 89, 19.10.1–19.10.21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Willmann, K.L.; Klaver, S.; Doğu, F.; Santos-Valente, E.; Garncarz, W.; Bilic, I.; Mace, E.; Salzer, E.; Domínguez Conde, C.; Sic, H.; et al. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun. 2014, 5, 5360. [Google Scholar] [CrossRef]
- Karabay, O.; Guney Eskiler, G.; Alkurt, U.; Hamarat, K.F.; Deveci Ozkan, A.; Aydin, A. The predictive role of NF-κB-mediated pro-inflammatory cytokine expression levels in hepatitis B vaccine response. J. Immunoass. Immunochem. 2023, 44, 192–203. [Google Scholar] [CrossRef]
- Duan, Z.; Xing, J.; Shi, H.; Wang, Y.; Zhao, C. The matrix protein of Newcastle disease virus inhibits inflammatory response through IRAK4/TRAF6/TAK1/NF-κB signaling pathway. Int. J. Biol. Macromol. 2022, 218, 295–309. [Google Scholar] [CrossRef]
- Zhang, D.; Ding, Z.; Xu, X. Pathologic Mechanisms of the Newcastle Disease Virus. Viruses 2023, 15, 864. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.B.; Miyauchi, S.; Xu, H.Z.; Liu, D.; Kim, L.J.; Burkart, C.; Cheng, H.; Arimoto, K.I.; Yan, M.; Zhou, Y.; et al. Type I Interferon Regulates a Coordinated Gene Network to Enhance Cytotoxic T Cell-Mediated Tumor Killing. Cancer Discov. 2020, 10, 382–393. [Google Scholar] [CrossRef]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoité, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.Y.; Feng, T.; et al. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.K.; An, Y.H.; Jang, S.H.; Ryu, G.; Jung, S.B.; Kim, S.; Kim, C.S.; Jang, H. The tumor suppressive effect and apoptotic mechanism of TRAIL gene-containing recombinant NDV in TRAIL-resistant colorectal cancer HT-29 cells and TRAIL-nonresistant HCT116 cells, with each cell bearing a mouse model. Cancer Med. 2023, 12, 20380–20395. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Burke, S.; Travers, J.; Rath, N.; Leinster, A.; Navarro, C.; Franks, R.; Leyland, R.; Mulgrew, K.; McGlinchey, K.; et al. Recombinant Newcastle Disease Virus Immunotherapy Drives Oncolytic Effects and Durable Systemic Antitumor Immunity. Mol. Cancer Ther. 2021, 20, 1723–1734. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, S.; Liao, T.; Wang, C.; Han, J.; Yang, Z.; Lu, X.; Hu, Z.; Hu, J.; Wang, X.; et al. The HN protein of Newcastle disease virus induces cell apoptosis through the induction of lysosomal membrane permeabilization. PLoS Pathog. 2024, 20, e1011981. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.; Wang, C.; Tang, Q.; Shi, X.; Gao, X.; Ma, J.; Lu, K.; Han, Q.; Jia, Y.; Wang, X.; et al. Newcastle Disease Virus V Protein Inhibits Cell Apoptosis and Promotes Viral Replication by Targeting CacyBP/SIP. Front. Cell. Infect. Microbiol. 2018, 8, 304. [Google Scholar] [CrossRef]
- Huang, Z.; Krishnamurthy, S.; Panda, A.; Samal, S.K. Newcastle disease virus V protein is associated with viral pathogenesis and functions as an alpha interferon antagonist. J. Virol. 2003, 77, 8676–8685. [Google Scholar] [CrossRef]
- Park, M.S.; García-Sastre, A.; Cros, J.F.; Basler, C.F.; Palese, P. Newcastle disease virus V protein is a determinant of host range restriction. J. Virol. 2003, 77, 9522–9532. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef]
- Melero, I.; Navarro, B.; Teijeira, A.; Coukos, G. Cancer immunotherapy full speed ahead. Ann. Oncol. 2017, 28 (Suppl. S12), xii1–xii2. [Google Scholar] [CrossRef]
- Martin, N.T.; Bell, J.C. Oncolytic Virus Combination Therapy: Killing One Bird with Two Stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2019, 176, 677. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Kohlhapp, F.J.; Zloza, A.; Kaufman, H.L. Talimogene laherparepvec (T-VEC) as cancer immunotherapy. Drugs Today 2015, 51, 549–558. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Feng, S.; Yi, G.; Jin, S.; Zhu, Y.; Liu, X.; Zhou, J.; Li, H. Genome-Wide Screening Reveals the Oncolytic Mechanism of Newcastle Disease Virus in a Human Colonic Carcinoma Cell Line. Viruses 2025, 17, 1043. https://doi.org/10.3390/v17081043
Zhang Y, Feng S, Yi G, Jin S, Zhu Y, Liu X, Zhou J, Li H. Genome-Wide Screening Reveals the Oncolytic Mechanism of Newcastle Disease Virus in a Human Colonic Carcinoma Cell Line. Viruses. 2025; 17(8):1043. https://doi.org/10.3390/v17081043
Chicago/Turabian StyleZhang, Yu, Shufeng Feng, Gaohang Yi, Shujun Jin, Yongxin Zhu, Xiaoxiao Liu, Jinsong Zhou, and Hai Li. 2025. "Genome-Wide Screening Reveals the Oncolytic Mechanism of Newcastle Disease Virus in a Human Colonic Carcinoma Cell Line" Viruses 17, no. 8: 1043. https://doi.org/10.3390/v17081043
APA StyleZhang, Y., Feng, S., Yi, G., Jin, S., Zhu, Y., Liu, X., Zhou, J., & Li, H. (2025). Genome-Wide Screening Reveals the Oncolytic Mechanism of Newcastle Disease Virus in a Human Colonic Carcinoma Cell Line. Viruses, 17(8), 1043. https://doi.org/10.3390/v17081043