
De Novo Expressed Vpr Stimulates HIV-1 Replication in T Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. HIV-1 Proviral Clones
2.2. Retroviral Expression Vectors
2.3. Cells Lines
2.4. Virus Production in HEK293T Cells
2.5. Production of HIV-1.R Virions Trans-Complemented with Vpr in HEK293T Cells
2.6. Virus Production in CEM.SS T Cells
2.7. Pairwise Replication Competition Assay (PRCA)
2.8. Cell and Virus Lysates, and Immunoblotting
2.9. Expression from Unintegrated HIV-1
2.10. Statistical Analysis
2.11. Reagents Obtained Through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH
3. Results
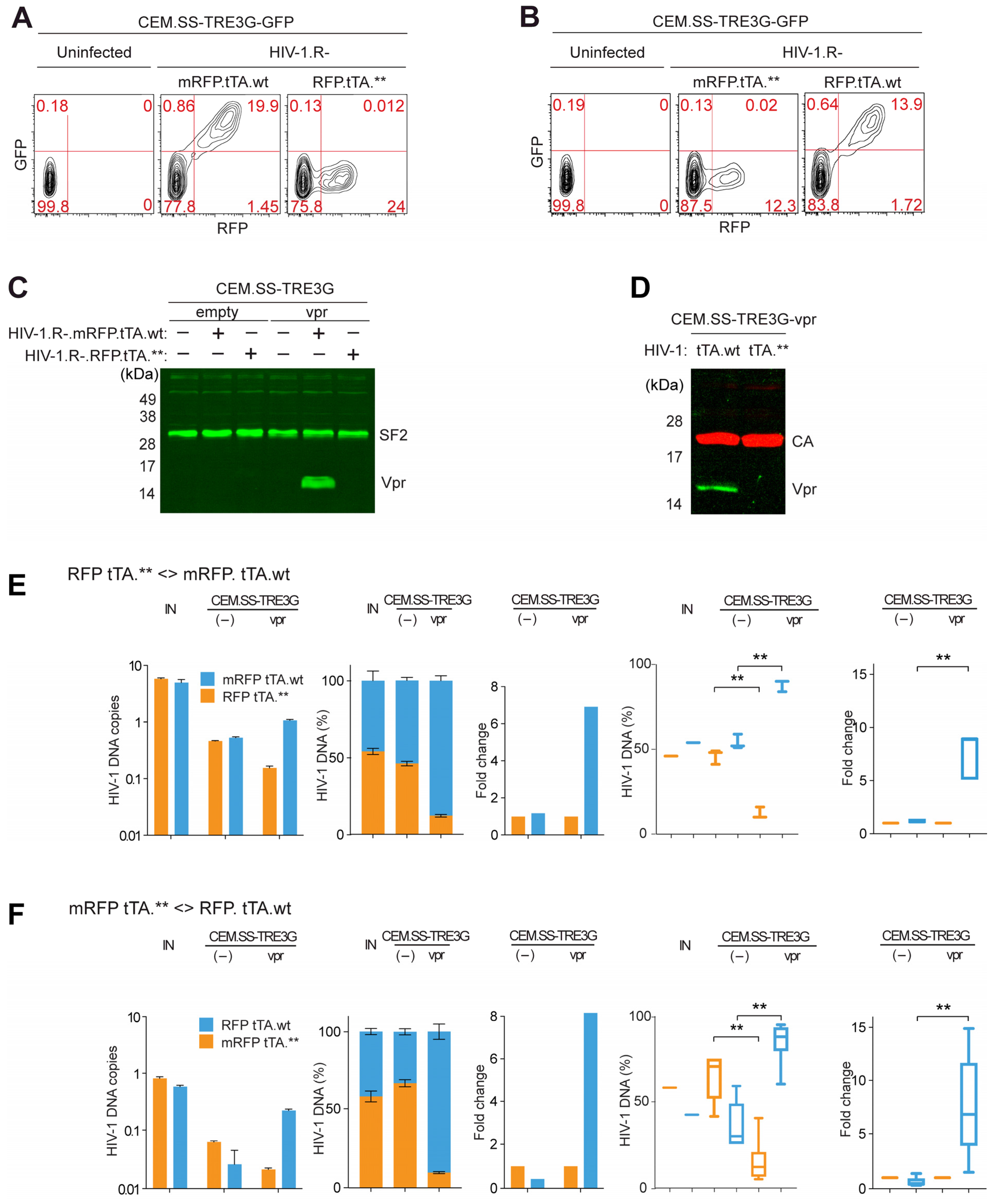
3.1. Inducible Expression System for Trans-Complementation of HIV-1 Particles Produced from a vpr-Defective Provirus with HIV-1 Vpr
3.2. Vpr Provided in Trans in Infected CEM.SS T Cells Stimulates HIV-1 Replication
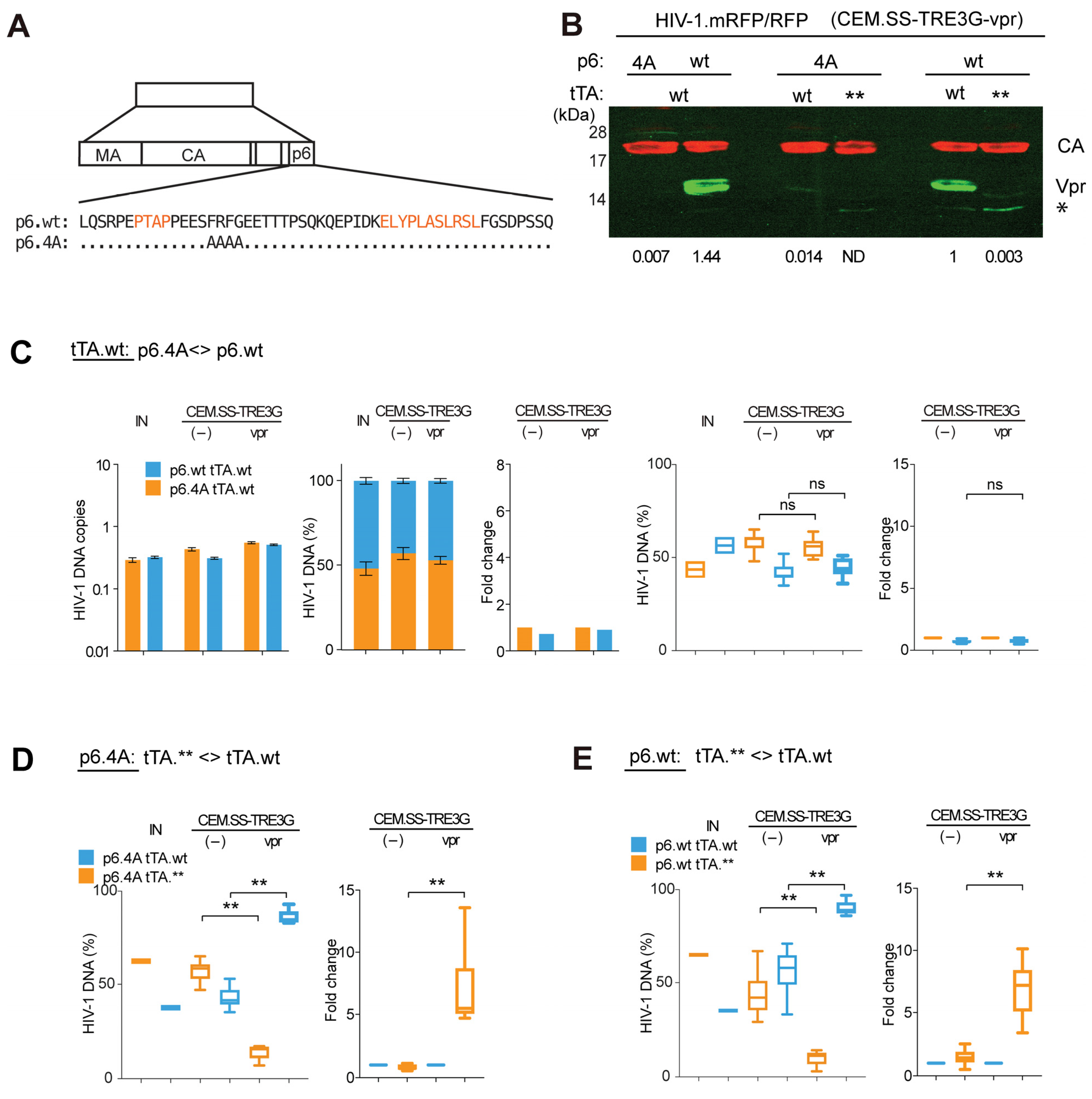
3.3. Virion-Associated Vpr Is Not Critical for Vpr’s Positive Effect on HIV-1 Replication
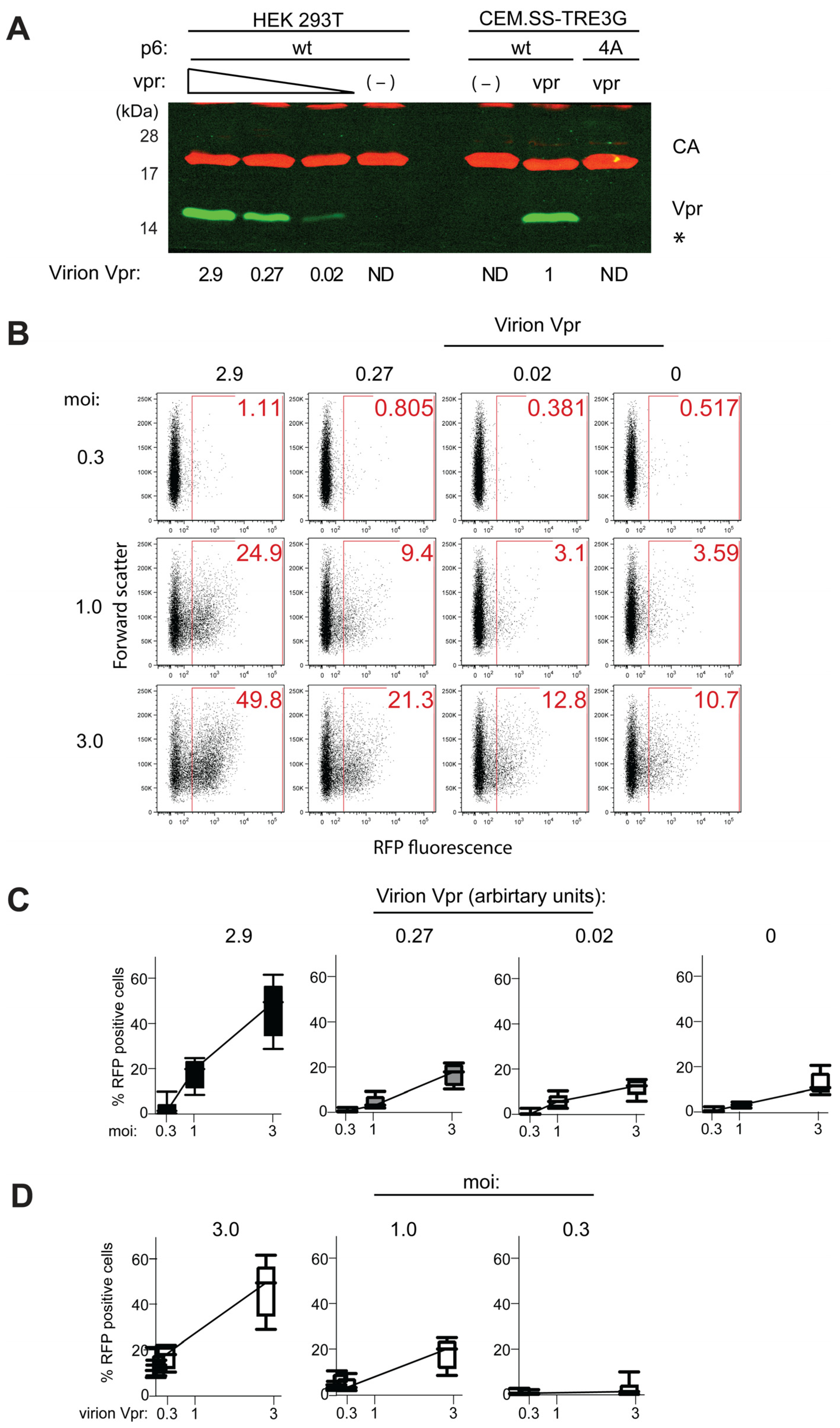
3.4. Vpr Antagonism of HIV-1 Preintegration Silencing Is Vpr-Dose Dependent
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rashid, F.; Xie, Z.; Suleman, M.; Shah, A.; Khan, S.; Luo, S. Roles and Functions of SARS-CoV-2 Proteins in Host Immune Evasion. Front. Immunol. 2022, 13, 940756. [Google Scholar] [CrossRef] [PubMed]
- Crow, M.S.; Lum, K.K.; Sheng, X.; Song, B.; Cristea, I.M. Diverse Mechanisms Evolved by DNA Viruses to Inhibit Early Host Defenses. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 452–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, T.; Duan, L.; Chen, H.; Hu, R.; Wang, X.; Jia, Y.; Chu, Z.; Liu, H.; Wang, X.; et al. Evasion of Host Antiviral Innate Immunity by Paramyxovirus Accessory Proteins. Front. Microbiol. 2022, 12, 790191. [Google Scholar] [CrossRef] [PubMed]
- Comar, C.E.; Otter, C.J.; Pfannenstiel, J.; Doerger, E.; Renner, D.M.; Tan, L.H.; Perlman, S.; Cohen, N.A.; Fehr, A.R.; Weiss, S.R. MERS-CoV Endoribonuclease and Accessory Proteins Jointly Evade Host Innate Immunity during Infection of Lung and Nasal Epithelial Cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2123208119. [Google Scholar] [CrossRef]
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses 2021, 13, 17. [Google Scholar] [CrossRef]
- Fan, D.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Zhao, X.; Chen, S.; Liu, M.; et al. The Role of VP16 in the Life Cycle of Alphaherpesviruses. Front. Microbiol. 2020, 11, 1910. [Google Scholar] [CrossRef]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef]
- Kmiec, D.; Kirchhoff, F. Antiviral Factors and Their Counteraction by HIV-1: Many Uncovered and More to Be Discovered. J. Mol. Cell Biol. 2024, 16, mjae005. [Google Scholar] [CrossRef]
- Laguette, N.; Benkirane, M. Shaping of the Host Cell by Viral Accessory Proteins. Front. Microbiol. 2015, 6, 142. [Google Scholar] [CrossRef]
- Li, Y.-L.; Langley, C.; Emerman, M.; Gross, J.D. APOBEC3G Antagonism by Vif, or When Structure Meets Biological and Evolutionary Studies. Annu. Rev. Virol. 2025, 12. [Google Scholar] [CrossRef]
- Barone, M.E.; Lim, A.; Woody, M.; Taklifi, P.; Yeasmin, F.; Wang, K.; Lewinski, M.K.; Singh, R.; Stoneham, C.A.; Jia, X.; et al. Adaptor Protein Complexes in HIV-1 Pathogenesis: Mechanisms and Therapeutic Potential. Viruses 2025, 17, 715. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Geiger, J.D. Role of Viral Protein U (Vpu) in HIV-1 Infection and Pathogenesis. Viruses 2021, 13, 1466. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Kremmer, E.; Mueller-Lantzsch, N. Evidence for the Association of Nef Protein with HIV-2 Virions. Virus Res. 1996, 46, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Pandori, M.W.; Fitch, N.J.; Craig, H.M.; Richman, D.D.; Spina, C.A.; Guatelli, J.C. Producer-Cell Modification of Human Immunodeficiency Virus Type 1: Nef Is a Virion Protein. J. Virol. 1996, 70, 4283–4290. [Google Scholar] [CrossRef]
- Welker, R.; Kottler, H.; Kalbitzer, H.R.; Kräusslich, H.G. Human Immunodeficiency Virus Type 1 Nef Protein Is Incorporated into Virus Particles and Specifically Cleaved by the Viral Proteinase. Virology 1996, 219, 228–236. [Google Scholar] [CrossRef]
- Schwartz, O.; Maréchal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of Major Histocompatibility Complex Class I Molecules Is Induced by the HIV-1 Nef Protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef Protein Protects Infected Primary Cells against Killing by Cytotoxic T Lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 from Virion Incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Kondo, E.; Mammano, F.; Cohen, E.A.; Göttlinger, H.G. The P6gag Domain of Human Immunodeficiency Virus Type 1 Is Sufficient for the Incorporation of Vpr into Heterologous Viral Particles. J. Virol. 1995, 69, 2759–2764. [Google Scholar] [CrossRef]
- Kondo, E.; Göttlinger, H.G. A Conserved LXXLF Sequence Is the Major Determinant in P6gag Required for the Incorporation of Human Immunodeficiency Virus Type 1 Vpr. J. Virol. 1996, 70, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.L.; Spearman, P.; Ratner, L. Human Immunodeficiency Virus Type 1 Viral Protein R Localization in Infected Cells and Virions. J. Virol. 1993, 67, 6542–6550. [Google Scholar] [CrossRef] [PubMed]
- Paxton, W.; Connor, R.I.; Landau, N.R. Incorporation of Vpr into Human Immunodeficiency Virus Type 1 Virions: Requirement for the P6 Region of Gag and Mutational Analysis. J. Virol. 1993, 67, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Orenstein, J.M.; Martin, M.A.; Freed, E.O. p6Gag Is Required for Particle Production from Full-Length Human Immunodeficiency Virus Type 1 Molecular Clones Expressing Protease. J. Virol. 1995, 69, 6810–6818. [Google Scholar] [CrossRef]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr Is Required for Efficient Replication of Human Immunodeficiency Virus Type-1 in Mononuclear Phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef]
- Goh, W.C.; Rogel, M.E.; Matthew Kinsey, C.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr Increases Viral Expression by Manipulation of the Cell Cycle: A Mechanism for Selection of Vpr in Vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef]
- Mashiba, M.; Collins, D.R.; Terry, V.H.; Collins, K.L. Vpr Overcomes Macrophage-Specific Restriction of HIV-1 Env Expression and Virion Production. Cell Host Microbe 2015, 17, 414. [Google Scholar] [CrossRef]
- Vermeire, J.; Roesch, F.; Sauter, D.; Rua, R.; Hotter, D.; Van Nuffel, A.; Vanderstraeten, H.; Naessens, E.; Iannucci, V.; Landi, A.; et al. HIV Triggers a cGAS-Dependent, Vpu- and Vpr-Regulated Type I Interferon Response in CD4+ T Cells. Cell Rep. 2016, 17, 413–424. [Google Scholar] [CrossRef]
- Yan, J.; Shun, M.C.; Hao, C.; Zhang, Y.; Qian, J.; Hrecka, K.; Delucia, M.; Monnie, C.; Ahn, J.; Skowronski, J. Hiv-1 Vpr Reprograms Clr4dcaf1 E3 Ubiquitin Ligase to Antagonize Exonuclease 1-Mediated Restriction of Hiv-1 Infection. mBio 2018, 9, e01732-18. [Google Scholar] [CrossRef]
- Yan, J.; Shun, M.C.; Zhang, Y.; Hao, C.; Skowronski, J. HIV-1 Vpr Counteracts HLTF-Mediated Restriction of HIV-1 Infection in T Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9568–9577. [Google Scholar] [CrossRef]
- Dupont, L.; Bloor, S.; Williamson, J.C.; Cuesta, S.M.; Shah, R.; Teixeira-Silva, A.; Naamati, A.; Greenwood, E.J.D.; Sarafianos, S.G.; Matheson, N.J.; et al. The SMC5/6 Complex Compacts and Silences Unintegrated HIV-1 DNA and Is Antagonized by Vpr. Cell Host Microbe 2021, 29, 792–805.e6. [Google Scholar] [CrossRef] [PubMed]
- Poon, B.; Chen, I.S.Y. Human Immunodeficiency Virus Type 1 (HIV-1) Vpr Enhances Expression from Unintegrated HIV-1 DNA. J. Virol. 2003, 77, 3962–3972. [Google Scholar] [CrossRef] [PubMed]
- Geis, F.K.; Goff, S.P. Unintegrated HIV-1 DNAs Are Loaded with Core and Linker Histones and Transcriptionally Silenced. Proc. Natl. Acad. Sci. USA 2019, 116, 23735–23742. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.P. Silencing of Unintegrated Retroviral DNAs. Viruses 2021, 13, 2248. [Google Scholar] [CrossRef]
- Felzien, L.K.; Woffendin, C.; Hottiger, M.O.; Subbramanian, R.A.; Cohen, E.A.; Nabel, G.J. HIV Transcriptional Activation by the Accessory Protein, Vpr, Is Mediated by the P300 Co-Activator. Proc. Natl. Acad. Sci. USA 1998, 95, 5281–5286. [Google Scholar] [CrossRef]
- Forget, J.; Yao, X.J.; Mercier, J.; Cohen, É.A. Human Immunodeficiency Virus Type 1 Vpr Protein Transactivation Function: Mechanism and Identification of Domains Involved. J. Mol. Biol. 1998, 284, 915–923. [Google Scholar] [CrossRef]
- Zhang, F.; Bieniasz, P.D. HIV-1 VPR Induces Cell Cycle Arrest and Enhances Viral Gene Expression by Depleting CCDC137. eLife 2020, 9, e55806. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight Control of Gene Expression in Mammalian Cells by Tetracycline-Responsive Promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef]
- Wanaguru, M.; Bishop, K.N. HIV-1 Gag Recruits Oligomeric Vpr via Two Binding Sites in P6, but Both Mature P6 and Vpr Are Rapidly Lost upon Target Cell Entry. J. Virol. 2021, 95, JVI0055421. [Google Scholar] [CrossRef]
- Szymczak, A.L.; Workman, C.J.; Wang, Y.; Vignali, K.M.; Dilioglou, S.; Vanin, E.F.; Vignali, D.A.A. Correction of Multi-Gene Deficiency in vivo Using a Single “self-Cleaving” 2A Peptide-Based Retroviral Vector. Nat. Biotechnol. 2004, 22, 589–594. [Google Scholar] [CrossRef]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpr Usurps Cul4-DDB1[VprBP] E3 Ubiquitin Ligase to Modulate Cell Cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Nara, P.L.; Hatch, W.C.; Dunlop, N.M.; Robey, W.G.; Arthur, L.O.; Gonda, M.A.; Fischinger, P.J. Simple, Rapid, Quantitative, Syncytium-Forming Microassay for the Detection of Human Immunodeficiency Virus Neutralizing Antibody. AIDS Res. Hum. Retroviruses 1987, 3, 283–302. [Google Scholar] [CrossRef]
- Nara, P.L.; Fischinger, P.J. Quantitative Infectivity Assay for HIV-1 and -2. Nature 1988, 332, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Foley, G.E.; Lazarus, H.; Farber, S.; Uzman, B.G.; Boone, B.A.; McCarthy, R.E. Continuous Culture of Human Lymphoblasts from Peripheral Blood of a Child with Acute Leukemia. Cancer 1965, 18, 522–529. [Google Scholar] [CrossRef]
- Pear, W.S.; Nolan, G.P.; Scott, M.L.; Baltimore, D. Production of High-Titer Helper-Free Retroviruses by Transient Transfection. Proc. Natl. Acad. Sci. USA 1993, 90, 8392–8396. [Google Scholar] [CrossRef]
- Janardhan, A.; Swigut, T.; Hill, B.; Myers, M.P.; Skowronski, J. HIV-1 Nef Binds the DOCK2-ELMO1 Complex to Activate Rac and Inhibit Lymphocyte Chemotaxis. PLoS Biol. 2004, 2, e6. [Google Scholar] [CrossRef]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpx Accessory Factor Targets VprBP/DCAF1 Substrate Adaptor for Cullin 4 E3 Ubiquitin Ligase to Enable Macrophage Infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Shun, M.C.; Kaur, S.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. HIV-1 and HIV-2 Exhibit Divergent Interactions with HLTF and UNG2 DNA Repair Proteins. Proc. Natl. Acad. Sci. USA 2016, 113, 201605023–201605030. [Google Scholar] [CrossRef]
- Field, J.; Nikawa, J.; Broek, D.; MacDonald, B.; Rodgers, L.; Wilson, I.A.; Lerner, R.A.; Wigler, M. Purification of a RAS-Responsive Adenylyl Cyclase Complex from Saccharomyces Cerevisiae by Use of an Epitope Addition Method. Mol. Cell. Biol. 1988, 8, 2159–2165. [Google Scholar] [CrossRef]
- Chesebro, B.; Wehrly, K.; Nishio, J.; Perryman, S. Macrophage-Tropic Human Immunodeficiency Virus Isolates from Different Patients Exhibit Unusual V3 Envelope Sequence Homogeneity in Comparison with T-Cell-Tropic Isolates: Definition of Critical Amino Acids Involved in Cell Tropism. J. Virol. 1992, 66, 6547–6554. [Google Scholar] [CrossRef]
- Khan, H.; Sumner, R.P.; Rasaiyaah, J.; Tan, C.P.; Rodriguez-Plata, M.T.; Van Tulleken, C.; Fink, D.; Zuliani-Alvarez, L.; Thorne, L.; Stirling, D.; et al. HIV-1 Vpr Antagonizes Innate Immune Activation by Targeting Karyopherin-Mediated NF-κB/IRF3 Nuclear Transport. eLife 2020, 9, e60821. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, M.C.; Ramnani, B.; Chen, T.; Disbennett, W.M.; Lubow, J.; Welch, J.D.; Collins, K.L. HIV-1 Vpr Combats the PU.1-Driven Antiviral Response in Primary Human Macrophages. Nat. Commun. 2024, 15, 5514. [Google Scholar] [CrossRef] [PubMed]
- Kibe, A.; Buck, S.; Gribling-Burrer, A.-S.; Gilmer, O.; Bohn, P.; Koch, T.; Mireisz, C.N.-M.; Schlosser, A.; Erhard, F.; Smyth, R.P.; et al. The Translational Landscape of HIV-1 Infected Cells Reveals Key Gene Regulatory Principles. Nat. Struct. Mol. Biol. 2025, 32, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Lapek, J.D.; Lewinski, M.K.; Wozniak, J.M.; Guatelli, J.; Gonzalez, D.J. Quantitative Temporal Viromics of an Inducible HIV-1 Model Yields Insight to Global Host Targets and Phospho-Dynamics Associated with Protein Vpr. Mol. Cell. Proteomics 2017, 16, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Tessmer, U.; Schubert, U.; Kräusslich, H.-G. Human Immunodeficiency Virus Type 1 Vpr Protein Is Incorporated into the Virion in Significantly Smaller Amounts than Gag and Is Phosphorylated in Infected Cells. J. Virol. 2000, 74, 10341–10348. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Belaïdouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. HIV1 Vpr Arrests the Cell Cycle by Recruiting DCAF1/VprBP, a Receptor of the Cul4-DDB1 Ubiquitin Ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Hakata, Y.; Landau, N.R. HIV-1 Vpr Function Is Mediated by Interaction with the Damage-Specific DNA-Binding Protein DDB1. Proc. Natl. Acad. Sci. USA 2007, 104, 4130–4135. [Google Scholar] [CrossRef]
- Forouzanfar, F.; Ali, S.; Wallet, C.; De Rovere, M.; Ducloy, C.; El Mekdad, H.; El Maassarani, M.; Aït-Ammar, A.; Van Assche, J.; Boutant, E.; et al. HIV-1 Vpr Mediates the Depletion of the Cellular Repressor CTIP2 to Counteract Viral Gene Silencing. Sci. Rep. 2019, 9, 13154. [Google Scholar] [CrossRef]
- Romani, B.; Baygloo, N.S.; Hamidi-Fard, M.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr Protein Induces Proteasomal Degradation of Chromatin-Associated Class I HDACs to Overcome Latent Infection of Macrophages. J. Biol. Chem. 2016, 291, 2696–2711. [Google Scholar] [CrossRef]
- Zhou, X.; Monnie, C.; DeLucia, M.; Ahn, J. HIV-1 Vpr Activates Host CRL4-DCAF1 E3 Ligase to Degrade Histone Deacetylase SIRT7. Virol. J. 2021, 18, 48. [Google Scholar] [CrossRef]
- Trinité, B.; Ohlson, E.C.; Voznesensky, I.; Rana, S.P.; Chan, C.N.; Mahajan, S.; Alster, J.; Burke, S.A.; Wodarz, D.; Levy, D.N. An HIV-1 Replication Pathway Utilizing Reverse Transcription Products That Fail to Integrate. J. Virol. 2013, 87, 12701–12720. [Google Scholar] [CrossRef] [PubMed]
- Van Nuffel, A.; Impens, F.; Baeyens, A.; Vanhee, S.; Witkowski, W.; Vandermarliere, E.; Vanderstraeten, H.; Naessens, E.; Vanlandeghem, K.; Vermaut, S.; et al. Vpr Content of HIV-1 Virions Determines Infection of Resting Peripheral Blood CD4+ Lymphocytes. Retrovirology 2013, 10, P92. [Google Scholar] [CrossRef]
- Guyader, M.; Emerman, M.; Sonigo, P.; Clavel, F.; Montagnier, L.; Alizon, M. Genome Organization and Transactivation of the Human Immunodeficiency Virus Type 2. Nature 1987, 326, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, V.M.; Dapolito, G.; Goeken, R.; Campbell, B.J. Phylogeny and Natural History of the Primate Lentiviruses, SIV and HIV. Curr. Opin. Genet. Dev. 1995, 5, 798–806. [Google Scholar] [CrossRef]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.M.; Morel, M.; Lahouassa, H.; Leduc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV Viral Protein X Counteracts HUSH Repressor Complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef]
- Henderson, L.E.; Sowder, R.C.; Copeland, T.D.; Benveniste, R.E.; Oroszlan, S. Isolation and Characterization of a Novel Protein (X-ORF Product) from SIV and HIV-2. Science 1988, 241, 199–201. [Google Scholar] [CrossRef]
- Pancio, H.A.; Ratner, L. Human Immunodeficiency Virus Type 2 Vpx-Gag Interaction. J. Virol. 1998, 72, 5271–5275. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate Immunodeficiency Virus Proteins Vpx and Vpr Counteract Transcriptional Repression of Proviruses by the HUSH Complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enya, B.; Skowronski, J. De Novo Expressed Vpr Stimulates HIV-1 Replication in T Cells. Viruses 2025, 17, 958. https://doi.org/10.3390/v17070958
Enya B, Skowronski J. De Novo Expressed Vpr Stimulates HIV-1 Replication in T Cells. Viruses. 2025; 17(7):958. https://doi.org/10.3390/v17070958
Chicago/Turabian StyleEnya, Blessing, and Jacek Skowronski. 2025. "De Novo Expressed Vpr Stimulates HIV-1 Replication in T Cells" Viruses 17, no. 7: 958. https://doi.org/10.3390/v17070958
APA StyleEnya, B., & Skowronski, J. (2025). De Novo Expressed Vpr Stimulates HIV-1 Replication in T Cells. Viruses, 17(7), 958. https://doi.org/10.3390/v17070958