

Regulation and Deregulation of Viral Gene Expression During High-Risk HPV Infection

Abstract
1. Introduction
2. Control of Gene Expression During the Productive Life Cycle
2.1. Basal Cells as a Reservoir of Infection
2.2. Genome Amplification and Virus Production
3. Deregulation of Viral Gene Expression
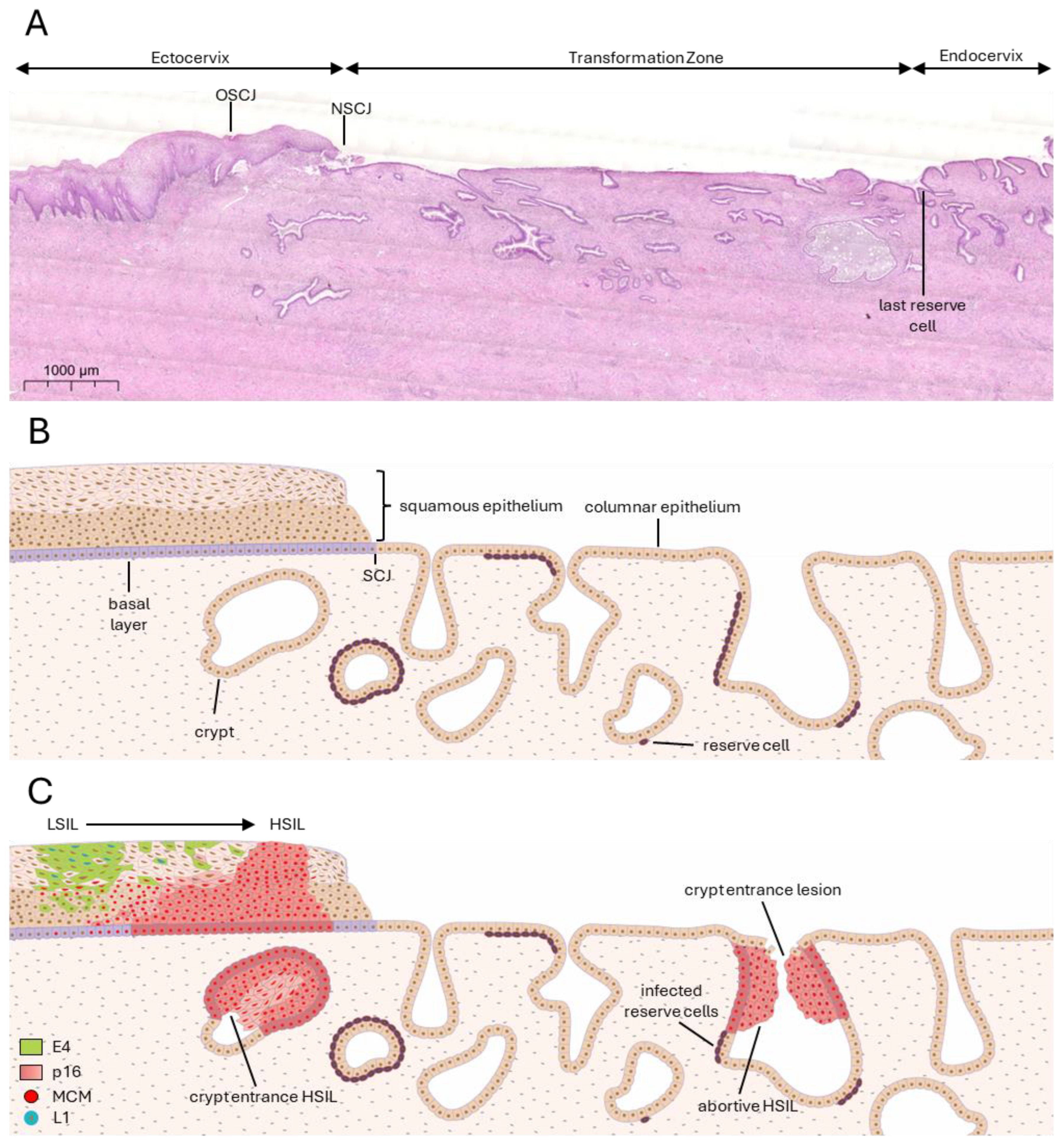
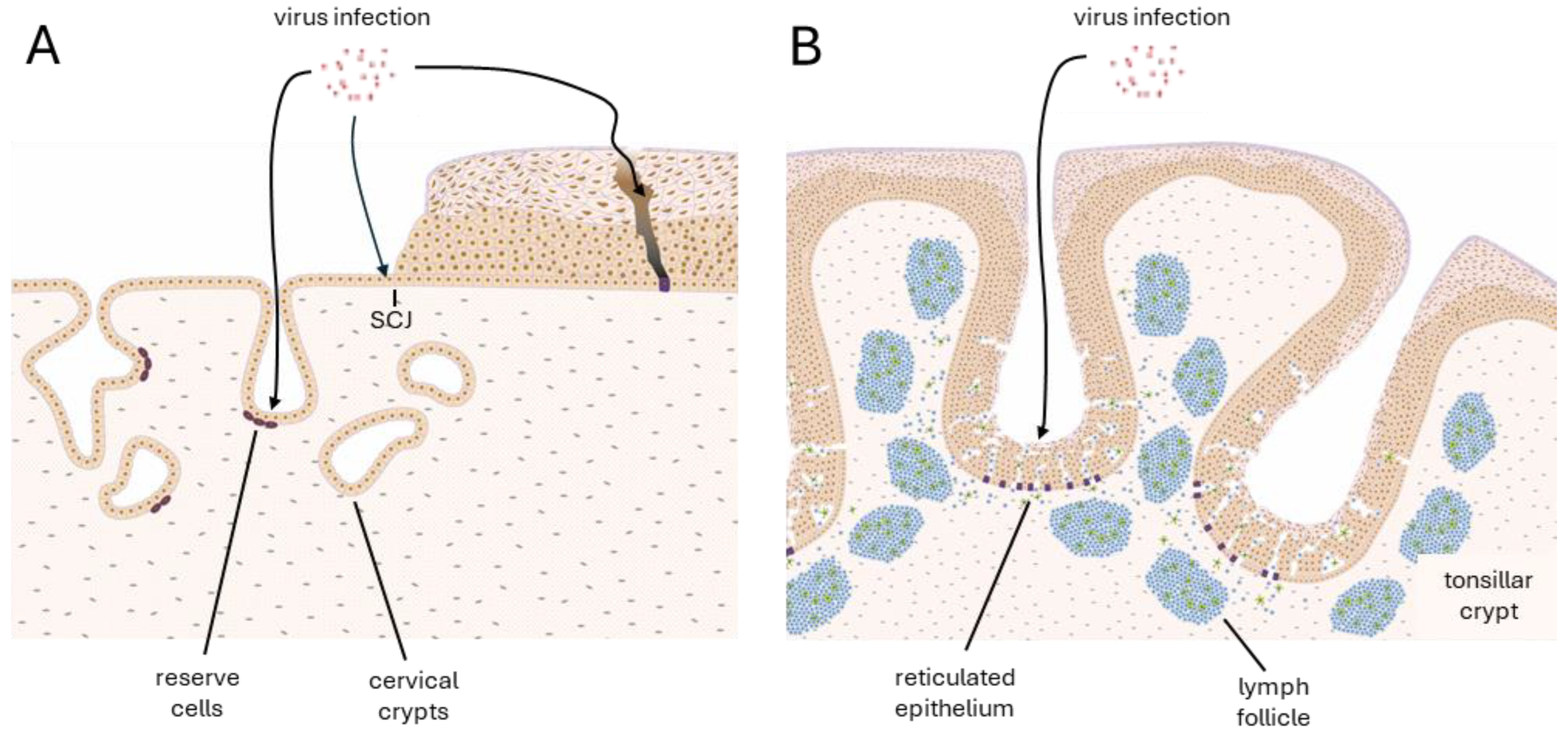
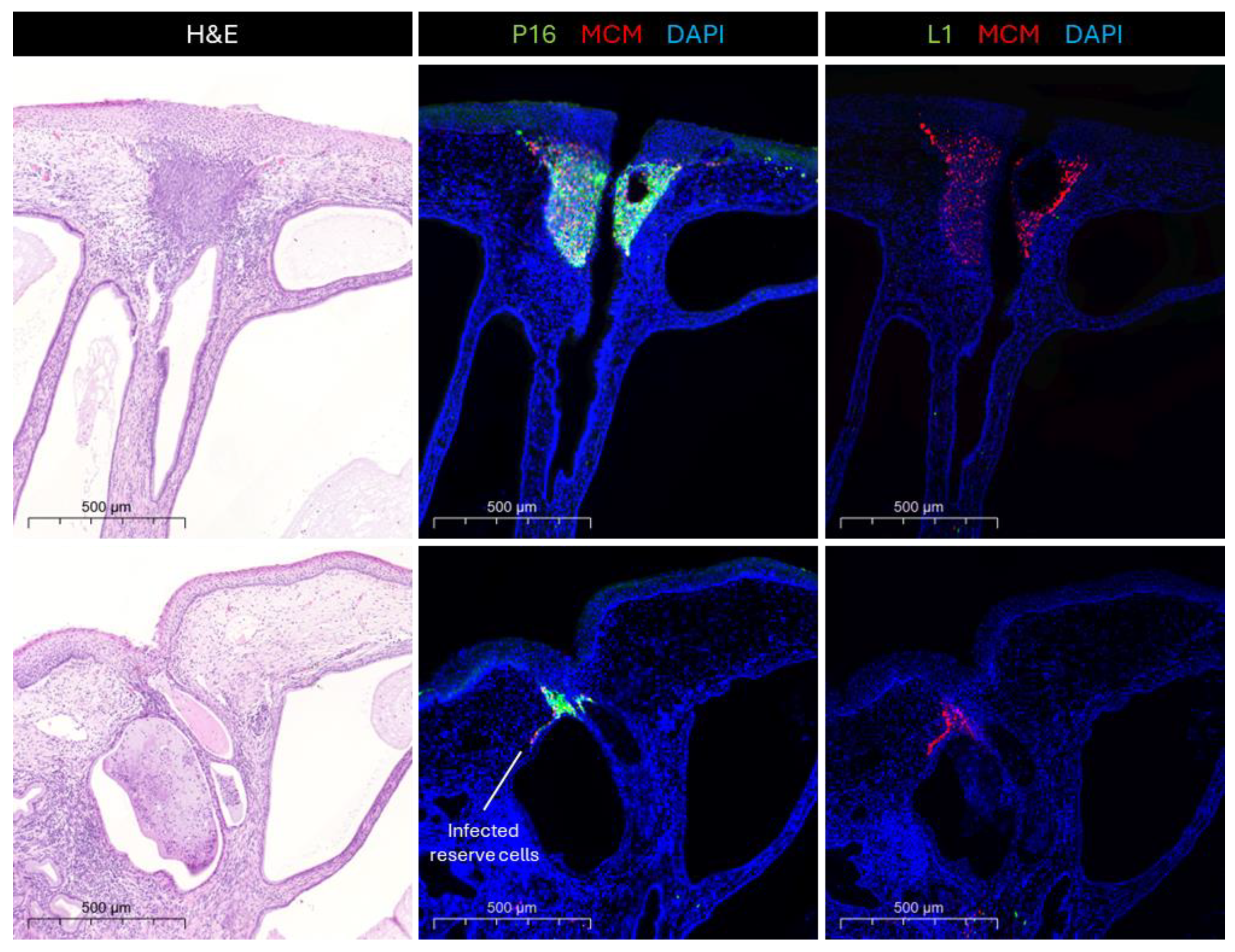
3.1. Deregulation Occurs at Specific Epithelial Sites—Hotspots
3.2. Deregulation and Why It Happens
3.3. Consequences of Deregulation
4. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muñoz, N.; Bosch, F.X.; De Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- IARC. Human Papillomaviruses; World Health Organization: Geneva, Switzerland, 2007; Volume 90. [Google Scholar]
- Marsh, M. Original site of cervical carcinoma: Topographical relationship of carcinoma of the cervix to the external os and to the squamocolumnar junction. Obstet. Gynecol. 1956, 7, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Regauer, S.; Reich, O. The origin of Human Papillomavirus (HPV)—Induced cervical squamous cancer. Curr. Opin. Virol. 2021, 51, 111–118. [Google Scholar] [CrossRef]
- Reich, O.; Regauer, S.; McCluggage, W.; Bergeron, C.; Redman, C. Defining the cervical transformation zone and squamocolumnar junction: Can we reach a common colposcopic and histologic definition? Int. J. Gynecol. Pathol. 2017, 36, 517–522. [Google Scholar] [CrossRef]
- Mcnairn, A.J.; Guasch, G. Epithelial transition zones: Merging microenvironments, niches, and cellular transformation. Eur. J. Dermatol. 2011, 21, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Huang, L.-H.; Chen, T.-M. Differential effects of progestins and estrogens on long control regions of human papillomavirus types 16 and 18. Biochem. Biophys. Res. Commun. 1996, 224, 651–659. [Google Scholar] [CrossRef]
- Accardi, R.; Rubino, R.; Scalise, M.; Gheit, T.; Shahzad, N.; Thomas, M.; Banks, L.; Indiveri, C.; Sylla, B.S.; Cardone, R.A. E6 and E7 from human papillomavirus type 16 cooperate to target the PDZ protein Na/H exchange regulatory factor 1. J. Virol. 2011, 85, 8208–8216. [Google Scholar] [CrossRef]
- Thomas, M.; Pim, D.; Banks, L. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 1999, 18, 7690–7700. [Google Scholar] [CrossRef]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef]
- Aiyenuro, A.; Griffin, H.; Schichl, K.; Omar, T.; Ordi, J.; Kelly, H.; Walker, C.; Del Pino, M.; Desai, K.; de Sanjosé, S. Role of Reserve Cells in Metaplasia and the Development of HPV-Associated High-Grade Squamous Intraepithelial Lesion (HSIL) at the Cervical Transformation Zone. Lab. Investig. 2025, 105, 104166. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, H. The so-called “reserve cells “of the human cervical. Arch. Gynäkol. 1975, 218, 205–217. [Google Scholar] [CrossRef]
- Martens, J.E.; Arends, J.; Van Der Linden, P.J.; De Boer, B.A.; Helmerhorst, T.J. Cytokeratin 17 and p63 are markers of the HPV target cell, the cervical stem cell. Anticancer Res. 2004, 24, 771–776. [Google Scholar]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Doorbar, J. The human Papillomavirus twilight zone–Latency, immune control and subclinical infection. Tumour Virus Res. 2023, 16, 200268. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Egawa, N.; Zheng, K.; Doorbar, J. How can HPV E6 manipulate host cell differentiation process to maintain the reservoir of infection. Tumour Virus Res. 2025, 19, 200313. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Kyo, S.; Klumpp, D.J.; Inoue, M.; Kanaya, T.; Laimins, L.A. Expression of AP1 during cellular differentiation determines human papillomavirus E6/E7 expression in stratified epithelial cells. J. Gen. Virol. 1997, 78, 401–411. [Google Scholar] [CrossRef]
- Groves, I.; Knight, E.; Ang, Q.; Scarpini, C.; Coleman, N. HPV16 oncogene expression levels during early cervical carcinogenesis are determined by the balance of epigenetic chromatin modifications at the integrated virus genome. Oncogene 2016, 35, 4773–4786. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Holleywood, C.; Libert, D.; Griffin, H.; Mahmood, R.; Isaacson, E.; Doorbar, J. Modulation of basal cell fate during productive and transforming HPV-16 infection is mediated by progressive E6-driven depletion of notch. J. Pathol. 2017, 242, 448–462. [Google Scholar] [CrossRef]
- Weijzen, S.; Zlobin, A.; Braid, M.; Miele, L.; Kast, W.M. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J. Cell. Physiol. 2003, 194, 356–362. [Google Scholar] [CrossRef]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, 7–15. [Google Scholar] [CrossRef]
- Li, J.-J.; Rhim, J.S.; Schlegel, R.; Vousden, K.H.; Colburn, N.H. Expression of dominant negative Jun inhibits elevated AP-1 and NF-κB transactivation and suppresses anchorage independent growth of HPV immortalized human keratinocytes. Oncogene 1998, 16, 2711–2721. [Google Scholar] [CrossRef]
- Bauknecht, T.; Angel, P.; Royer, H.; Zur Hausen, H. Identification of a negative regulatory domain in the human papillomavirus type 18 promoter: Interaction with the transcriptional repressor YY1. EMBO J. 1992, 11, 4607–4617. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Tan, S.-H.; Tan, C.-H.; Bernard, H.-U. YY1 represses human papillomavirus type 16 transcription by quenching AP-1 activity. J. Virol. 1996, 70, 6529–6539. [Google Scholar] [CrossRef] [PubMed]
- Warowicka, A.; Broniarczyk, J.; Węglewska, M.; Kwaśniewski, W.; Goździcka-Józefiak, A. Dual role of YY1 in HPV life cycle and cervical cancer development. Int. J. Mol. Sci. 2022, 23, 3453. [Google Scholar] [CrossRef]
- Eyermann, C.E.; Chen, X.; Somuncu, O.S.; Li, J.; Joukov, A.N.; Chen, J.; Alexandrova, E.M. ΔNp63 regulates homeostasis, stemness, and suppression of inflammation in the adult epidermis. J. Investig. Dermatol. 2024, 144, 73–83.e10. [Google Scholar] [CrossRef]
- Eldakhakhny, S.; Zhou, Q.; Crosbie, E.J.; Sayan, B.S. Human papillomavirus E7 induces p63 expression to modulate DNA damage response. Cell Death Dis. 2018, 9, 127. [Google Scholar] [CrossRef]
- Dooley, K.E.; Warburton, A.; McBride, A.A. Tandemly integrated HPV16 can form a Brd4-dependent super-enhancer-like element that drives transcription of viral oncogenes. MBio 2016, 7. [Google Scholar] [CrossRef]
- Gauson, E.J.; Wang, X.; Dornan, E.S.; Herzyk, P.; Bristol, M.; Morgan, I.M. Failure to interact with Brd4 alters the ability of HPV16 E2 to regulate host genome expression and cellular movement. Virus Res. 2016, 211, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Schweiger, M.-R.; Martinez-Noel, G.; Zheng, L.; Smith, J.A.; Harper, J.W.; Howley, P.M. Brd4 regulation of papillomavirus protein E2 stability. J. Virol. 2009, 83, 8683–8692. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-S.; Tam, J.K.; Wang, A.-F.; Hegde, R.S. The structural basis of DNA target discrimination by papillomavirus E2 proteins. J. Biol. Chem. 2000, 275, 31245–31254. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef]
- Dreer, M.; Fertey, J.; van de Poel, S.; Straub, E.; Madlung, J.; Macek, B.; Iftner, T.; Stubenrauch, F. Interaction of NCOR/SMRT repressor complexes with papillomavirus E8^ E2C proteins inhibits viral replication. PLoS Pathog. 2016, 12, e1005556. [Google Scholar] [CrossRef]
- Dreer, M.; Van De Poel, S.; Stubenrauch, F. Control of viral replication and transcription by the papillomavirus E8^ E2 protein. Virus Res. 2017, 231, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Hummel, M.; Iftner, T.; Laimins, L.A. The E8^ E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 2000, 74, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Yugawa, T.; Handa, K.; Narisawa-Saito, M.; Ohno, S.-i.; Fujita, M.; Kiyono, T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell. Biol. 2007, 27, 3732–3742. [Google Scholar] [CrossRef]
- Saunders-Wood, T.; Egawa, N.; Zheng, K.; Giaretta, A.; Griffin, H.M.; Doorbar, J. Role of E6 in maintaining the basal cell reservoir during productive papillomavirus infection. J. Virol. 2022, 96, e01181-01121. [Google Scholar] [CrossRef]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Münger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef]
- Lechler, T.; Fuchs, E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005, 437, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Zwolinska, K.; Keiffer, T.; Scott, R.S.; Sapp, M. Human papillomavirus genome copy number is maintained by S-phase amplification, genome loss to the cytosol during mitosis, and degradation in G1 phase. J. Virol. 2023, 97, e01879-01822. [Google Scholar] [CrossRef]
- Kirk, A.; Graham, S.V. The human papillomavirus late life cycle and links to keratinocyte differentiation. J. Med. Virol. 2024, 96, e29461. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef]
- Pentland, I.; Campos-León, K.; Cotic, M.; Davies, K.-J.; Wood, C.D.; Groves, I.J.; Burley, M.; Coleman, N.; Stockton, J.D.; Noyvert, B. Disruption of CTCF-YY1–dependent looping of the human papillomavirus genome activates differentiation-induced viral oncogene transcription. PLoS Biol. 2018, 16, e2005752. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.-M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef]
- Chumduri, C.; Gurumurthy, R.K.; Berger, H.; Dietrich, O.; Kumar, N.; Koster, S.; Brinkmann, V.; Hoffmann, K.; Drabkina, M.; Arampatzi, P. Opposing Wnt signals regulate cervical squamocolumnar homeostasis and emergence of metaplasia. Nat. Cell Biol. 2021, 23, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Vargas, S.O.; Yamamoto, Y.; Howitt, B.E.; Nucci, M.R.; Hornick, J.L.; Mckeon, F.D.; Xian, W.; Crum, C.P. A novel blueprint for ‘top down’differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. J. Pathol. 2013, 229, 460–468. [Google Scholar] [CrossRef]
- Huang, E.C.; Tomic, M.M.; Hanamornroongruang, S.; Meserve, E.E.; Herfs, M.; Crum, C.P. p16ink4 and cytokeratin 7 immunostaining in predicting HSIL outcome for low-grade squamous intraepithelial lesions: A case series, literature review and commentary. Mod. Pathol. 2016, 29, 1501–1510. [Google Scholar] [CrossRef]
- Deng, H.; Hillpot, E.; Mondal, S.; Khurana, K.K.; Woodworth, C.D. HPV16-immortalized cells from human transformation zone and endocervix are more dysplastic than ectocervical cells in organotypic culture. Sci. Rep. 2018, 8, 15402. [Google Scholar] [CrossRef] [PubMed]
- Moghissi, K.S. The function of the cervix in fertility. Fertil. Steril. 1972, 23, 295–306. [Google Scholar] [CrossRef]
- Bernardo, G.M.; Lozada, K.L.; Miedler, J.D.; Harburg, G.; Hewitt, S.C.; Mosley, J.D.; Godwin, A.K.; Korach, K.S.; Visvader, J.E.; Kaestner, K.H. FOXA1 is an essential determinant of ERα expression and mammary ductal morphogenesis. Development 2010, 137, 2045–2054. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Du, L.; Chen, Z.; Tang, Y.; Chen, L.; Liu, X.; You, L.; Zhang, Y.; Fu, X. Foxa1 mediates eccrine sweat gland development through transcriptional regulation of Na-K-ATPase expression. Braz. J. Med. Biol. Res. 2022, 55, e12149. [Google Scholar] [CrossRef]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol. Oncol. 2010, 117, S5–S10. [Google Scholar] [CrossRef]
- Lyford-Pike, S.; Peng, S.; Young, G.D.; Taube, J.M.; Westra, W.H.; Akpeng, B.; Bruno, T.C.; Richmon, J.D.; Wang, H.; Bishop, J.A. Evidence for a role of the PD-1: PD-L1 pathway in immune resistance of HPV-associated head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Giannini, S.L.; Hubert, P.; Doyen, J.; Boniver, J.; Delvenne, P. Influence of the mucosal epithelium microenvironment on Langerhans cells: Implications for the development of squamous intraepithelial lesions of the cervix. Int. J. Cancer 2002, 97, 654–659. [Google Scholar] [CrossRef]
- Chaiwongkot, A.; Vinokurova, S.; Pientong, C.; Ekalaksananan, T.; Kongyingyoes, B.; Kleebkaow, P.; Chumworathayi, B.; Patarapadungkit, N.; Reuschenbach, M.; von Knebel Doeberitz, M. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int. J. Cancer 2013, 132, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F.; Yaniv, M. The BPV1-E2 trans-acting protein can be either an activator or a repressor of the HPV18 regulatory region. EMBO J. 1987, 6, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Egawa, N.; Zheng, K.; Griffin, H.; Tian, P.; Aiyenuro, A.; Bornstein, J.; Doorbar, J. HPV E6 inhibits E6AP to regulate epithelial homeostasis by modulating keratinocyte differentiation commitment and YAP1 activation. PLoS Pathog. 2023, 19, e1011464. [Google Scholar] [CrossRef]
- Thain, A.; Jenkins, O.; Clarke, A.R.; Gaston, K. CpG methylation directly inhibits binding of the human papillomavirus type 16 E2 protein to specific DNA sequences. J. Virol. 1996, 70, 7233–7235. [Google Scholar] [CrossRef]
- Kim, K.; Garner-Hamrick, P.A.; Fisher, C.; Lee, D.; Lambert, P.F. Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J. Virol. 2003, 77, 12450–12459. [Google Scholar] [CrossRef]
- Vandermark, E.R.; Deluca, K.A.; Gardner, C.R.; Marker, D.F.; Schreiner, C.N.; Strickland, D.A.; Wilton, K.M.; Mondal, S.; Woodworth, C.D. Human papillomavirus type 16 E6 and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB promotes cell growth and immortalization. Virology 2012, 425, 53–60. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV transcription. Clinics 2018, 73, e486s. [Google Scholar] [CrossRef]
- Sichero, L.; Sobrinho, J.S.; Villa, L.L. Identification of novel cellular transcription factors that regulate early promoters of human papillomavirus types 18 and 16. J. Infect. Dis. 2012, 206, 867–874. [Google Scholar] [CrossRef]
- Karpathiou, G.; Da Cruz, V.; Casteillo, F.; Mobarki, M.; Dumollard, J.M.; Chauleur, C.; Forest, F.; Prades, J.M.; Peoc’h, M. FOXA1 in HPV associated carcinomas: Its expression in carcinomas of the head and neck and of the uterine cervix. Exp. Mol. Pathol. 2017, 102, 230–236. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef]
- Kupryjanczyk, J.; Möller, P. Estrogen receptor distribution in the normal and pathologically changed human cervix uteri: An immunohistochemical study with use of monoclonal anti-ER antibody. Int. J. Gynecol. Pathol. 1988, 7, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, B. Functional association of oestrogen receptors with HPV infection in cervical carcinogenesis. Endocr.-Relat. Cancer 2017, 24, R99–R108. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Oncogenic human papillomaviruses. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160273. [Google Scholar] [CrossRef]
- Andersson, S.; Mints, M.; Wilander, E. Results of cytology and high-risk human papillomavirus testing in females with cervical adenocarcinoma in situ. Oncol. Lett. 2013, 6, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Münger, K. Human papillomaviruses and centrosome duplication errors: Modeling the origins of genomic instability. Oncogene 2002, 21, 6241–6248. [Google Scholar] [CrossRef]
- White, A.E.; Livanos, E.M.; Tlsty, T.D. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994, 8, 666–677. [Google Scholar] [CrossRef]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.-o.; Gonzalez, S.; Crum, C.P.; Münger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef]
- Pett, M.R.; Alazawi, W.O.; Roberts, I.; Dowen, S.; Smith, D.I.; Stanley, M.A.; Coleman, N. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res. 2004, 64, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.-M.; Wang, X. Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2011, 1809, 668–677. [Google Scholar] [CrossRef]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulation of microRNA-mRNA target pairs by human papillomavirus 16 oncoproteins. MBio 2017, 8. [Google Scholar] [CrossRef]
- Hu, J.; Liao, D.; Sun, Z.; Ren, W.; Zhao, L.; Fang, Y.; Hu, K.; Yu, H.; Liu, S.; Zhou, L. The HPV16 E6, E7/miR-23b-3p/ICAT signaling axis promotes proliferation, migration, invasion and EMT of cervical cancer cells. Carcinogenesis 2023, 44, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Dürst, M.; Kleinheinz, A.; Hotz, M.; Gissmann, L. The physical state of human papillomavirus type 16 DNA in benign and malignant genital tumours. J. Gen. Virol. 1985, 66, 1515–1522. [Google Scholar] [CrossRef]
- Collins, S.I.; Constandinou-Williams, C.; Wen, K.; Young, L.S.; Roberts, S.; Murray, P.G.; Woodman, C.B. Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: A longitudinal cohort study. Cancer Res. 2009, 69, 3828–3832. [Google Scholar] [CrossRef]
- Lagström, S.; Løvestad, A.H.; Umu, S.U.; Ambur, O.H.; Nygård, M.; Rounge, T.B.; Christiansen, I.K. HPV16 and HPV18 type-specific APOBEC3 and integration profiles in different diagnostic categories of cervical samples. Tumour Virus Res. 2021, 12, 200221. [Google Scholar] [CrossRef]
- Tindle, R.W. Immune evasion in human papillomavirus-associated cervical cancer. Nat. Rev. Cancer 2002, 2, 59–64. [Google Scholar] [CrossRef]
- Roy-Biswas, S.; Hibma, M. The Epithelial Immune Response to Human Papillomavirus Infection. Pathogens 2025, 14, 464. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Münger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- López-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar] [CrossRef]
- Tischer, E.; Mitchell, R.; Hartman, T.; Silva, M.; Gospodarowicz, D.; Fiddes, J.; Abraham, J. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1991, 266, 11947–11954. [Google Scholar] [CrossRef]
- Cullen, A.P.; Reid, R.; Campion, M.; Lörincz, A. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Epithelial Layer | Key Regulatory Mechanisms | Outcomes |
|---|---|---|
| Basal Layer (stem-like cells, site of HPV infection) | Transcriptional Control: Early gene expression (E6/E7) is kept low to prevent immune activation [15,16]. E1 and E2 initiate episomal maintenance and modulate genome partitioning [15]. Epigenetic Regulation: DNA is tightly packed with histones to minimise viral gene expression [17]. Epithelial Homeostasis: E6 and E7 increase longevity of infected cells [18,19]. E6 degrades p53 via E6AP, inhibiting cell delamination [20]. E7 modulates cell cycle progression [15]. Viral Genome Maintenance: The viral genome is maintained as low copy number episome [15,21]. | HPV establishes reservoir that is not detected by the immune system. Viral genomes are copied and passed to daughter cells. |
| Parabasal Layers (early differentiating cells) | Transcriptional Control: Early gene expression can decline. Cell division declines [22]. Epigenetic Regulation: DNA methylation patterns change to allow viral replication [17,23]. Cell Signalling Modulation: HPV alters Notch and Wnt pathways, delaying terminal differentiation [24,25]. | Infected cells continue to undergo cell division, ensuring viral genome replication. |
| Mid-epithelial Layers (late differentiating cells) | Early Gene Expression: Increased E7 expression to maintain replication-competent environment [18]. E5 enhances amplification via EGFR signalling and MAP kinase activation [15]. Late Gene Activation: Expression of viral structural proteins (L1, L2) through alternative splicing, bypassing early polyadenylation sites [15]. Cytoskeletal Remodelling: E4 reorganises keratin network to favour virion assembly [26]. | High levels of viral genome amplification. Initiation of late gene expression. |
| Upper-epithelial Layers (fully differentiated keratinocytes) | Virion Assembly: L2 is recruited to replication foci by E2, then L1 and L2 encapsidate viral genomes [15]. Virus Maturation: Superficial keratinocytes undergo redox state change to facilitate capsid maturation. Disulphide bonding between L1 proteins [15]. Immune Evasion: Terminally differentiated keratinocytes lack immune surveillance, allowing mostly undetected viral shedding [15,27]. | HPV virions are assembled and shed with keratinocytes. The infection remains undetected by the immune system. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schichl, K.; Doorbar, J. Regulation and Deregulation of Viral Gene Expression During High-Risk HPV Infection. Viruses 2025, 17, 937. https://doi.org/10.3390/v17070937
Schichl K, Doorbar J. Regulation and Deregulation of Viral Gene Expression During High-Risk HPV Infection. Viruses. 2025; 17(7):937. https://doi.org/10.3390/v17070937
Chicago/Turabian StyleSchichl, Konstanze, and John Doorbar. 2025. "Regulation and Deregulation of Viral Gene Expression During High-Risk HPV Infection" Viruses 17, no. 7: 937. https://doi.org/10.3390/v17070937
APA StyleSchichl, K., & Doorbar, J. (2025). Regulation and Deregulation of Viral Gene Expression During High-Risk HPV Infection. Viruses, 17(7), 937. https://doi.org/10.3390/v17070937