
Integrated Analysis of the 2022 SARS-CoV-2 Omicron Lineage Replacement Dynamics in Connecticut, US

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Data Sources
2.2.1. Sample Collection and Processing
2.2.2. Individual Metadata
2.2.3. Population Vaccination Trends
2.2.4. Population Variant Trends
2.2.5. Variant Rt and Immunity Estimates
2.3. Analyses
2.3.1. Variant Rt Ratios
2.3.2. Variant Emergence Periods and Logistic Growth Rates
2.3.3. Variant Ct Values over Time and in Periods of Emergence
2.3.4. Mixed Effect Multivariable Logistic Regression Models
2.3.5. Factors Impacting XBB.1.5 Ct Values
2.3.6. Conceptual SIR Model
2.3.7. Statistical Analysis and Data Availability
3. Results
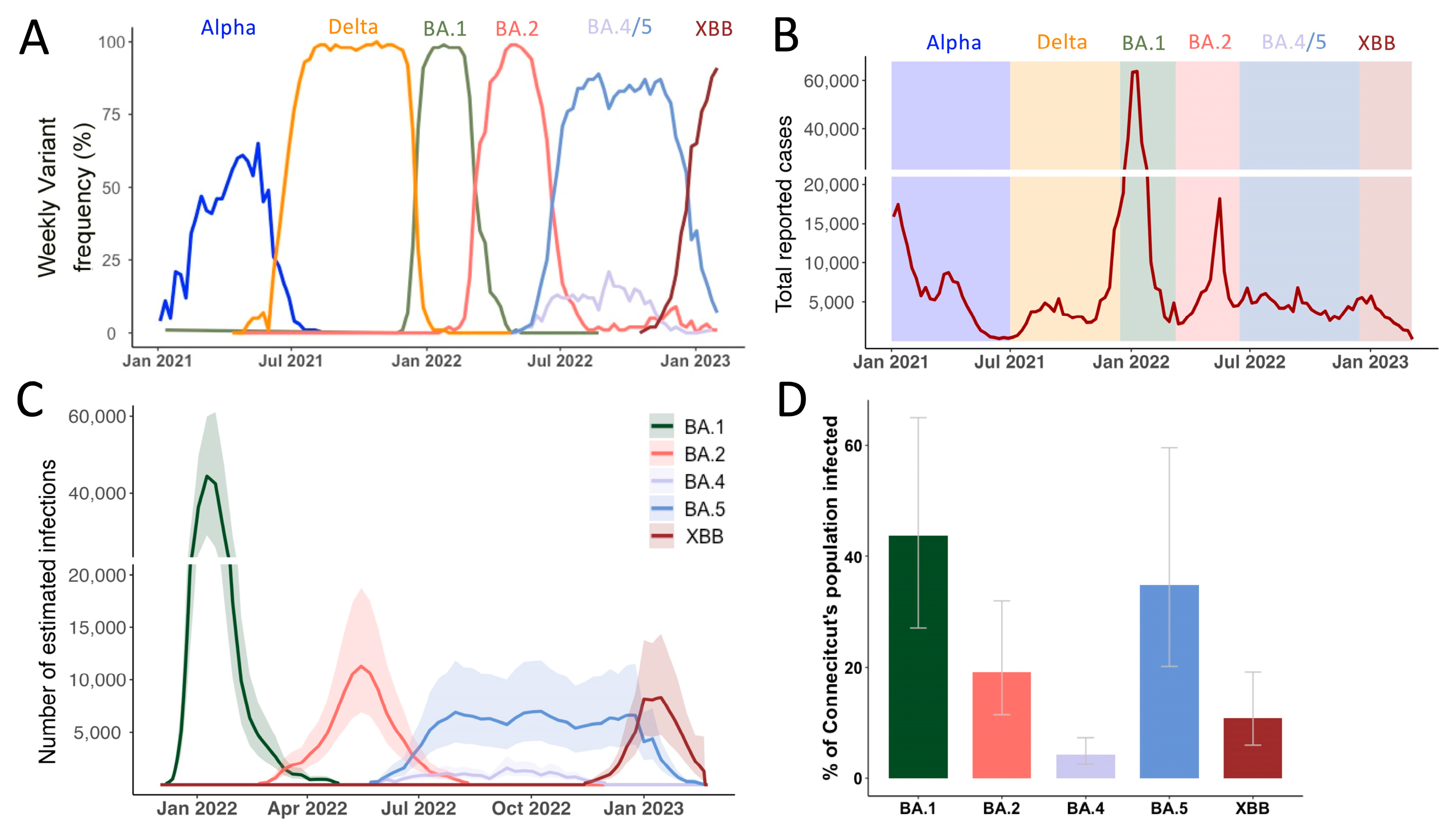
3.1. Continuous Omicron Lineage Replacement Causes High Levels of Community Transmission Throughout 2022
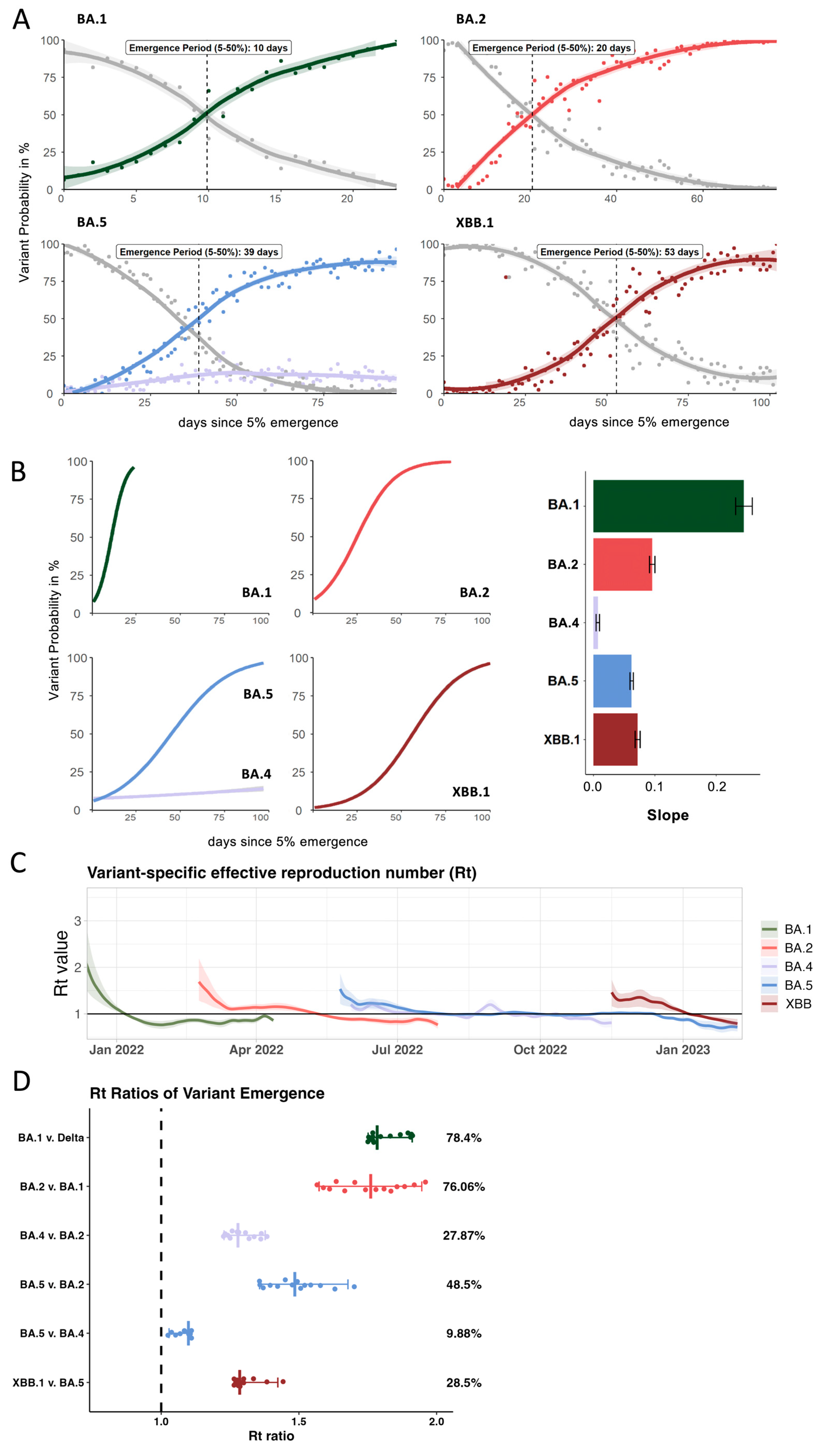
3.2. Growth Advantage of Emerging Omicron Lineages Shrinks Towards the End of 2022
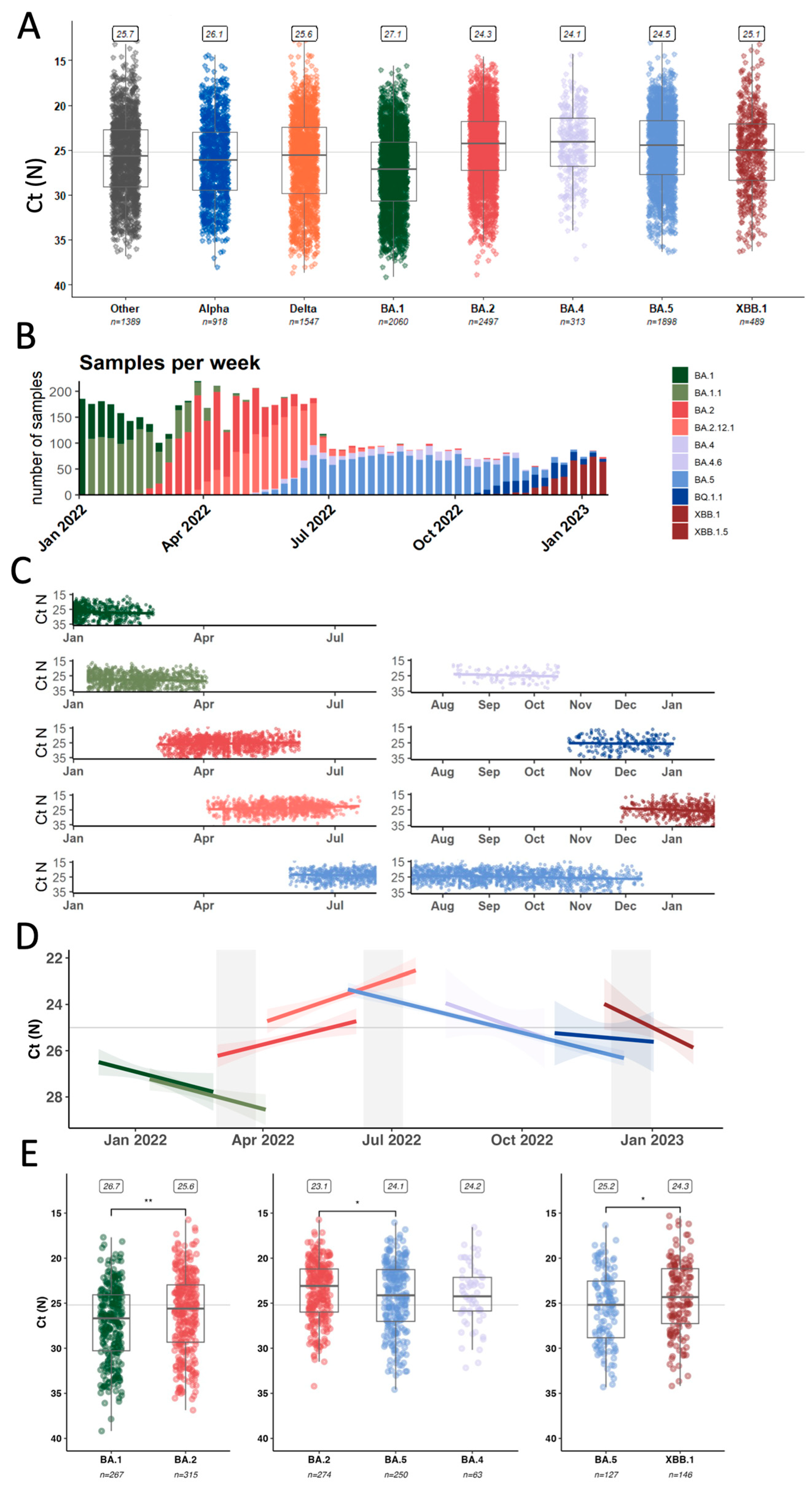
3.3. Average Inter- and Intra-Lineage Viral Copy Numbers Vary over Time Only Partially Explaining Lineage Replacement Patterns
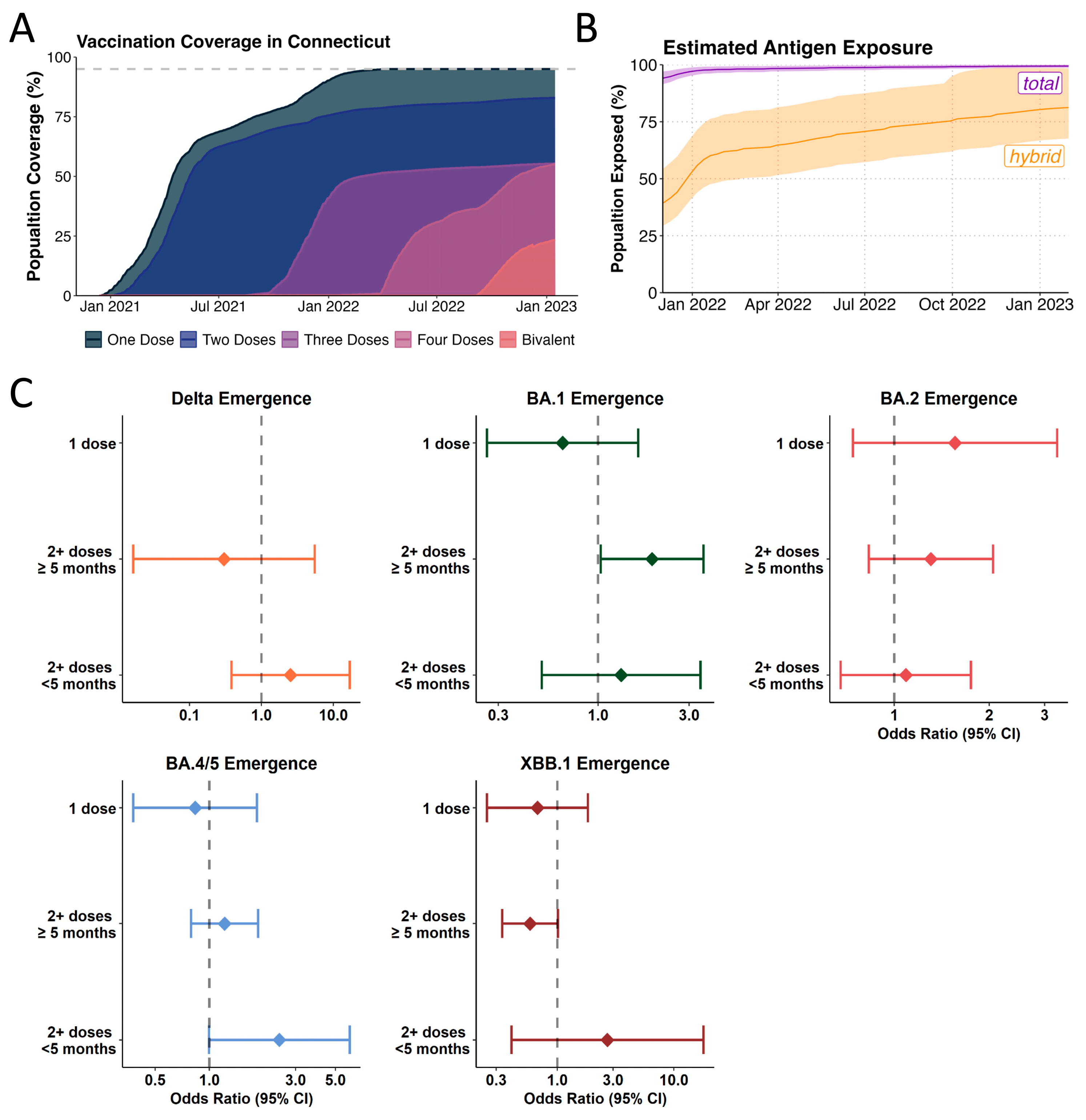
3.4. Likelihood of Incoming Lineages to Cause Breakthrough Infections in Recent Vaccinees Varies Across Different Omicron Lineages
3.5. Omicron Lineage Replacements in a Highly Antigen-Experienced Population
4. Discussion
5. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brito, A.F.; Semenova, E.; Dudas, G.; Hassler, G.W.; Kalinich, C.C.; Kraemer, M.U.G.; Ho, J.; Tegally, H.; Githinji, G.; Agoti, C.N.; et al. Global disparities in SARS-CoV-2 genomic surveillance. Nat. Commun. 2022, 13, 7003. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.; Du Plessis, L.; Peacock, T.P.; Aggarwal, D.; Colquhoun, R.; Carabelli, A.M.; Ellaby, N.; Gallagher, E.; Groves, N.; Jackson, B.; et al. The origins and molecular evolution of SARS-CoV-2 lineage B.1.1.7 in the UK. Virus Evol. 2022, 8, veac080. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Virological.org 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 1 July 2025).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- WHO. COVID-19 Weekly Epidemiological Update—27 April 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---27-april-2021 (accessed on 30 October 2023).
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Tsui, J.L.; Moir, M.; Martin, D.; Brito, A.F.; Giovanetti, M.; Khan, K.; Huber, C.; Bogoch, I.I.; et al. Dispersal patterns and influence of air travel during the global expansion of SARS-CoV-2 variants of concern. Cell 2023, 186, 3277–3290.E16. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes de Jesus, J.; et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- CDC. COVID-19 Vaccination Trends in the United States, National and Jurisdictional|Data. Available online: www.cdc.gov (accessed on 30 March 2023).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, F.; Chitwood, M.H.; Cohen, T.; Pitzer, V.E.; Russi, M.; Swartwood, N.A.; Salomon, J.A.; Menzies, N.A. Changes in Population Immunity Against Infection and Severe Disease from Severe Acute Respiratory Syndrome Coronavirus 2 Omicron Variants in the United States Between December 2021 and November 2022. Clin. Infect. Dis. 2023, 77, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, F.; Chitwood, M.H.; Cohen, T.; Pitzer, V.E.; Russi, M.; Swartwood, N.A.; Salomon, J.A.; Menzies, N.A. Population Immunity to Pre-Omicron and Omicron Severe Acute Respiratory Syndrome Coronavirus 2 Variants in US States and Counties Through 1 December 2021. Clin. Infect. Dis. 2023, 76, e350–e359. [Google Scholar] [CrossRef] [PubMed]
- Chitwood, M.H.; Russi, M.; Gunasekera, K.; Havumaki, J.; Klaassen, F.; Pitzer, V.E.; Salomon, J.A.; Swartwood, N.A.; Warren, J.L.; Weinberger, D.M.; et al. Reconstructing the course of the COVID-19 epidemic over 2020 for US states and counties: Results of a Bayesian evidence synthesis model. PLoS Comput. Biol. 2022, 18, e1010465. [Google Scholar] [CrossRef] [PubMed]
- Nouvellet, P.; Cori, A.; Garske, T.; Blake, I.M.; Dorigatti, I.; Hinsley, W.; Jombart, T.; Mills, H.L.; Nedjati-Gilani, G.; Van Kerkhove, M.D.; et al. A simple approach to measure transmissibility and forecast incidence. Epidemics 2018, 22, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Earnest, R.; Uddin, R.; Matluk, N.; Renzette, N.; Turbett, S.E.; Siddle, K.J.; Loreth, C.; Adams, G.; Tomkins-Tinch, C.H.; Petrone, M.E.; et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. Cell Rep. Med. 2022, 3, 100583. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org (accessed on 5 July 2025).
- Goh, A.X.C.; Chae, S.R.; Chiew, C.J.; Tang, N.; Pang, D.; Lin, C.; Tan, K.B.; Lee, V.J.; Ho, Z.J.M. Characteristics of the omicron XBB subvariant wave in Singapore. Lancet 2023, 401, 1261–1262. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.C.; Shirk, P.; Lambrou, A.S.; Hassell, N.; Zheng, X.Y.; Payne, A.B.; Ali, A.R.; Batra, D.; Caravas, J.; Chau, R.; et al. Genomic Surveillance for SARS-CoV-2 Variants: Circulation of Omicron Lineages—United States, January 2022–May 2023. MMWR Morb. Mortal. Wkly. Rep. 2023, 72, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.; Pham, K.; Klaassen, F.; Chitwood, M.H.; Hahn, A.M.; Redmond, S.; Swartwood, N.A.; Salomon, J.A.; Menzies, N.A.; Cohen, T.; et al. Combining genomic data and infection estimates to characterize the complex dynamics of SARS-CoV-2 Omicron variants in the US. Cell Rep. 2024, 43, 114451. [Google Scholar] [CrossRef] [PubMed]
- Petrone, M.E.; Rothman, J.E.; Breban, M.I.; Ott, I.M.; Russell, A.; Lasek-Nesselquist, E.; Badr, H.; Kelly, K.; Omerza, G.; Renzette, N.; et al. Combining genomic and epidemiological data to compare the transmissibility of SARS-CoV-2 variants Alpha and Iota. Commun. Biol. 2022, 5, 439. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Cobey, S.; Metcalf, C.J.E.; Levine, J.M.; Grenfell, B.T. Predicting pathogen mutual invasibility and co-circulation. Science 2024, 386, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.A.; Kennedy-Shaffer, L.; Kanjilal, S.; Lennon, N.J.; Gabriel, S.B.; Lipsitch, M.; Mina, M.J. Estimating epidemiologic dynamics from cross-sectional viral load distributions. Science 2021, 373, eabh0635. [Google Scholar] [CrossRef] [PubMed]
- Musalkova, D.; Piherova, L.; Kwasny, O.; Dindova, Z.; Stancik, L.; Hartmannova, H.; Slama, O.; Peckova, P.; Pargac, J.; Minarik, G.; et al. Trends in SARS-CoV-2 cycle threshold values in the Czech Republic from April 2020 to April 2022. Sci. Rep. 2023, 13, 6156. [Google Scholar] [CrossRef] [PubMed]
- Lentini, A.; Pereira, A.; Winqvist, O.; Reinius, B. Monitoring of the SARS-CoV-2 Omicron BA.1/BA.2 lineage transition in the Swedish population reveals increased viral RNA levels in BA.2 cases. Med 2022, 3, 636–643.e634. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chaguza, C.; Stamp, J.; Chew, Y.T.; Chen, N.F.G.; Ferguson, D.; Pandya, S.; Kerantzas, N.; Schulz, W.; Yale, S.-C.-G.S.I.; et al. Genome-wide association study between SARS-CoV-2 single nucleotide polymorphisms and virus copies during infections. PLoS Comput. Biol. 2024, 20, e1012469. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.A.; Kissler, S.M.; Fauver, J.R.; Mack, C.; Tai, C.G.; Samant, R.M.; Connolly, S.; Anderson, D.J.; Khullar, G.; MacKay, M.; et al. Quantifying the impact of immune history and variant on SARS-CoV-2 viral kinetics and infection rebound: A retrospective cohort study. eLife 2022, 11, e81849. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.P.; Stramer, S.L.; Stone, M.; Yu, E.A.; Grebe, E.; Notari, E.; Saa, P.; Ferg, R.; Manrique, I.M.; Weil, N.; et al. Population-Weighted Seroprevalence from Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection, Vaccination, and Hybrid Immunity Among US Blood Donations from January to December 2021. Clin. Infect. Dis. 2022, 75, S254–S263. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Opsomer, J.D.; Stone, M.; Benoit, T.; Ferg, R.A.; Stramer, S.L.; Busch, M.P. Updated US Infection- and Vaccine-Induced SARS-CoV-2 Seroprevalence Estimates Based on Blood Donations, July 2020–December 2021. JAMA 2022, 328, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Rossler, A.; Netzl, A.; Knabl, L.; Schafer, H.; Wilks, S.H.; Bante, D.; Falkensammer, B.; Borena, W.; von Laer, D.; Smith, D.J.; et al. BA.2 and BA.5 omicron differ immunologically from both BA.1 omicron and pre-omicron variants. Nat. Commun. 2022, 13, 7701. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, A.Z.; Fouchier, R.A.; Haagmans, B.L. Antigenic evolution of SARS coronavirus 2. Curr. Opin. Virol. 2023, 62, 101349. [Google Scholar] [CrossRef] [PubMed]
- Mykytyn, A.Z.; Rosu, M.E.; Kok, A.; Rissmann, M.; van Amerongen, G.; Geurtsvankessel, C.; de Vries, R.D.; Munnink, B.B.O.; Smith, D.J.; Koopmans, M.P.G.; et al. Antigenic mapping of emerging SARS-CoV-2 omicron variants BM.1.1.1, BQ.1.1, and XBB.1. Lancet Microbe 2023, 4, e294–e295. [Google Scholar] [CrossRef] [PubMed]
- Chaguza, C.; Coppi, A.; Earnest, R.; Ferguson, D.; Kerantzas, N.; Warner, F.; Young, H.P.; Breban, M.I.; Billig, K.; Koch, R.T.; et al. Rapid emergence of SARS-CoV-2 Omicron variant is associated with an infection advantage over Delta in vaccinated persons. Med 2022, 3, 325–334.E4. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Zhang, L.; Rocha, C.; Sidarovich, A.; Kempf, A.; Schulz, S.; Cossmann, A.; Manger, B.; Baier, E.; Tampe, B.; et al. Comparable neutralisation evasion of SARS-CoV-2 omicron subvariants BA.1, BA.2, and BA.3. Lancet Infect. Dis. 2022, 22, 766–767. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.W.; Chia, W.N.; Zhu, F.; Young, B.E.; Chantasrisawad, N.; Hwa, S.H.; Yeoh, A.Y.; Lim, B.L.; Yap, W.C.; Pada, S.; et al. SARS-CoV-2 Omicron variant emerged under immune selection. Nat. Microbiol. 2022, 7, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Kurhade, C.; Zou, J.; Xia, H.; Cai, H.; Yang, Q.; Cutler, M.; Cooper, D.; Muik, A.; Jansen, K.U.; Xie, X.; et al. Neutralization of Omicron BA.1, BA.2, and BA.3 SARS-CoV-2 by 3 doses of BNT162b2 vaccine. Nat. Commun. 2022, 13, 3602. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Wong, L.R.; Arora, P.; Zhang, L.; Rocha, C.; Odle, A.; Nehlmeier, I.; Kempf, A.; Richter, A.; Halwe, N.J.; et al. Omicron subvariant BA.5 efficiently infects lung cells. Nat. Commun. 2023, 14, 3500. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Kempf, A.; Nehlmeier, I.; Schulz, S.R.; Cossmann, A.; Stankov, M.V.; Jack, H.M.; Behrens, G.M.N.; Pohlmann, S.; Hoffmann, M. Augmented neutralisation resistance of emerging omicron subvariants BA.2.12.1, BA.4, and BA.5. Lancet Infect. Dis. 2022, 22, 1117–1118. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.; Stowe, J.; Kirsebom, F.; Toffa, S.; Rickeard, T.; Gallagher, E.; Gower, C.; Kall, M.; Groves, N.; O’Connell, A.M.; et al. Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. N. Engl. J. Med. 2022, 386, 1532–1546. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; St Denis, K.J.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466.E4. [Google Scholar] [CrossRef] [PubMed]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G.; et al. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Schwartz, O. Towards SARS-CoV-2 serotypes? Nat. Rev. Microbiol. 2022, 20, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Schaefer-Babajew, D.; Wang, Z.; Muecksch, F.; Cho, A.; Loewe, M.; Cipolla, M.; Raspe, R.; Johnson, B.; Canis, M.; DaSilva, J.; et al. Antibody feedback regulates immune memory after SARS-CoV-2 mRNA vaccination. Nature 2023, 613, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Muecksch, F.; Wang, Z.; Cho, A.; Gaebler, C.; Ben Tanfous, T.; DaSilva, J.; Bednarski, E.; Ramos, V.; Zong, S.; Johnson, B.; et al. Increased memory B cell potency and breadth after a SARS-CoV-2 mRNA boost. Nature 2022, 607, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Pinto, D.; Walls, A.C.; Liu, Z.; De Marco, A.; Benigni, F.; Zatta, F.; Silacci-Fregni, C.; Bassi, J.; Sprouse, K.R.; et al. Imprinted antibody responses against SARS-CoV-2 Omicron sublineages. Science 2022, 378, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sukhova, K.; Peacock, T.P.; McKay, P.F.; Brown, J.C.; Frise, R.; Baillon, L.; Moshe, M.; Kugathasan, R.; Shattock, R.J.; et al. Omicron breakthrough infections in vaccinated or previously infected hamsters. Proc. Natl. Acad. Sci. USA 2023, 120, e2308655120. [Google Scholar] [CrossRef] [PubMed]
- Andeweg, S.P.; de Gier, B.; Vennema, H.; van Walle, I.; van Maarseveen, N.; Kusters, N.E.; de Melker, H.E.; Hahne, S.J.; van den Hof, S.; Eggink, D.; et al. Higher risk of SARS-CoV-2 Omicron BA.4/5 infection than of BA.2 infection after previous BA.1 infection, the Netherlands, 2 May to 24 July 2022. Euro Surveill. 2023, 28, 2200724. [Google Scholar] [CrossRef] [PubMed]
- Malato, J.; Ribeiro, R.M.; Fernandes, E.; Leite, P.P.; Casaca, P.; Antunes, C.; Fonseca, V.R.; Gomes, M.C.; Graca, L. Stability of hybrid versus vaccine immunity against BA.5 infection over 8 months. Lancet Infect. Dis. 2023, 23, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023, 29, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Springer, D.N.; Medits, I.; Weseslindtner, L.; Stiasny, K.; Aberle, J.H. SARS-CoV-2 neutralising antibody response to bivalent booster after omicron infection. Lancet Microbe 2024, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Nehlmeier, I.; Kempf, A.; Cossmann, A.; Schulz, S.R.; Morillas Ramos, G.; Manthey, L.A.; Jack, H.M.; Behrens, G.M.N.; et al. Profound neutralization evasion and augmented host cell entry are hallmarks of the fast-spreading SARS-CoV-2 lineage XBB.1.5. Cell Mol. Immunol. 2023, 20, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Raharinirina, N.A.; Gubela, N.; Bornigen, D.; Smith, M.R.; Oh, D.Y.; Budt, M.; Mache, C.; Schillings, C.; Fuchs, S.; Durrwald, R.; et al. SARS-CoV-2 evolution on a dynamic immune landscape. Nature 2025, 639, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Meijers, M.; Ruchnewitz, D.; Eberhardt, J.; Luksza, M.; Lassig, M. Population immunity predicts evolutionary trajectories of SARS-CoV-2. Cell 2023, 186, 5151–5164.E13. [Google Scholar] [CrossRef] [PubMed]
- Fryer, H.R.; Golubchik, T.; Hall, M.; Fraser, C.; Hinch, R.; Ferretti, L.; Thomson, L.; Nurtay, A.; Pellis, L.; House, T.; et al. Viral burden is associated with age, vaccination, and viral variant in a population-representative study of SARS-CoV-2 that accounts for time-since-infection-related sampling bias. PLoS Pathog. 2023, 19, e1011461. [Google Scholar] [CrossRef]
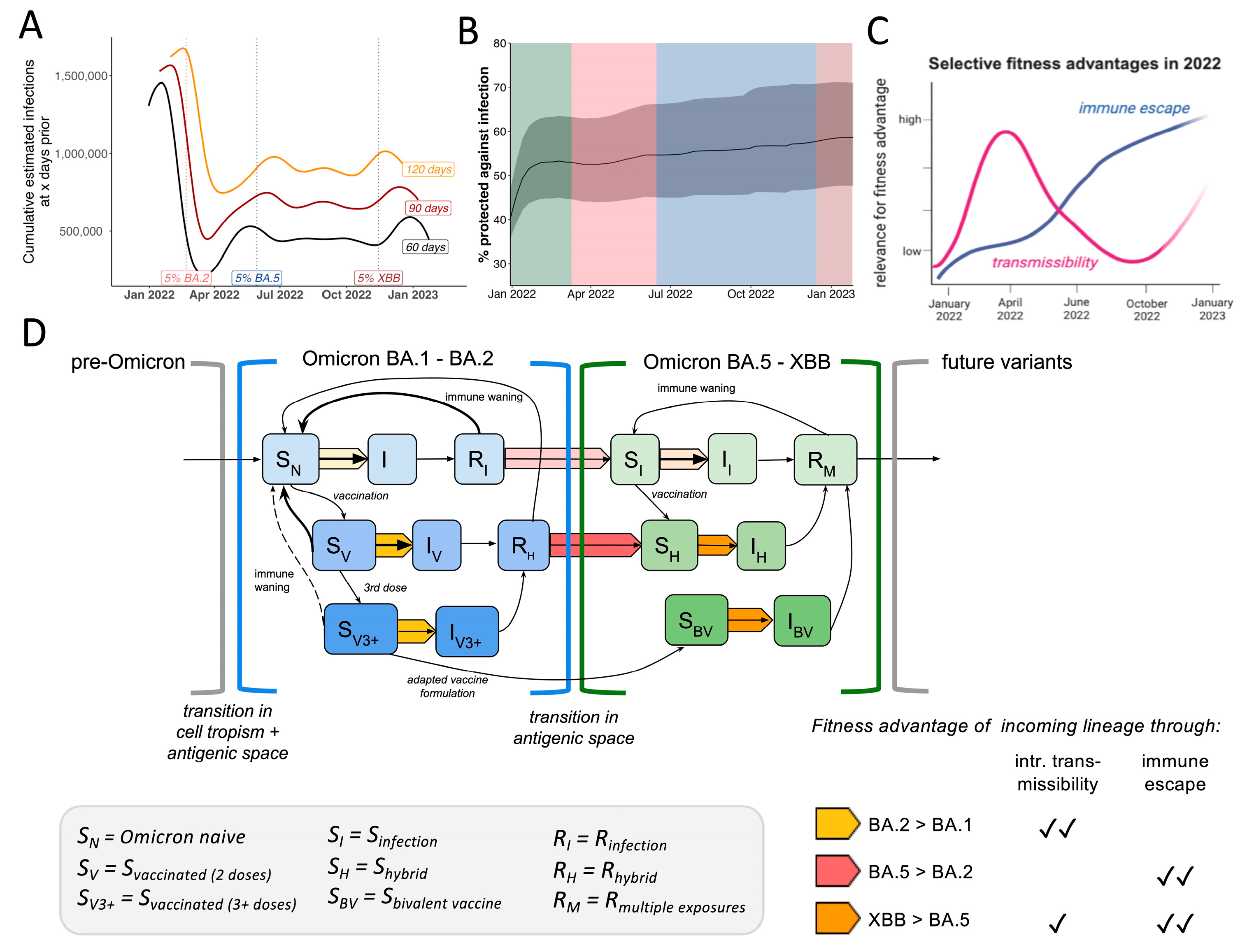

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | First Detected Globally | First Detected in CT | Major Sub-Lineages in Connecticut (+Key Amino Acid Changes in Spike) |
|---|---|---|---|
| BA.1 | October 2021 | November 2021 | BA.1.1 (+R346K) |
| BA.2 | November 2021 | January 2022 | BA.2.12.1 (+L452Q, S704L) |
| BA.4 | November 2021 | May 2022 | BA.4.6 (+R346T, N658S) |
| BA.5 | November 2021 | May 2022 | BQ.1.1 (+R346T, K444T, N460K) BF.7 (+R346T) |
| XBB (BA.2.10*x BA.2.75*) | September 2022 | October 2022 | XBB.1.5 (+F486P) |
| Lineage | Median Ct Early Samples | Median Ct Late Samples | Regression Coefficient (Ct/Day) (p-Value) |
|---|---|---|---|
| BA.1 | 27.297 (IQR: 24.179–30.918) | 27.799 (IQR: 26.139–28.760) | +0.016 (0.045) |
| BA.1.1 | 25.637 (IQR: 22.8 – 28.15) | 28.399 (IQR: 25.563, 30–184) | +0.016 (0.011) |
| BA.2 | 26.681, IQR: 22.557–29.128 | 22.544 (IQR: 20.304, 25.718) | −0.015 (0.002) |
| BA.2.12.1 | 22.972 (IQR: 22.928–22.540) | 23.335 (IQR: 22.928–22.540) | −0.021 (<0.001) |
| BA.4 | 21.2 (IQR: 18.944–23.75) | 19.379 (IQR: 19.088–21.132) | +0.022 (0.227) |
| BA.5 | 23.077 (IQR: 19.61–25.606) | 25.131 (IQR: 22.750–28.133) | +0.015 (<0.001) |
| BQ.1.1 | 23.231 (IQR: 21.580–24.561) | 25.828 (IQR: 23.446–29.863) | +0.005 (0.758) |
| XBB | 21.794 (IQR: 20.573–26.42) | 24.753 (IQR: 19.846–27.566) | +0.03 (0.024) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, N.F.G.; Pham, K.; Chaguza, C.; Lopes, R.; Klaassen, F.; Kalinich, C.C.; Yale SARS-CoV-2 Genomic Surveillance Initiative; Kerantzas, N.; Pandya, S.; Ferguson, D.; et al. Integrated Analysis of the 2022 SARS-CoV-2 Omicron Lineage Replacement Dynamics in Connecticut, US. Viruses 2025, 17, 1020. https://doi.org/10.3390/v17071020
Chen NFG, Pham K, Chaguza C, Lopes R, Klaassen F, Kalinich CC, Yale SARS-CoV-2 Genomic Surveillance Initiative, Kerantzas N, Pandya S, Ferguson D, et al. Integrated Analysis of the 2022 SARS-CoV-2 Omicron Lineage Replacement Dynamics in Connecticut, US. Viruses. 2025; 17(7):1020. https://doi.org/10.3390/v17071020
Chicago/Turabian StyleChen, Nicholas F. G., Kien Pham, Chrispin Chaguza, Rafael Lopes, Fayette Klaassen, Chaney C. Kalinich, Yale SARS-CoV-2 Genomic Surveillance Initiative, Nicholas Kerantzas, Sameer Pandya, David Ferguson, and et al. 2025. "Integrated Analysis of the 2022 SARS-CoV-2 Omicron Lineage Replacement Dynamics in Connecticut, US" Viruses 17, no. 7: 1020. https://doi.org/10.3390/v17071020
APA StyleChen, N. F. G., Pham, K., Chaguza, C., Lopes, R., Klaassen, F., Kalinich, C. C., Yale SARS-CoV-2 Genomic Surveillance Initiative, Kerantzas, N., Pandya, S., Ferguson, D., Schulz, W., Weinberger, D. M., Pitzer, V. E., Warren, J. L., Grubaugh, N. D., & Hahn, A. M. (2025). Integrated Analysis of the 2022 SARS-CoV-2 Omicron Lineage Replacement Dynamics in Connecticut, US. Viruses, 17(7), 1020. https://doi.org/10.3390/v17071020