
A More Rapid Method for Culturing LUHMES-Derived Neurons Provides Greater Cell Numbers and Facilitates Studies of Multiple Viruses
 , ,
, ,  , ,
, ,  , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
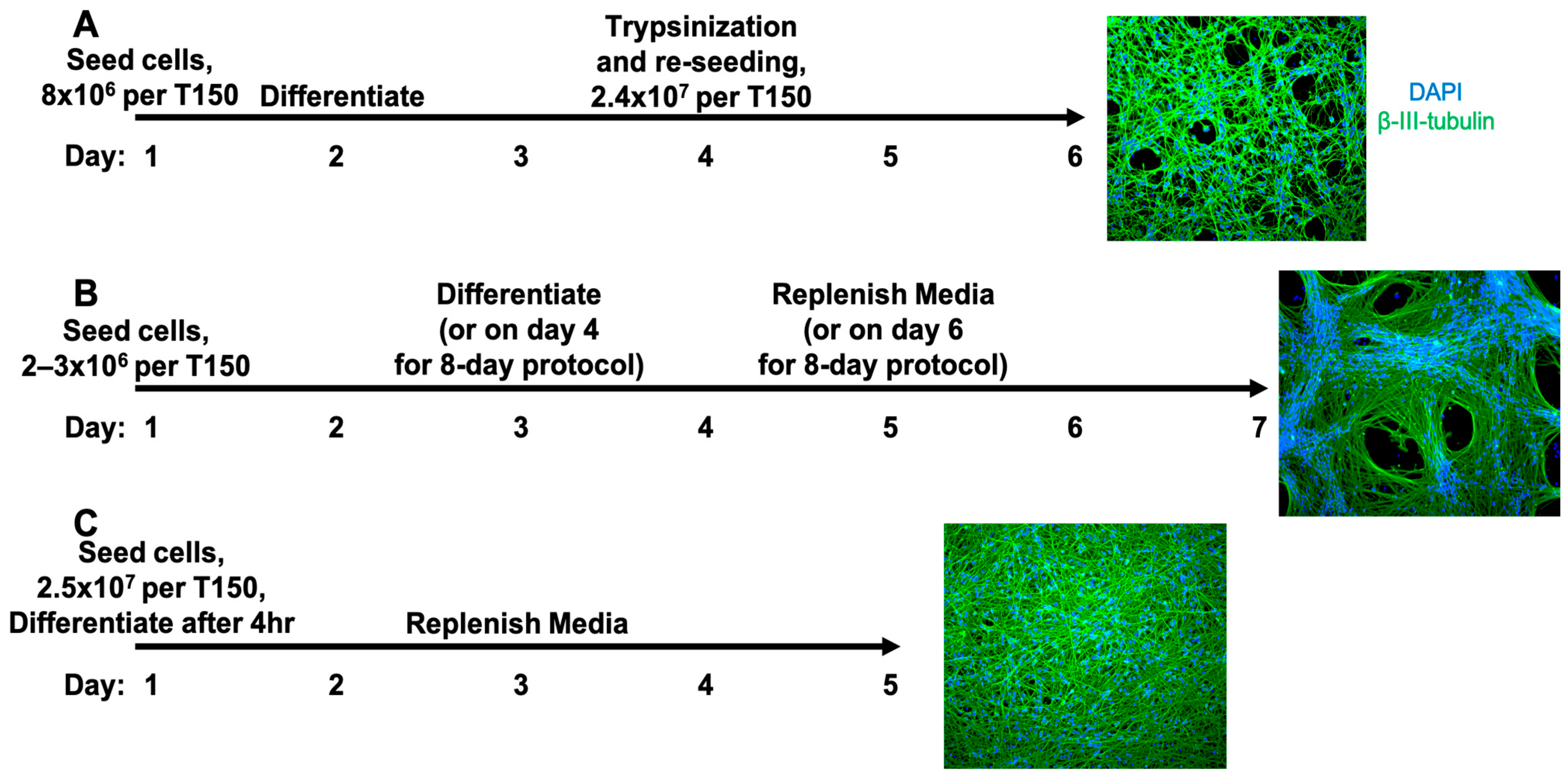
2.1. Cell Culture
2.2. Viruses
2.3. Immunofluorescence
2.4. qRT-PCR
2.5. Luciferase Assay
2.6. RNA Sequencing and Data Analysis
3. Results
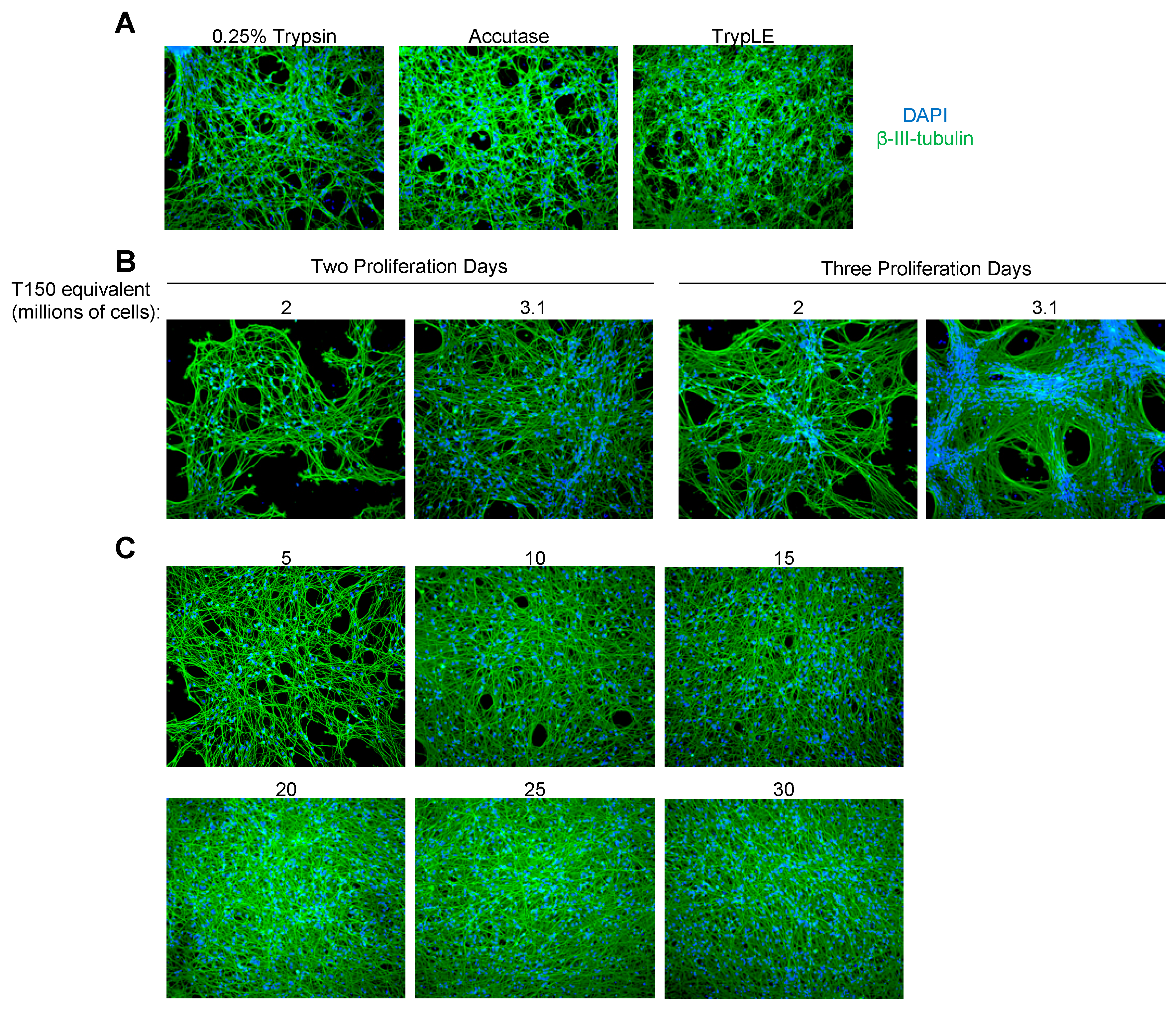
3.1. Evaluation of Three LUHMES Differentiation Methods
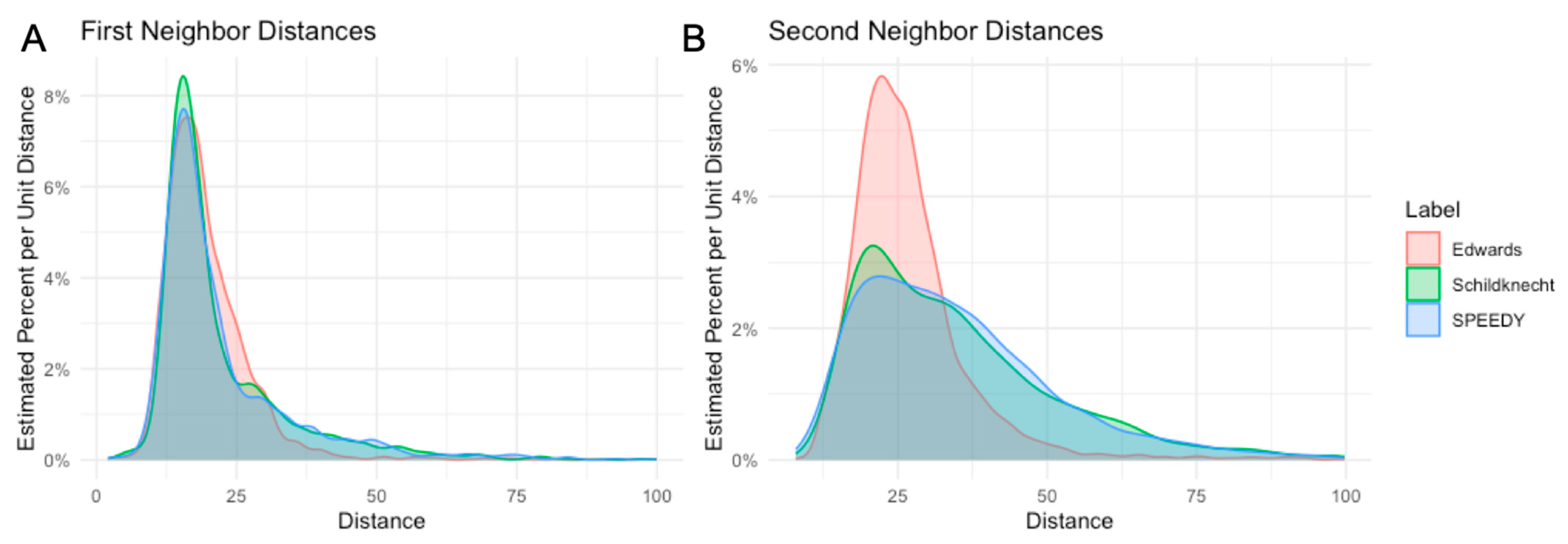
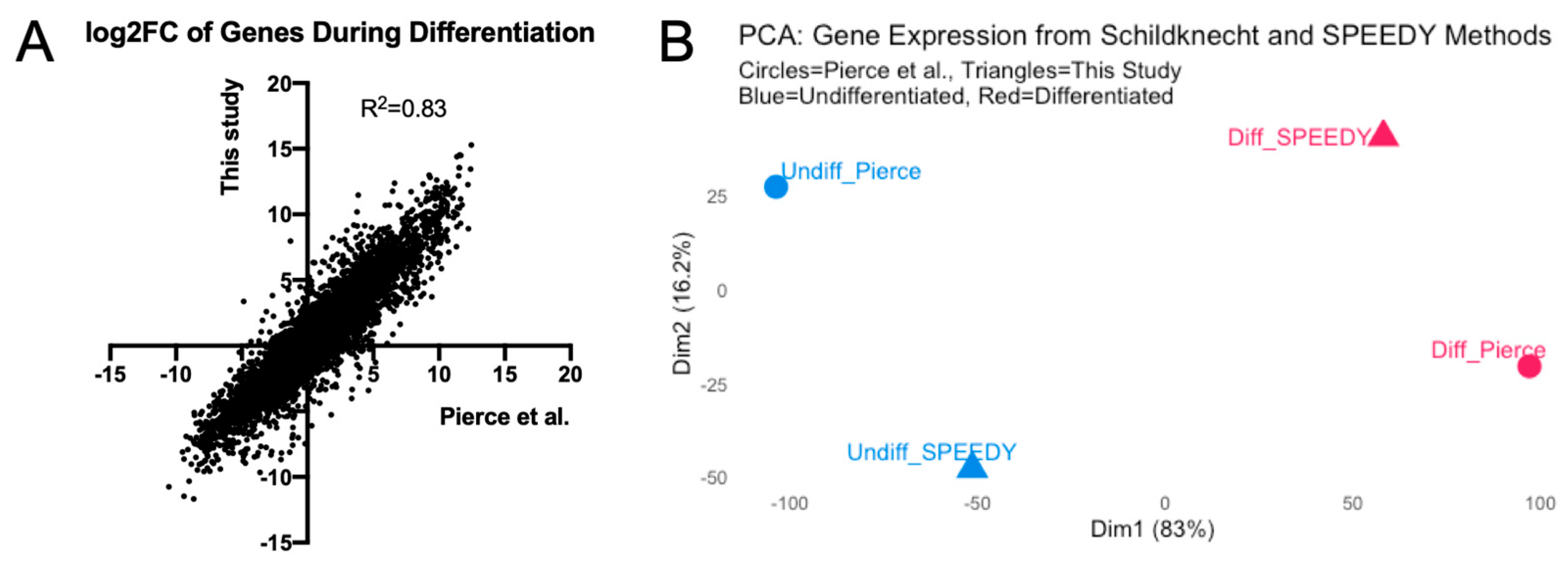
3.2. Transcriptome Analysis of LUHMES Differentiation
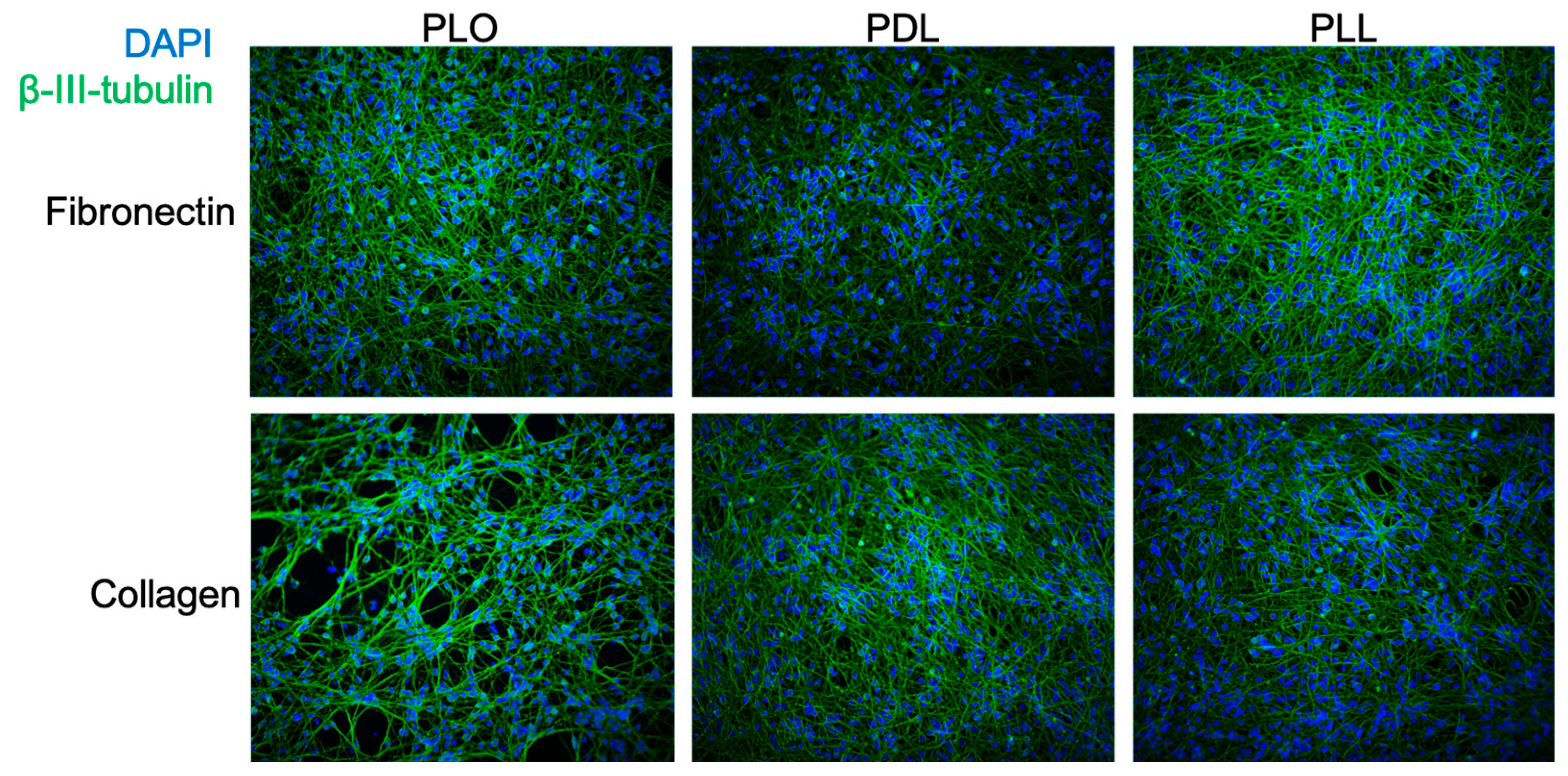
3.3. Evaluation of Coating Conditions for the SPEEDY Method
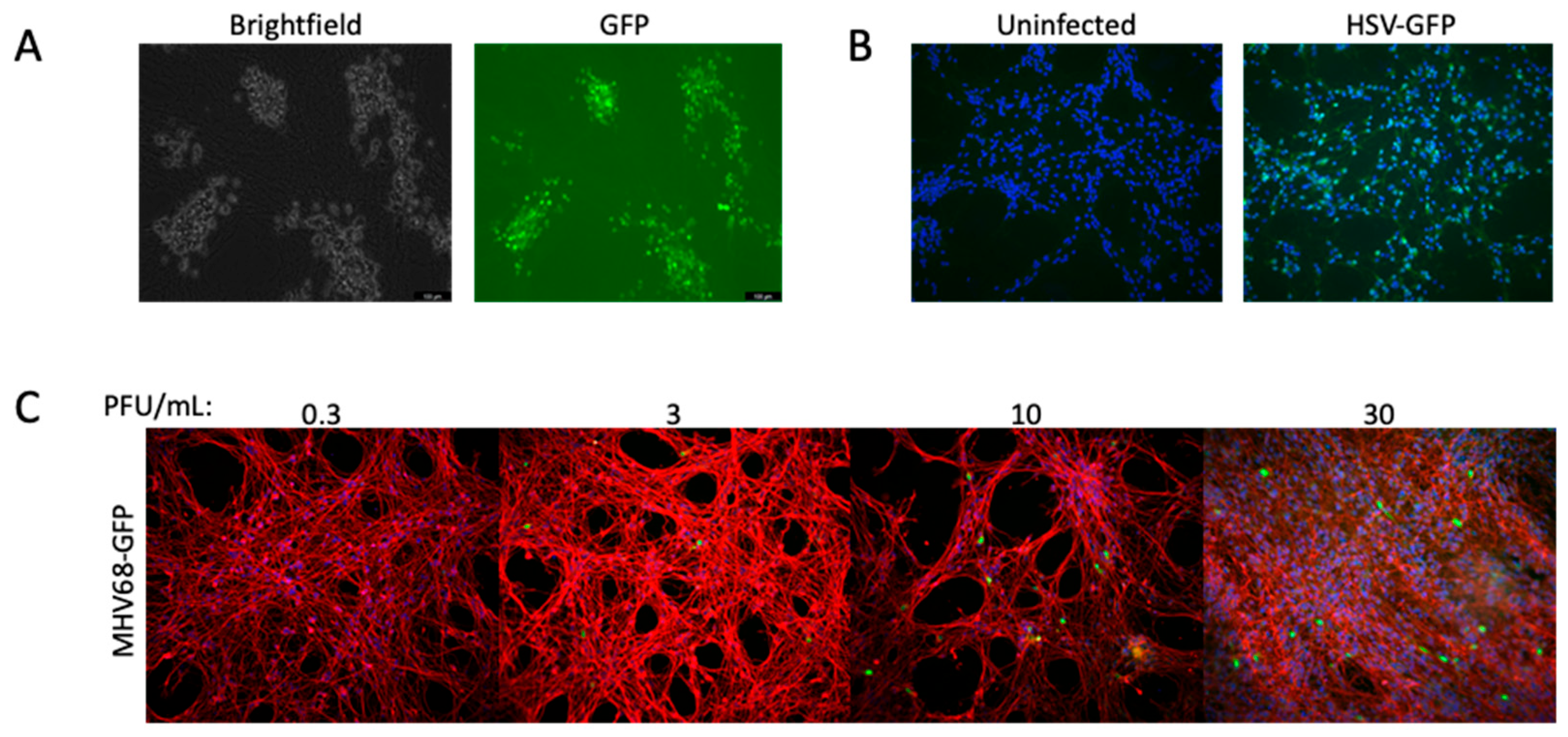
3.4. High-Density LUHMES Cultures as a Model for Multiple Viruses
3.4.1. Entry via Glycoproteins of Hemorrhagic Fever Viruses
3.4.2. Childhood Encephalitis Viruses
3.4.3. Herpesviruses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry Factor. | Virus | This Study (FPKM) | Pierce et al. TMM-Normalized log2(CPM) |
|---|---|---|---|
| Tyro3 | Zika, Lassa, Marburg, Ebola | 4.80 | 6.42 |
| DAG1 | Lassa | 7.88 | 7.19 |
| EPHA7 | MHV68 | 2.77 | 4.44 |
| Nectin 1 | HSV-1, Measles | 2.37 | 5.97 |
| CADM1 | Measles | 40.93 | 9.18 |
| CADM2 | Measles | 5.24 | 8.38 |
References
- Soriano, J. Neuronal Cultures: Exploring Biophysics, Complex Systems, and Medicine in a Dish. Biophysica 2023, 3, 181–202. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef]
- Lokai, T.; Albin, B.; Qubbaj, K.; Tiwari, A.; Adhikari, P.; Yang, I. A review on current brain organoid technologies from a biomedical engineering perspective. Exp. Neurol. 2023, 367, 114461. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Falsig, J.; van Beek, J.; Payne, S.; Dringen, R.; Brundin, P.; Leist, M. Progressive Degeneration of Human Mesencephalic Neuron-Derived Cells Triggered by Dopamine-Dependent Oxidative Stress Is Dependent on the Mixed-Lineage Kinase Pathway. J. Neurosci. 2005, 25, 6329–6342. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Barg, S.; Wiekop, P.; Lundberg, C.; Raymon, H.K.; Brundin, P. Effect of mutant α-synuclein on dopamine homeostasis in a new human mesencephalic cell line. J. Biol. Chem. 2002, 277, 38884–38894. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.B.; Hogberg, H.; Kuo, D.; Sakamuru, S.; Xia, M.; Smirnova, L.; Hartung, T.; Gerhold, D. Characterization of three human cell line models for high-throughput neuronal cytotoxicity screening. J. Appl. Toxicol. 2017, 37, 167–180. [Google Scholar] [CrossRef]
- Gutbier, S.; Spreng, A.S.; Delp, J.; Schildknecht, S.; Karreman, C.; Suciu, I.; Brunner, T.; Groettrup, M.; Leist, M. Prevention of neuronal apoptosis by astrocytes through thiol-mediated stress response modulation and accelerated recovery from proteotoxic stress. Cell Death Differ. 2018, 25, 2101–2117. [Google Scholar] [CrossRef]
- Calamini, B.; Geyer, N.; Huss-Braun, N.; Bernhardt, A.; Harsanyi, V.; Rival, P.; Cindhuchao, M.; Hoffmann, D.; Gratzer, S. Development of a physiologically relevant and easily scalable LUHMES cell-based model of G2019S LRRK2-driven Parkinson’s disease. DMM Dis. Model. Mech. 2021, 14, dmm048017. [Google Scholar] [CrossRef]
- Schildknecht, S.; Pöltl, D.; Nagel, D.M.; Matt, F.; Scholz, D.; Lotharius, J.; Schmieg, N.; Salvo-Vargas, A.; Leist, M. Requirement of a dopaminergic neuronal phenotype for toxicity of low concentrations of 1-methyl-4-phenylpyridinium to human cells. Toxicol. Appl. Pharmacol. 2009, 241, 23–35. [Google Scholar] [CrossRef]
- Edwards, T.G.; Bloom, D.C. Lund Human Mesencephalic (LUHMES) Neuronal Cell Line Supports Herpes Simplex Virus 1 Latency In Vitro. J. Virol. 2019, 93, 10.1128. [Google Scholar] [CrossRef]
- Whisnant, A.W.; Dyck Dionisi, O.; Salazar Sanchez, V.; Rappold, J.M.; Djakovic, L.; Grothey, A.; Marante, A.L.; Fischer, P.; Peng, S.; Wolf, K.; et al. Herpes simplex virus 1 inhibits phosphorylation of RNA polymerase II CTD serine-7. J. Virol. 2024, 98, e01178-24. [Google Scholar] [CrossRef] [PubMed]
- Tüshaus, J.; Kataka, E.S.; Zaucha, J.; Frishman, D.; Müller, S.A.; Lichtenthaler, S.F. Neuronal Differentiation of LUHMES Cells Induces Substantial Changes of the Proteome. Proteomics 2021, 21, 2000174. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.J.; Shtanko, O.; Vijay, R.; Mallinger, L.N.; Joyner, C.J.; Galinski, M.R.; Butler, N.S.; Maury, W. Acute Plasmodium Infection Promotes Interferon-Gamma-Dependent Resistance to Ebola Virus Infection. Cell Rep. 2020, 30, 4041–4051. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, R.B.; Phillips, E.K.; Patel, R.; Mahauad-Fernandez, W.; Moller-Tank, S.; Rogers, K.J.; Dillard, J.A.; Cooney, A.L.; Martinez-Sobrido, L.; Okeoma, C.; et al. TIM-1 Mediates Dystroglycan-Independent Entry of Lassa Virus. J. Virol. 2018, 92, e00093-18. [Google Scholar] [CrossRef]
- Lennemann, N.J.; Rhein, B.A.; Ndungo, E.; Chandran, K.; Qiu, X.; Maury, W. Comprehensive functional analysis of N-linked glycans on ebola virus GP1. MBio 2014, 5, e00862-13. [Google Scholar] [CrossRef]
- Kaufman, J.W.; Singh, B.K.; Durnell, L.A.; Sinn, P.L. Representative measles virus infection requires appropriate airway epithelia culture conditions. J. Virol. 2023, 97, e01051-23. [Google Scholar] [CrossRef]
- Tucker, J.M.; Schaller, A.M.; Willis, I.; Glaunsinger, B.A. Alteration of the premature tRNA landscape by gammaherpesvirus infection. MBio 2020, 11, e02664-20. [Google Scholar] [CrossRef]
- Devadas, D.; Koithan, T.; Diestel, R.; Prank, U.; Sodeik, B.; Döhner, K. Herpes Simplex Virus Internalization into Epithelial Cells Requires Na+/H+ Exchangers and p21-Activated Kinases but neither Clathrin- nor Caveolin-Mediated Endocytosis. J. Virol. 2014, 88, 13378–13395. [Google Scholar] [CrossRef]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 29 April 2025).
- Bonfert, T.; Kirner, E.; Csaba, G.; Zimmer, R.; Friedel, C.C. ContextMap 2: Fast and accurate context-based RNA-seq mapping. BMC Bioinform. 2015, 16, 122. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Kluge, M.; Friedel, C.C. Watchdog—A workflow management system for the distributed analysis of large-scale experimental data. BMC Bioinform. 2018, 19, 97. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Smirnova, L.; Harris, G.; Delp, J.; Valadares, M.; Pamies, D.; Hogberg, H.T.; Waldmann, T.; Leist, M.; Hartung, T. A LUHMES 3D dopaminergic neuronal model for neurotoxicity testing allowing long-term exposure and cellular resilience analysis. Arch. Toxicol. 2015, 90, 2725–2743. [Google Scholar] [CrossRef]
- Pierce, S.E.; Tyson, T.; Booms, A.; Prahl, J.; Coetzee, G.A. Parkinson’s disease genetic risk in a midbrain neuronal cell line. Neurobiol. Dis. 2018, 114, 53–64. [Google Scholar] [CrossRef]
- Höllerhage, M.; Moebius, C.; Melms, J.; Chiu, W.H.; Goebel, J.N.; Chakroun, T.; Koeglsperger, T.; Oertel, W.H.; Rösler, T.W.; Bickle, M.; et al. Protective efficacy of phosphodiesterase-1 inhibition against alpha-synuclein toxicity revealed by compound screening in LUHMES cells. Sci. Rep. 2017, 7, 11469. [Google Scholar] [CrossRef]
- Harris, G.; Hogberg, H.; Hartung, T.; Smirnova, L. 3D Differentiation of LUHMES Cell Line to Study Recovery and Delayed Neurotoxic Effects. Curr. Protoc. Toxicol. 2017, 73, 11–23. [Google Scholar] [CrossRef]
- Ko, K.R.; Tam, N.W.; Teixeira, A.G.; Frampton, J.P. SH-SY5Y and LUHMES cells display differential sensitivity to MPP+, tunicamycin, and epoxomicin in 2D and 3D cell culture. Biotechnol. Prog. 2020, 36, E2942. [Google Scholar] [CrossRef]
- Dinh, N.D.; Chiang, Y.Y.; Hardelauf, H.; Baumann, J.; Jackson, E.; Waide, S.; Sisnaiske, J.; Frimat, J.P.; van Triel, C.; Janasek, D.; et al. Microfluidic construction of minimalistic neuronal co-cultures. Lab Chip 2013, 13, 1402–1412. [Google Scholar] [CrossRef]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar] [CrossRef]
- Sahoo, B.R.; Pattnaik, A.; Annamalai, A.S.; Franco, R.; Pattnaik, A.K. Mechanistic Target of Rapamycin Signaling Activation Antagonizes Autophagy To Facilitate Zika Virus Replication. J. Virol. 2020, 94, e01575-20. [Google Scholar] [CrossRef]
- Meertens, L.; Labeau, A.; Dejamac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Charpentier, T.L.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef]
- Grams, T.R.; Edwards, T.G.; Bloom, D.C. Herpes Simplex Virus 1 Strains 17syn+ and KOS(M) Differ Greatly in Their Ability To Reactivate from Human Neurons In Vitro. J. Virol. 2020, 94, e00796-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tibbetts, S.A.; Krug, L.T. Conquering the Host: Determinants of Pathogenesis Learned from Murine Gammaherpesvirus 68. Annu. Rev. Virol. 2021, 8, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Terry, L.A.; Stewart, J.P.; Nash, A.A.; Fazakerley, J.K. Murine gammaherpesvirus-68 infection of and persistence in the central nervous system. J. Gen. Virol. 2000, 81, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, S.; Kwak, S.E.; Kang, T.C.; Kim, H.S.; Kwon, H.J.; Kim, Y.W.; Kim, Y.S.; Choi, E.K.; Song, M.J. Age-dependent pathogenesis of murine gammaherpesvirus 68 infection of the central nervous system. Mol. Cells 2009, 27, 105–111. [Google Scholar] [CrossRef]
- Yousaf, I.; Domanico, L.; Nambara, T.; Yadav, K.; Kelly, L.K.; Trejo-Lopez, J.; Shieh, W.; Rota, P.A.; Devaux, P.; Kanekiyo, T.; et al. The measles virus matrix F50S mutation from a lethal case of subacute sclerosing panencephalitis promotes receptor-independent neuronal spread. J. Virol. 2025, 99, e01750-24. [Google Scholar] [CrossRef]
- Gutbier, S.; May, P.; Berthelot, S.; Krishna, A.; Trefzer, T.; Behbehani, M.; Efremova, L.; Delp, J.; Gstraunthaler, G.; Waldmann, T.; et al. Major changes of cell function and toxicant sensitivity in cultured cells undergoing mild, quasi-natural genetic drift. Arch. Toxicol. 2018, 92, 3487–3503. [Google Scholar] [CrossRef]
- Stirling, D.R.; Swain-Bowden, M.J.; Lucas, A.M.; Carpenter, A.E.; Cimini, B.A.; Goodman, A. CellProfiler 4: Improvements in speed, utility and usability. BMC Bioinform. 2021, 22, 433. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whisnant, A.W.; Clark, S.E.; Aguilar-Briseño, J.A.; Durnell, L.A.; Grothey, A.; Miller, A.M.; Varga, S.M.; Meier, J.L.; Grose, C.; Sinn, P.L.; et al. A More Rapid Method for Culturing LUHMES-Derived Neurons Provides Greater Cell Numbers and Facilitates Studies of Multiple Viruses. Viruses 2025, 17, 1001. https://doi.org/10.3390/v17071001
Whisnant AW, Clark SE, Aguilar-Briseño JA, Durnell LA, Grothey A, Miller AM, Varga SM, Meier JL, Grose C, Sinn PL, et al. A More Rapid Method for Culturing LUHMES-Derived Neurons Provides Greater Cell Numbers and Facilitates Studies of Multiple Viruses. Viruses. 2025; 17(7):1001. https://doi.org/10.3390/v17071001
Chicago/Turabian StyleWhisnant, Adam W., Stephanie E. Clark, José Alberto Aguilar-Briseño, Lorellin A. Durnell, Arnhild Grothey, Ann M. Miller, Steven M. Varga, Jeffery L. Meier, Charles Grose, Patrick L. Sinn, and et al. 2025. "A More Rapid Method for Culturing LUHMES-Derived Neurons Provides Greater Cell Numbers and Facilitates Studies of Multiple Viruses" Viruses 17, no. 7: 1001. https://doi.org/10.3390/v17071001
APA StyleWhisnant, A. W., Clark, S. E., Aguilar-Briseño, J. A., Durnell, L. A., Grothey, A., Miller, A. M., Varga, S. M., Meier, J. L., Grose, C., Sinn, P. L., Tucker, J. M., Friedel, C. C., Maury, W. J., Price, D. H., & Dölken, L. (2025). A More Rapid Method for Culturing LUHMES-Derived Neurons Provides Greater Cell Numbers and Facilitates Studies of Multiple Viruses. Viruses, 17(7), 1001. https://doi.org/10.3390/v17071001