
Novel Viral Sequences in a Patient with Cryptogenic Liver Cirrhosis Revealed by Serum Virome Sequencing


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Patient Samples
2.2. Serum Virome Sequencing
2.3. Viral Categorization and Discovery
2.4. Validation of Unknown Sequences
2.5. Machine-Learning Analysis for the Origin of Unknown Sequences
2.6. Genome Walking and Strand Attribute Determination
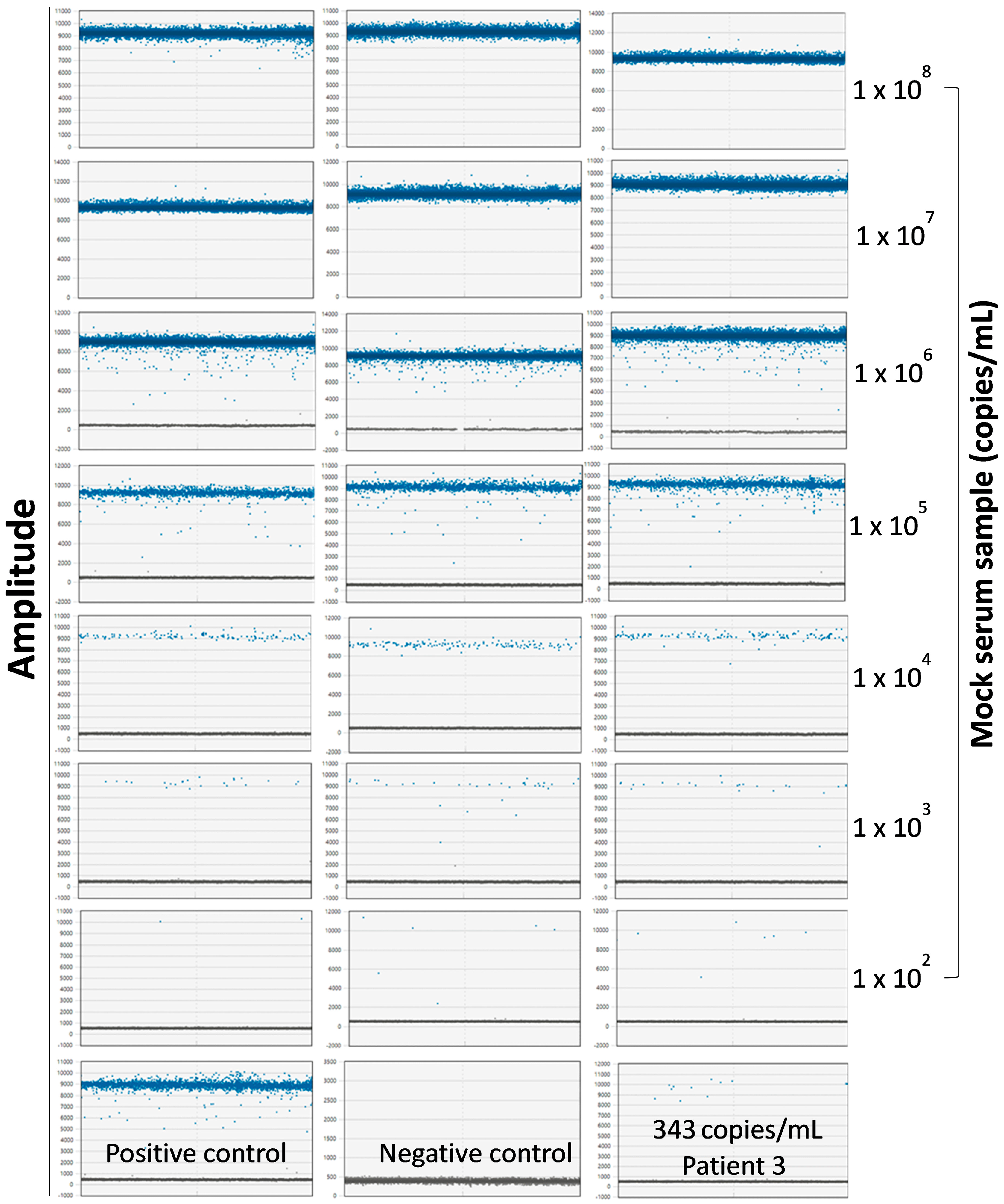
2.7. Determination of Copy Numbers of Seq260 Using Digital Droplet PCR
2.8. Detection of Seq260 in the Screened Cohort
2.9. In Silico Screening of Unknown Sequences
3. Results
3.1. Discovery of Novel Virus-like Sequences in a Patient with Cryptogenic Liver Cirrhosis
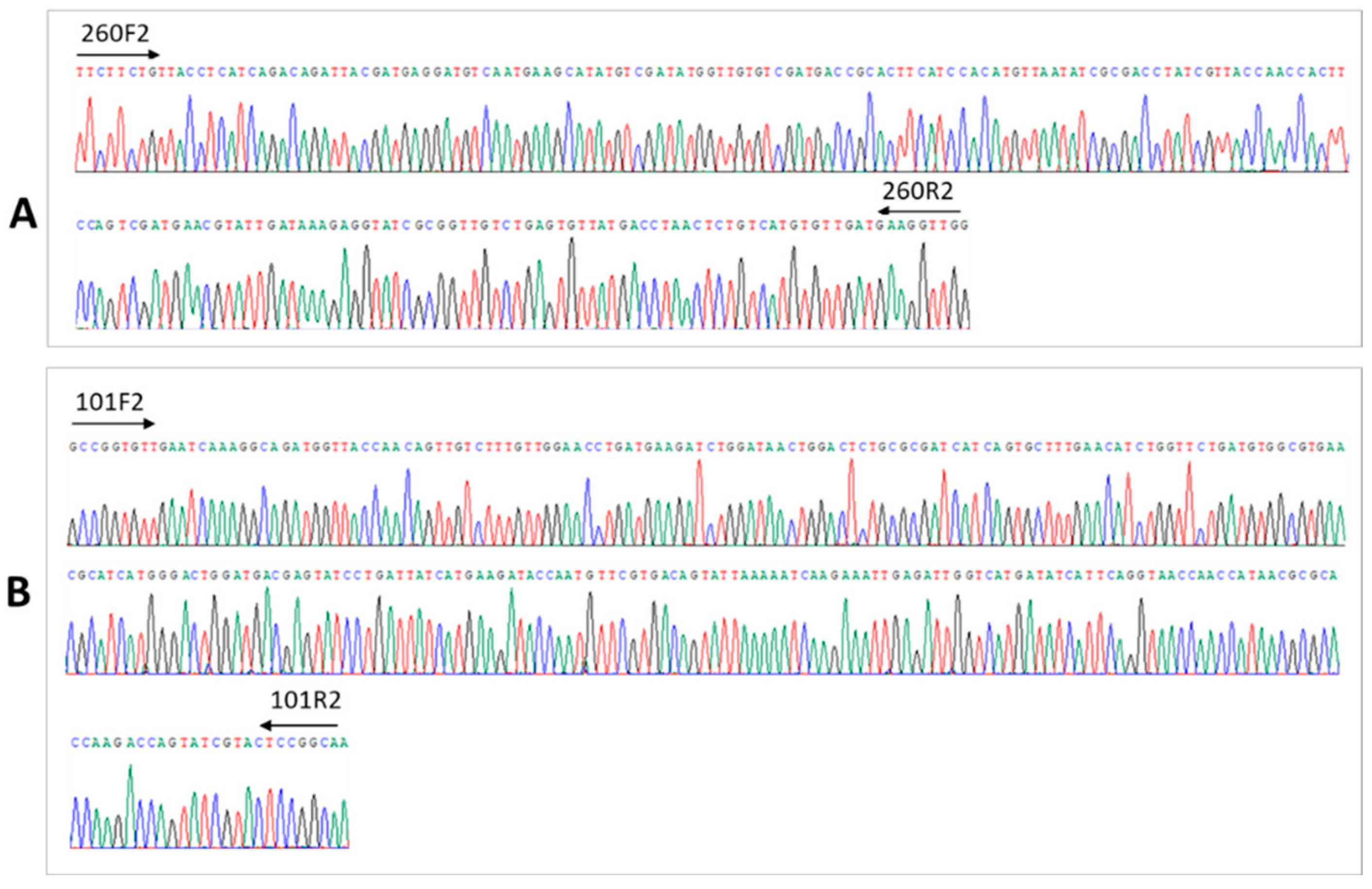
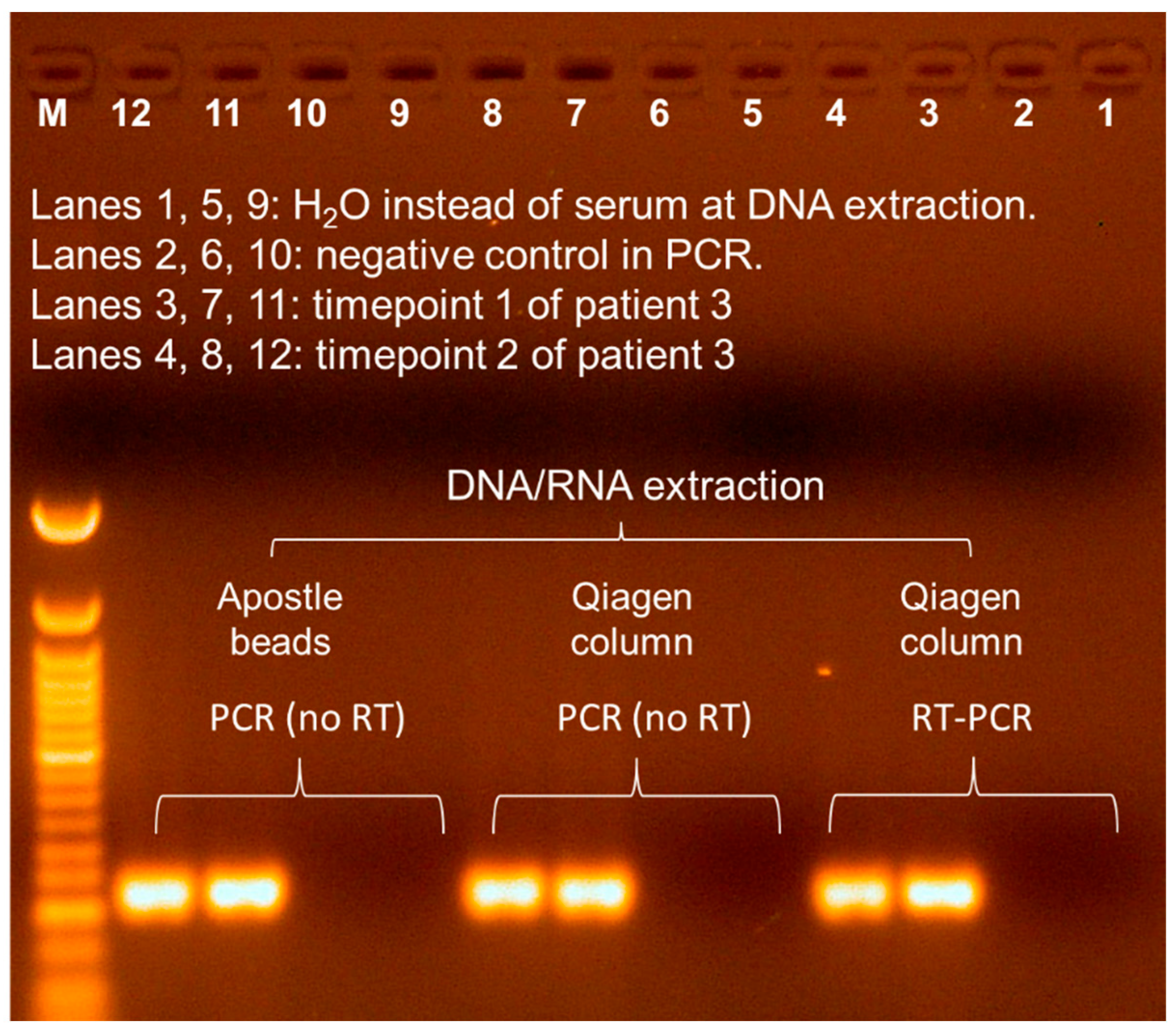
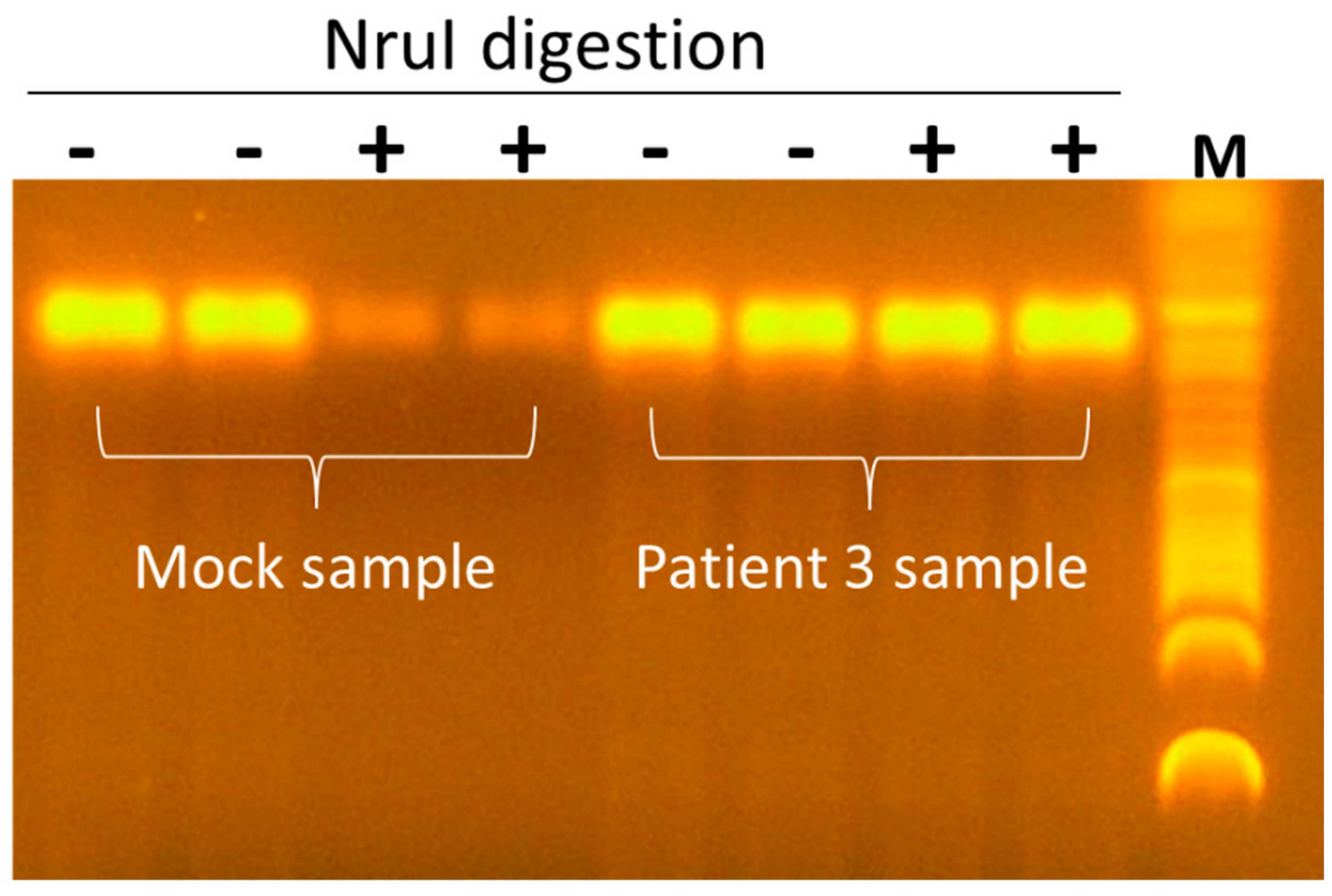
3.2. Seq260 (Contig xx03_260) from Patient 3 Is Not a Contaminant
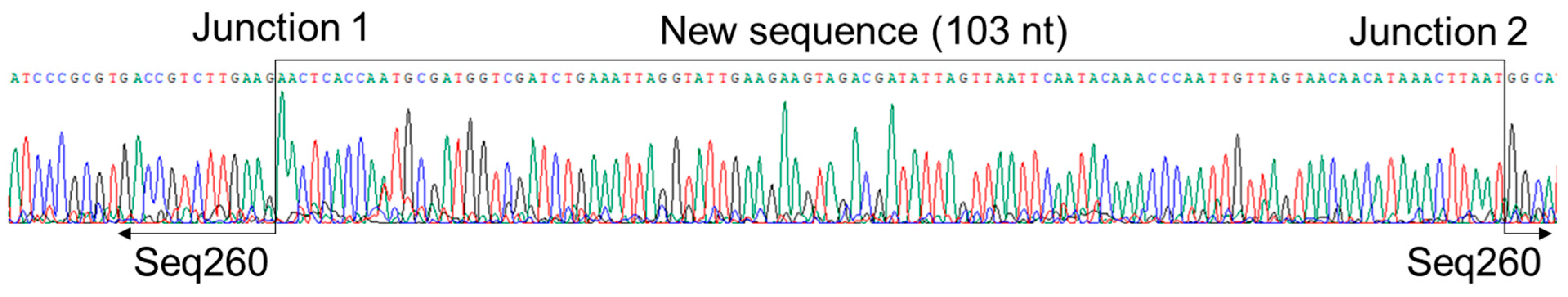
3.3. A Net Extension of 107 nt at the 3′ End of Seq260 by Genome Walking
3.4. Seq260 Is Not Detected in Other Patients and Next-Generation Sequencing Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gan, C.; Yuan, Y.; Shen, H.; Gao, J.; Kong, X.; Che, Z.; Guo, Y.; Wang, H.; Dong, E.; Xiao, J. Liver diseases: Epidemiology, causes, trends and predictions. Signal Transduct. Target Ther. 2025, 10, 33. [Google Scholar] [PubMed]
- Rane, S.V.; Jain, S.; Debnath, P.; Deshmukh, R.; Nair, S.; Chandnani, S.; Kamat, R.; Rathi, P. A comparative study of uncomplicated acute non-A-E hepatitis with acute viral hepatitis and acute onset autoimmune hepatitis. Indian J. Gastroenterol. 2024, 43, 443–451. [Google Scholar] [CrossRef]
- Vaz, J.; Eriksson, B.; Strömberg, U.; Buchebner, D.; Midlöv, P. Incidence, aetiology and related comorbidities of cirrhosis: A Swedish population-based cohort study. BMC Gastroenterol. 2020, 20, 84. [Google Scholar] [CrossRef]
- Nagaoki, Y.; Hyogo, H.; Ando, Y.; Kosaka, Y.; Uchikawa, S.; Nishida, Y.; Teraoka, Y.; Morio, K.; Fujino, H.; Ono, A.; et al. Increasing incidence of non-HBV- and non-HCV-related hepatocellular carcinoma: Single-institution 20-year study. BMC Gastroenterol. 2021, 21, 306. [Google Scholar] [CrossRef] [PubMed]
- Ganger, D.R.; Rule, J.; Rakela, J.; Bass, N.; Reuben, A.; Stravitz, R.T.; Sussman, N.; Larson, A.M.; James, L.; Chiu, C.; et al. Acute liver failure of indeterminate etiology: A comprehensive systematic approach by an expert committee to establish causality. Am. J. Gastroenterol. 2018, 113, 1319. [Google Scholar] [CrossRef]
- Mercado-Irizarry, A.; Torres, E.A. Cryptogenic cirrhosis: Current knowledge and future directions. Clin. Liver Dis. 2016, 7, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Okamoto, H.; Konishi, K.; Yoshizawa, H.; Miyakawa, Y.; Mayumi, M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem. Biophys. Res. Commun. 1997, 241, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Akiba, J.; Umemura, T.; Alter, H.J.; Kojiro, M.; Tabor, E. SEN virus: Epidemiology and characteristics of a transfusion-transmitted virus. Transfusion 2005, 45, 1084–1088. [Google Scholar] [CrossRef]
- Linnen, J.; Wages, J., Jr.; Zhang-Keck, Z.Y.; Fry, K.E.; Krawczynski, K.Z.; Alter, H.; Koonin, E.; Gallagher, M.; Alter, M.; Hadziyannis, S.; et al. Molecular cloning and disease association of hepatitis G virus: A transfusion-transmissible agent. Science 1996, 271, 505–508. [Google Scholar] [CrossRef]
- Simons, J.N.; Leary, T.P.; Dawson, G.J.; Pilot-Matias, T.J.; Muerhoff, A.S.; Schlauder, G.G.; Desai, S.M.; Mushahwar, I.K. Isolation of novel virus-like sequences associated with human hepatitis. Nat. Med. 1995, 1, 564–569. [Google Scholar] [CrossRef]
- Berg, M.G.; Lee, D.; Coller, K.; Frankel, M.; Aronsohn, A.; Cheng, K.; Forberg, K.; Marcinkus, M.; Naccache, S.N.; Dawson, G.; et al. Discovery of a novel human pegivirus in blood associated with hepatitis C virus co-infection. PLoS Pathog. 2015, 11, e1005325. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Kumar, A.; Simmonds, P.; Bhuva, N.; Singh Chauhan, L.; Lee, B.; Sall, A.A.; Jin, Z.; Morse, S.S.; Shaz, B.; et al. Virome analysis of transfusion recipients reveals a novel human virus that shares genomic features with hepaciviruses and pegiviruses. mBio 2015, 6, e01466-15. [Google Scholar] [CrossRef]
- Satoh, K.; Iwata-Takakura, A.; Osada, N.; Yoshikawa, A.; Hoshi, Y.; Miyakawa, K.; Gotanda, Y.; Satake, M.; Tadokoro, K.; Mizoguchi, H. Novel DNA sequence isolated from blood donors with high transaminase levels. Hepatol. Res. 2011, 41, 971–981. [Google Scholar] [CrossRef]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Unraveling the viral dark matter through viral metagenomics. Front. Immunol. 2022, 13, 1005107. [Google Scholar] [CrossRef]
- Abecassis, M.M.; Fisher, R.A.; Olthoff, K.M.; Freise, C.E.; Rodrigo, D.R.; Samstein, B.; Kam, I.; Merion, R.M.; A2ALL Study Group. Complications of living donor hepatic lobectomy--a comprehensive report. Am. J. Transplant. 2012, 12, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Clark, J.M.; Bass, N.M.; Van Natta, M.L.; Unalp-Arida, A.; Tonascia, J.; Zein, C.O.; Brunt, E.M.; Kleiner, D.E.; McCullough, A.J.; et al. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology 2010, 52, 913–924. [Google Scholar] [CrossRef]
- Li, G.; Zhou, Z.; Yao, L.; Xu, Y.; Wang, L.; Fan, X. Full annotation of serum virome in Chinese blood donors with elevated alanine aminotransferase levels. Transfusion 2019, 59, 3177–3185. [Google Scholar] [CrossRef]
- Ren, Y.; Xu, Y.; Lee, W.M.; Di Bisceglie, A.M.; Fan, X. In-depth serum virome analysis in patients with acute liver failure with indeterminate etiology. Arch. Virol. 2020, 165, 127–135. [Google Scholar] [CrossRef]
- Wang, W.; Ren, Y.; Lu, Y.; Xu, Y.; Crosby, S.D.; Di Bisceglie, A.M.; Fan, X. Template-dependent multiple displacement amplification for profiling human circulating RNA. Biotechniques 2017, 63, 21–27. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Niu, B.; Fu, L.; Sun, S.; Li, W. Artificial and natural duplicates in pyrosequencing reads of metagenomic data. BMC Bioinform. 2010, 11, 187. [Google Scholar] [CrossRef]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Peng, P.; Xu, Y.; Aurora, R.; Di Bisceglie, A.M.; Fan, X. Within-host quantitation of anellovirus genome complexity from clinical samples. J. Virol. Methods 2022, 302, 114493. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- The SRA Toolkit Development Team. Available online: https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software (accessed on 1 May 2023).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Morfopoulou, S.; Buddle, S.; Torres Montaguth, O.E.; Atkinson, L.; Guerra-Assunção, J.A.; Moradi Marjaneh, M.; Zennezini Chiozzi, R.; Storey, N.; Campos, L.; Hutchinson, J.C.; et al. Genomic investigations of unexplained acute hepatitis in children. Nature 2023, 617, 564–573. [Google Scholar] [CrossRef]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J., Jr.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef]
- Anani, H.; Destras, G.; Bulteau, S.; Castain, L.; Semanas, Q.; Burfin, G.; Petrier, M.; Martin, F.P.; Poulain, C.; Dickson, R.P.; et al. Lung Virome Convergence Precedes Hospital-Acquired Pneumonia in Intubated Critically Ill Patients. Preprint 2024. Available online: https://ssrn.com/abstract=5012218 (accessed on 1 October 2024).
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. BMC Infect. Dis. 2018, 18, 537. [Google Scholar] [CrossRef] [PubMed]
- Stegmüller, S.; Fraefel, C.; Kubacki, J. Genome sequence of alongshan virus from ixodes ricinus ticks collected in Switzerland. Microbiol. Resour. Announc. 2023, 12, e0128722. [Google Scholar] [CrossRef] [PubMed]
- Cordey, S.; Laubscher, F.; Hartley, M.A.; Junier, T.; Keitel, K.; Docquier, M.; Guex, N.; Iseli, C.; Vieille, G.; Le Mercier, P.; et al. Blood virosphere in febrile Tanzanian children. Emerg. Microbes Infect. 2021, 10, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Pingoud, A.; Jeltsch, A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001, 29, 3705–3727. [Google Scholar] [CrossRef]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001, 11, 1095–1099. [Google Scholar] [CrossRef]
- Brani, P.; Manzoor, H.Z.; Spezia, P.G.; Vigezzi, A.; Ietto, G.; Dalla Gasperina, D.; Minosse, C.; Bosi, A.; Giaroni, C.; Carcano, G.; et al. Torque Teno Virus: Lights and shades. Viruses 2025, 17, 334. [Google Scholar] [CrossRef]
- Somasekar, S.; Lee, D.; Rule, J.; Naccache, S.N.; Stone, M.; Busch, M.P.; Sanders, C.; Lee, W.M.; Chiu, C.Y. Viral surveillance in serum samples from patients with acute liver failure by metagenomic next-generation sequencing. Clin. Infect. Dis. 2017, 65, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowska, J.; van der Hoek, L. Human anelloviruses: Diverse, omnipresent and commensal members of the virome. FEMS Microbiol. Rev. 2020, 44, 305–313. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Martini, F.; Maritati, M.; Caselli, E.; Gallenga, C.E.; Guarino, M.; De Giorgio, R.; Mazziotta, C.; Tramarin, M.L.; Badiale, G.; et al. Advanced molecular and immunological diagnostic methods to detect SARS-CoV-2 infection. Microorganisms 2022, 10, 1193. [Google Scholar] [CrossRef]
- Qiu, J.; Söderlund-Venermo, M.; Young, N.S. Human parvoviruses. Clin. Microbiol. Rev. 2017, 30, 43–113. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort for Viral Discovery | ||||||
| Patient # | Sex | Age | Diagnosis (CLD) | Outcome | Specimen | Source |
| 1 | F | 64 | Cirrhosis | NA | Serum | SLULC |
| 2 | F | 73 | Hepatitis | NA | Serum | |
| 3 | F | 60 | Cirrhosis | OLT, deceased | Serum | |
| 4 | F | 42 | Cirrhosis | NA | Serum | |
| 5 | F | 68 | Cirrhosis | deceased | Serum | |
| 6 | M | 40 | Cirrhosis | NA | Serum | |
| 7 | M | 59 | Cirrhosis | OLT, deceased | Serum | |
| 8 | M | 65 | Cirrhosis | NA | Serum | |
| 9 | M | 54 | Hepatitis | NA | Serum | |
| Cohort for PCR Screening | ||||||
| Group # | Diagnosis | Number | Specimen | Source | ||
| 1 | Chronic HCV infection | 100 | Serum | SLULC | ||
| Chronic HBV infection | 10 | Serum | ||||
| 2 | Blood donors | 200 | Serum | ARC | ||
| 3 | Acute liver failure | 50 * | Serum | NIDDK- CR | ||
| Liver transplantation | 40 * | Serum | ||||
| Cirrhosis | 25 * | Serum | ||||
| Application | Target | Primer Name | Polarity | Sequence (5′→3′) | Amplicon Size | |
|---|---|---|---|---|---|---|
| RT-PCR/PCR Validation | Contig xx12_260 (Seq260) | F1 | Forward | tccttgatgcaagccattg | 237 bp | |
| R1 | Reverse | gcgggataccaacaacaac | ||||
| F2 | Forward | atgtcactggcatccttcttc | ||||
| R2 | Reverse | taccaacaacaacccaacc | ||||
| Contig xx12_101 | F1 | Forward | gatggtgtccccactacagc | 305 bp | ||
| R1 | Reverse | acaactcacgaccaggaacc | ||||
| F2 | Forward | tttaagcagtggtatgccggt | ||||
| R2 | Reverse | accatgttggtaattgccgga | ||||
| Contig xx01_23 | F1 | Forward | cgatcaagtactctcgccga | 237 bp | ||
| R1 | Reverse | gccatcacatgcatcaggaa | ||||
| F2 | Forward | gtactctcgccgatacgtct | ||||
| R2 | Reverse | agcatcaaccgaaaagccag | ||||
| Genome walking | Seq260 | 5′ end | PAWR | Reverse | phos-catcgactggaagtggttgg | NA |
| AWF1 | Forward | gaccgcacttcatccacatg | ||||
| AWR1 | Reverse | atgcttcattgacatcctcatc | ||||
| AWF2 | Forward | cgcgacctatcgttaccaac | ||||
| AWR2 | Reverse | cagaagaaggatgccagtgac | ||||
| AWF7bp | Forward | taatc*c*g | ||||
| AWR7bp | Reverse | ggata*c*c | ||||
| 3′ end | PBWF | Forward | phos-ggcatccttcttctgttacctc | NA | ||
| BWF1 | Forward | cctatcgttaccaaccacttcc | ||||
| BWR1 | Reverse | atgaagtgcggtcatcgac | ||||
| BWF2 | Forward | aggtatcgcggttgtctgag | ||||
| BWR2 | Reverse | atgcttcattgacatcctcatc | ||||
| BWF7bp | Forward | ctaac*t*c | ||||
| ddPCR | Seq260 | 260F | Forward | cagacagattacgatgaggatgt | 100 bp | |
| 260R | Reverse | ggtaacgataggtcgcgatatt | ||||
| 260P | Reverse | FAM-tcgatgaccgcacttcatccacat-BHQ1 | ||||
| MDA | C28 | NA | /5Sp18/nnn*n*n | 20 kb | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Fan, I.X.; Xu, Y.; Rule, J.; Tse, L.P.V.; Pourkarim, M.R.; Lee, W.M.; Di Bisceglie, A.M.; Fan, X. Novel Viral Sequences in a Patient with Cryptogenic Liver Cirrhosis Revealed by Serum Virome Sequencing. Viruses 2025, 17, 812. https://doi.org/10.3390/v17060812
Zhang X, Fan IX, Xu Y, Rule J, Tse LPV, Pourkarim MR, Lee WM, Di Bisceglie AM, Fan X. Novel Viral Sequences in a Patient with Cryptogenic Liver Cirrhosis Revealed by Serum Virome Sequencing. Viruses. 2025; 17(6):812. https://doi.org/10.3390/v17060812
Chicago/Turabian StyleZhang, Xiaoan, Ida X. Fan, Yanjuan Xu, Jody Rule, Long Ping Victor Tse, Mahmoud Reza Pourkarim, William M. Lee, Adrian M. Di Bisceglie, and Xiaofeng Fan. 2025. "Novel Viral Sequences in a Patient with Cryptogenic Liver Cirrhosis Revealed by Serum Virome Sequencing" Viruses 17, no. 6: 812. https://doi.org/10.3390/v17060812
APA StyleZhang, X., Fan, I. X., Xu, Y., Rule, J., Tse, L. P. V., Pourkarim, M. R., Lee, W. M., Di Bisceglie, A. M., & Fan, X. (2025). Novel Viral Sequences in a Patient with Cryptogenic Liver Cirrhosis Revealed by Serum Virome Sequencing. Viruses, 17(6), 812. https://doi.org/10.3390/v17060812