
Virtual Screening and Molecular Dynamics Simulation Targeting the ATP Domain of African Swine Fever Virus Type II DNA Topoisomerase

 ,
,
Abstract
1. Introduction
2. Results
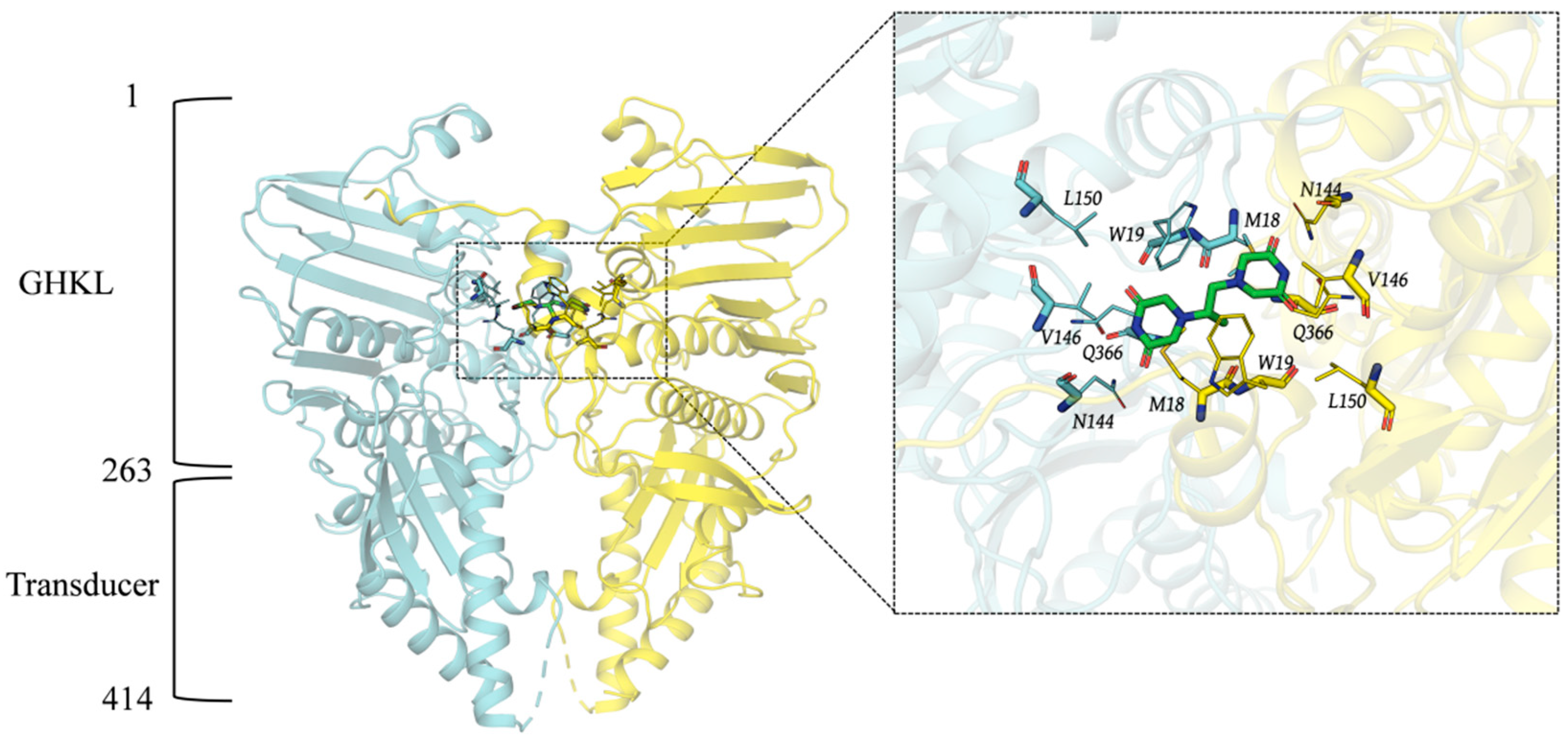
2.1. Three-Dimensional Structure of Topo II ATP Domain and Pocket Analysis
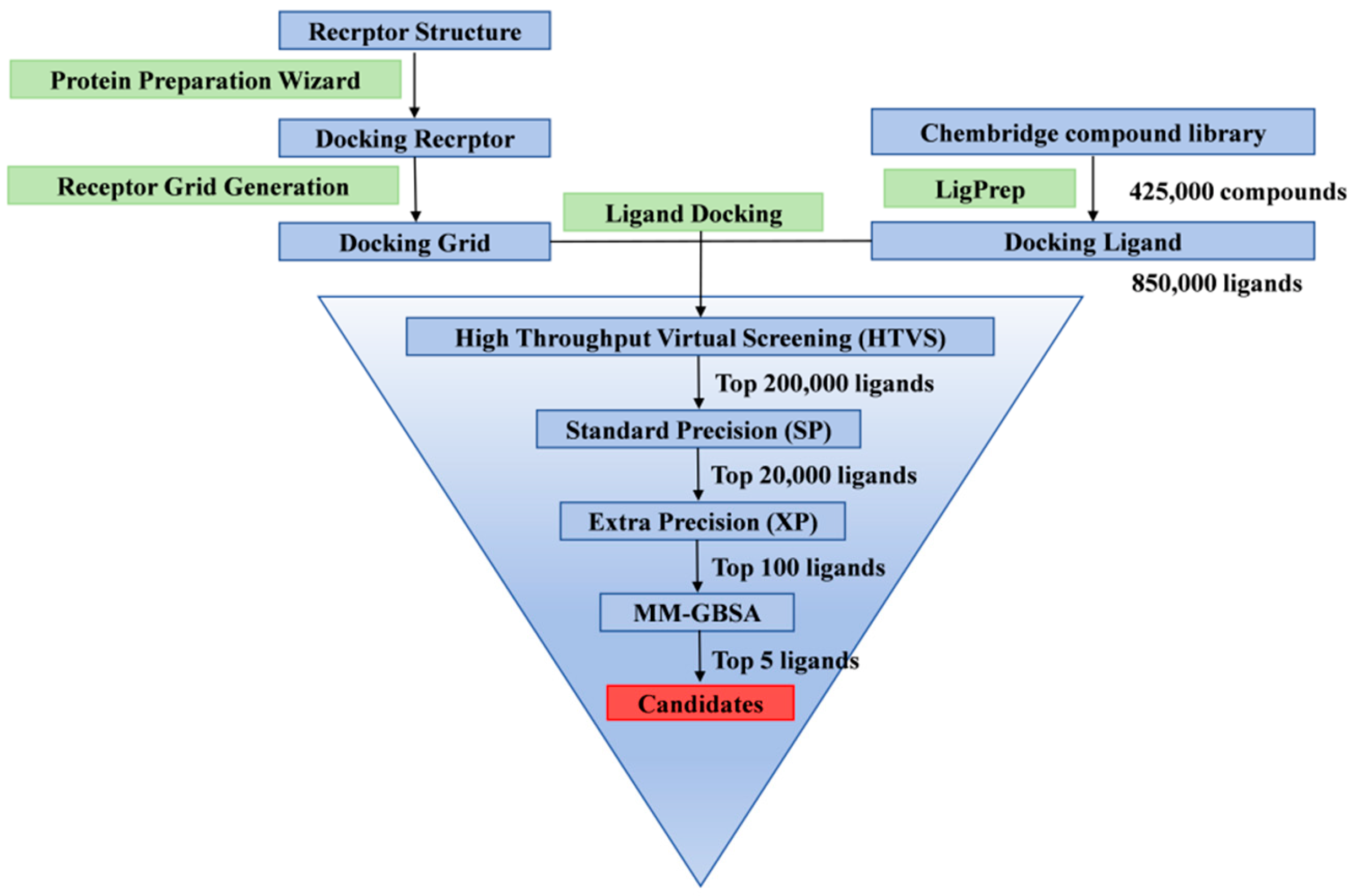
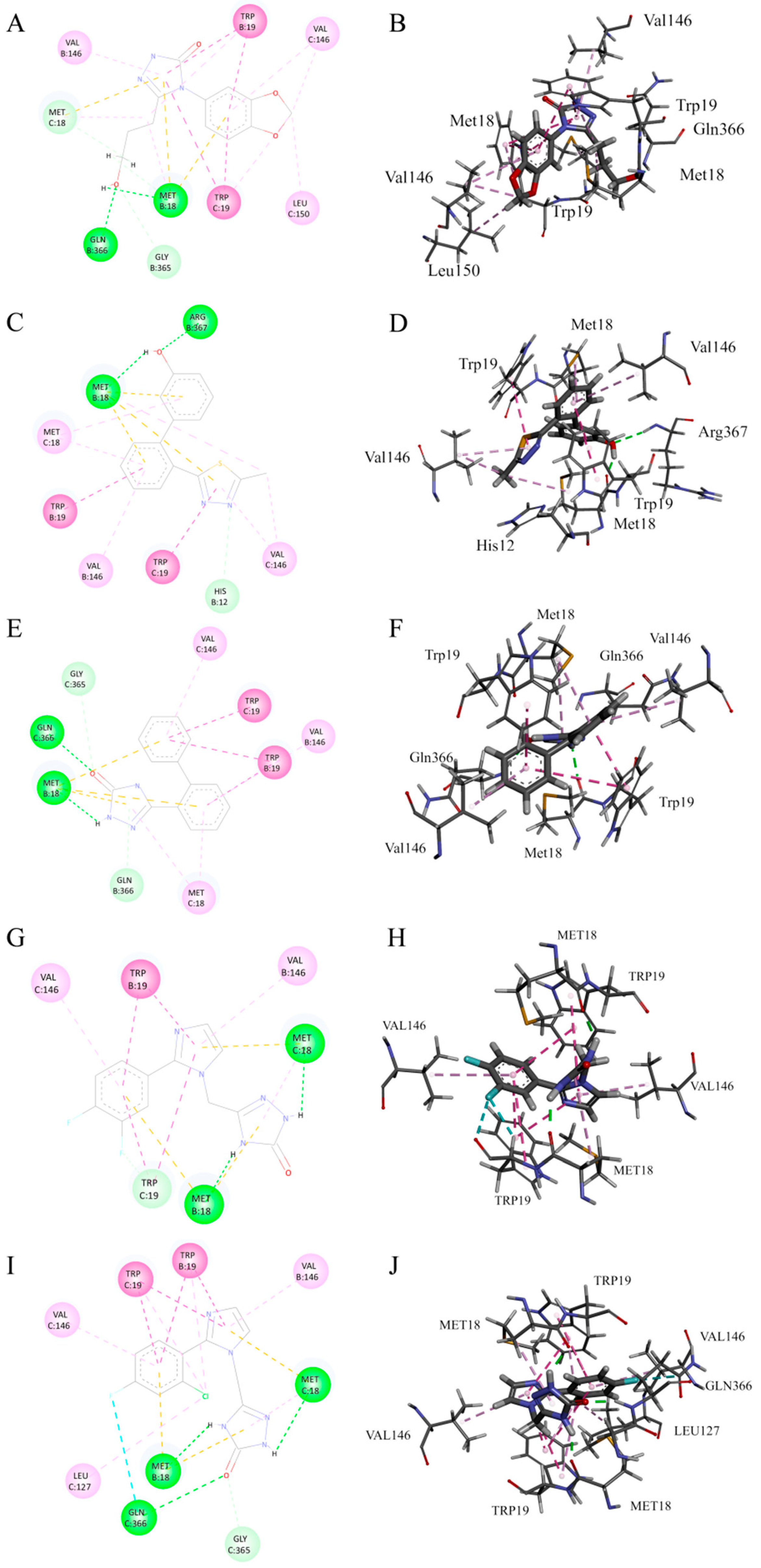
2.2. Virtual Screening Results from Triple Docking
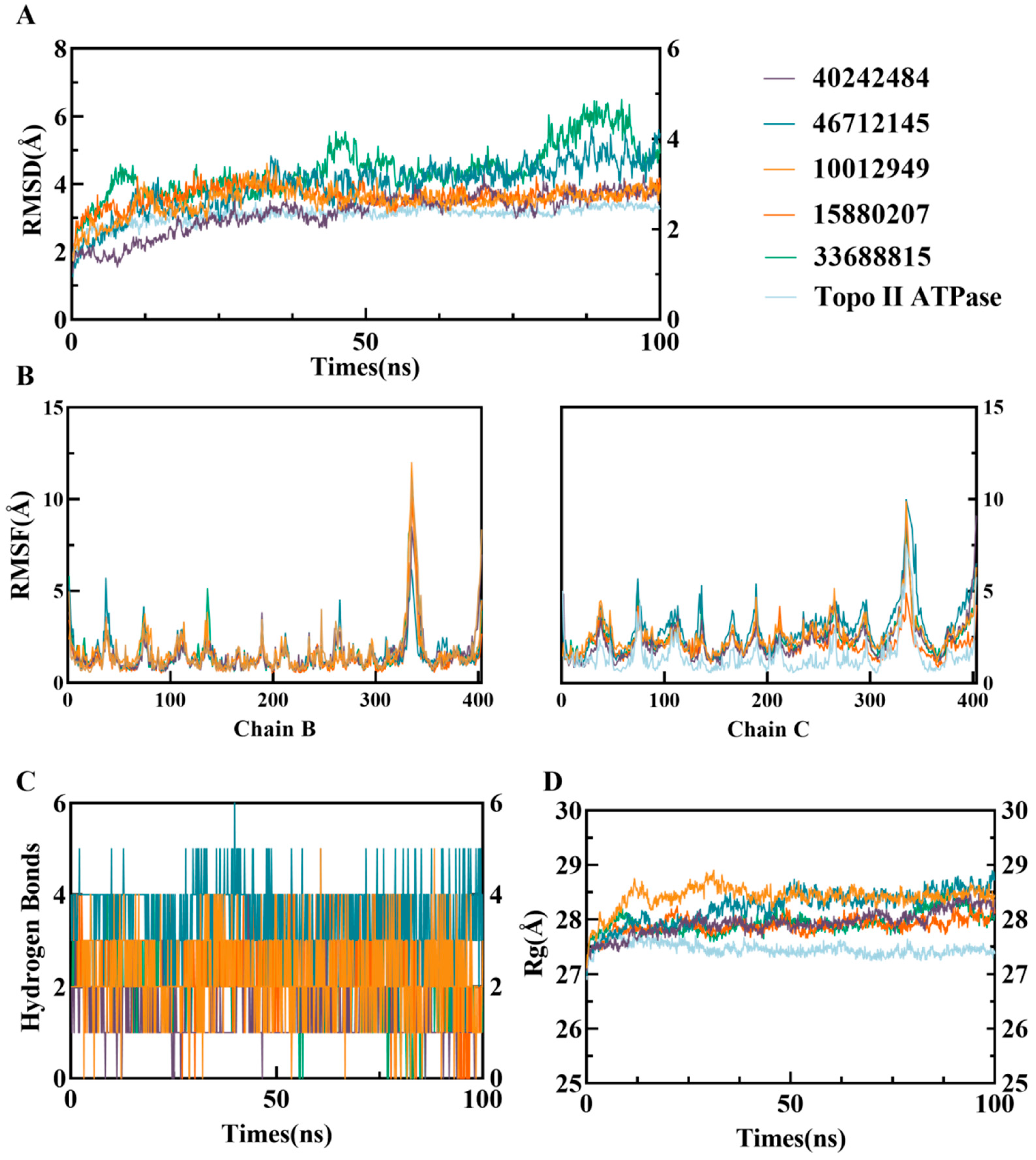
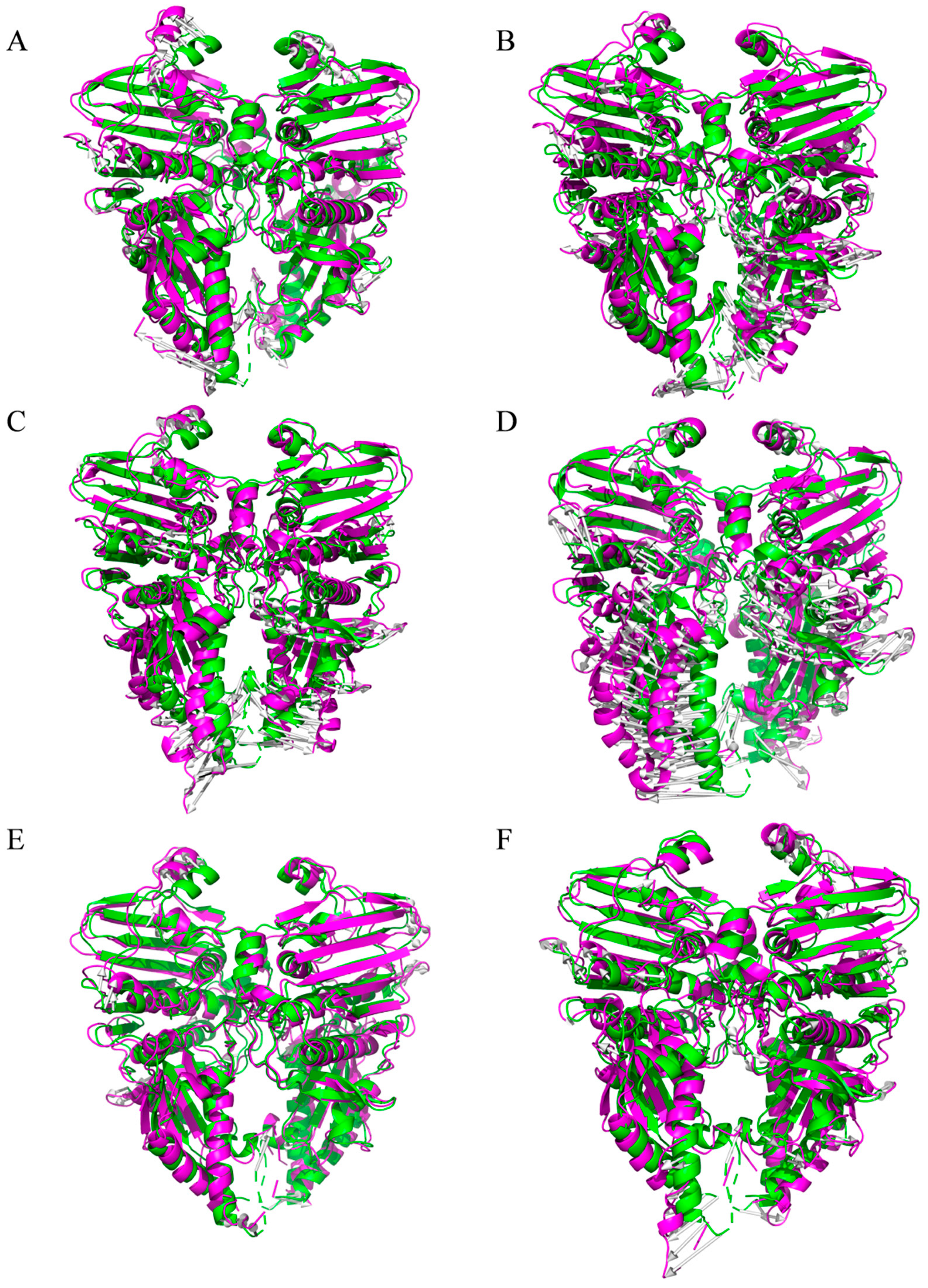
2.3. Molecular Dynamics Simulation Analysis of Candidate Small Molecules with ATP Domain Complexes
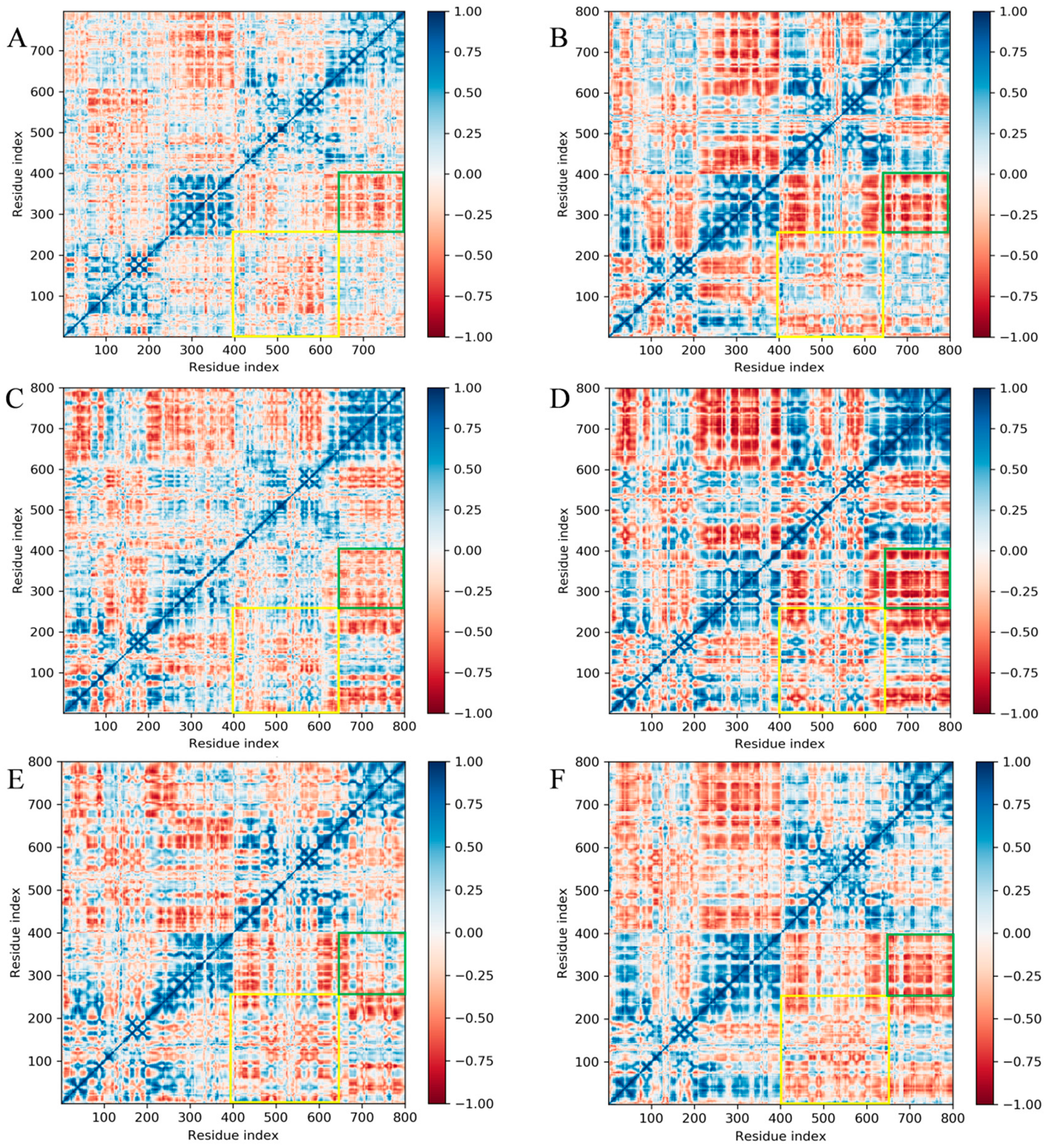
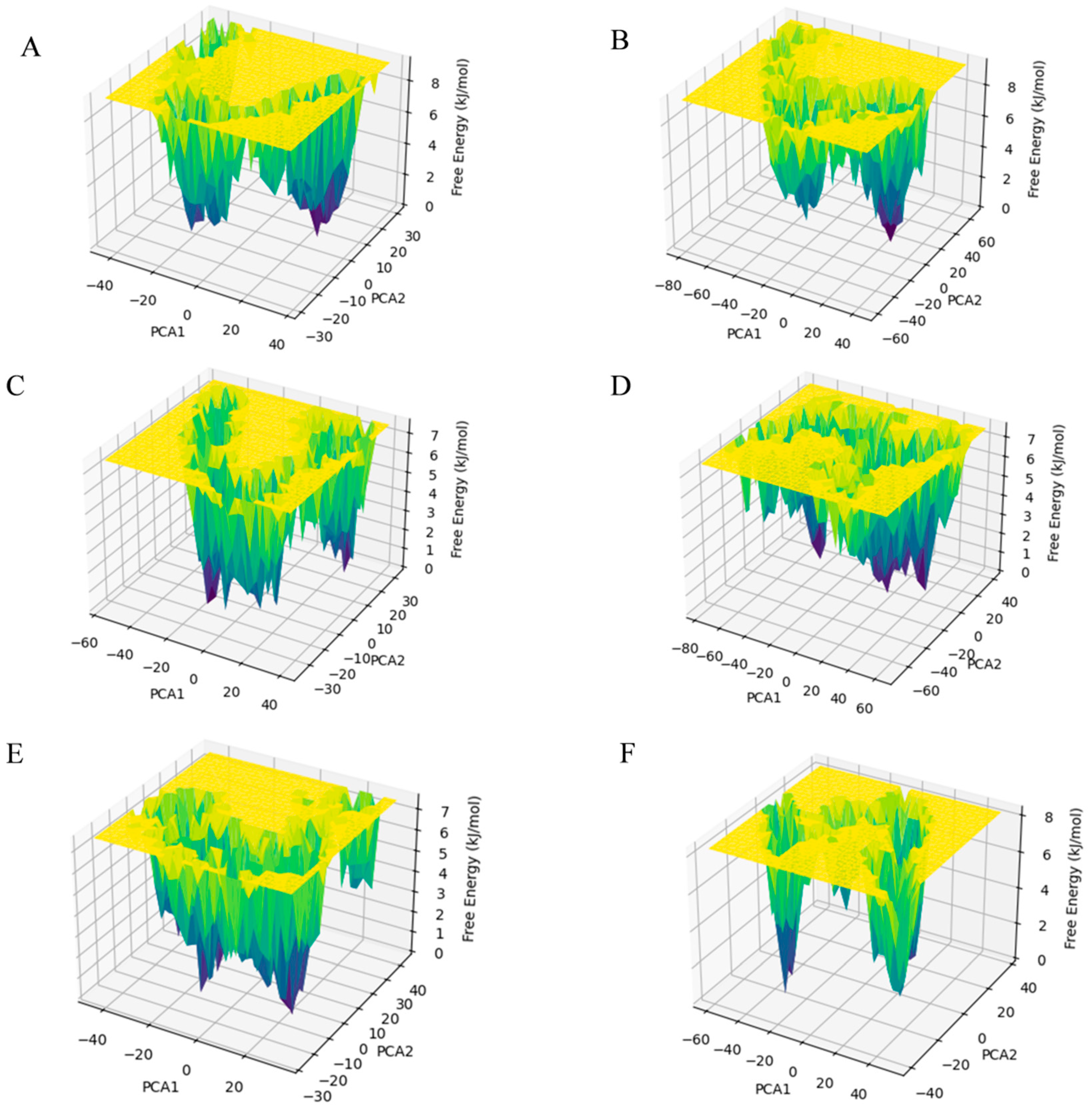
2.4. Free Energy Landscape and Dynamic Cross-Correlation Matrix Analysis
2.5. ADME Prediction
3. Discussion
4. Materials and Methods
4.1. Three-Dimensional Structure of ASFV Topo II ATP Domain and Pocket Analysis
4.2. Compound Library Preparation and Generation of Multiple Conformations
4.3. Molecular Docking and MMGBSA-Based Screening of Small Molecule Compounds
4.4. Molecular Dynamics Simulations of Docked Complexes
4.5. Principal Component Analysis, Free Energy Landscape, and Dynamic Cross-Correlation Matrix Analysis
4.6. In Silico ADME Prediction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African Swine Fever Virus and Implications for Viral Assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Z.; Tesfagaber, W.; Zhang, J.; Li, F.; Sun, E.; Tang, L.; Bu, Z.; Zhu, Y.; Zhao, D. Establishment of an Indirect Immunofluorescence Assay for the Detection of African Swine Fever Virus Antibodies. J. Integr. Agric. 2024, 23, 228–238. [Google Scholar] [CrossRef]
- Ding, L.; Ren, T.; Huang, L.; Tesfagaber, W.; Zhu, Y.; Li, F.; Sun, E.; Bu, Z.; Zhao, D. Developing a Duplex ARMS-qPCR Method to Differentiate Genotype I and II African Swine Fever Viruses Based on Their B646L Genes. J. Integr. Agric. 2023, 22, 1603–1607. [Google Scholar] [CrossRef]
- Yu, Q.; Fu, W.; Zhang, Z.; Liang, D.; Wang, L.; Zhu, Y.; Sun, E.; Li, F.; Bu, Z.; Chen, Y.; et al. The P15 Protein Is a Promising Immunogen for Developing Protective Immunity against African Swine Fever Virus. Protein Cell 2025, pwaf026. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.; Leitão, A. The African Swine Fever Virus (ASFV) Topoisomerase II as a Target for Viral Prevention and Control. Vaccines 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.B.; Frouco, G.; Martins, C.; Leitão, A.; Ferreira, F. In Vitro Inhibition of African Swine Fever Virus-Topoisomerase II Disrupts Viral Replication. Antiviral Res. 2016, 134, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.M.; Wang, S.-C.; Wang, C.-H.; Pang, A.H.; Yang, C.-H.; Chang, Y.-K.; Wu, W.-J.; Tsai, M.-D. A Unified View on Enzyme Catalysis by Cryo-EM Study of a DNA Topoisomerase. Commun. Chem. 2024, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Xin, Y.; Kang, H.; Yang, Y.; Wang, C.; Zhao, D.; Li, X.; Rao, Z.; Chen, Y. Structural Insights into the DNA Topoisomerase II of the African Swine Fever Virus. Nat. Commun. 2024, 15, 4607. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.M.; Baslé, A.; Cowell, I.G.; Van Den Berg, B.; Blower, T.R.; Austin, C.A. A Comprehensive Structural Analysis of the ATPase Domain of Human DNA Topoisomerase II Beta Bound to AMPPNP, ADP, and the Bisdioxopiperazine, ICRF193. Structure 2022, 30, 1129–1145.e3. [Google Scholar] [CrossRef] [PubMed]
- Classen, S.; Olland, S.; Berger, J.M. Structure of the Topoisomerase II ATPase Region and Its Mechanism of Inhibition by the Chemotherapeutic Agent ICRF-187. Proc. Natl. Acad. Sci. USA 2003, 100, 10629–10634. [Google Scholar] [CrossRef] [PubMed]
- Kuang, W.; Zhao, Y.; Li, J.; Deng, Z. Structure-Function Analysis of the ATPase Domain of African Swine Fever Virus Topoisomerase. mBio 2024, 15, e03086-23. [Google Scholar] [CrossRef] [PubMed]
- Walunj, D.; Egarmina, K.; Zipin-Roitman, A.; Muddineni, S.S.N.A.; Tkachenko, I.; Mitra, P.; Tobi, D.; Bazylevich, A.; Shpilberg, O.; Milyavsky, M.; et al. Novel Dual Action Chimera Doxorubizen Demonstrates Superior Efficacy to Doxorubicin in Acute Leukemia. Sci. Rep. 2025, 15, 10607. [Google Scholar] [CrossRef] [PubMed]
- Said, A.I.; Ewes, W.A.; Hamdi, A.; El-Rashedy, A.A.; Ahmed, M. New Pyrrolo[3,4-d] Isoxazolidines Hybrid with Furan as Antitumor Agents and Multi-Target Enzyme Inhibitors: Synthesis and in Silico Study. Bioorg. Chem. 2025, 159, 108377. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.K.J.; Miller, R.J.; Novak, B.; Wang, Z.; Ponder, J.W. Accurate Host–Guest Binding Free Energies Using the AMOEBA Polarizable Force Field. J. Chem. Inf. Model. 2023, 63, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Gervasio, F.L. Metadynamics: A Method to Simulate Rare Events and Reconstruct the Free Energy in Biophysics, Chemistry and Material Science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Arabyan, E.; Hakobyan, A.; Kotsinyan, A.; Karalyan, Z.; Arakelov, V.; Arakelov, G.; Nazaryan, K.; Simonyan, A.; Aroutiounian, R.; Ferreira, F.; et al. Genistein Inhibits African Swine Fever Virus Replication in Vitro by Disrupting Viral DNA Synthesis. Antiviral Res. 2018, 156, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Sun, J.; Li, L.-F.; Cheng, Y.; Li, M.; Fu, L.; Li, S.; Peng, G.; Wang, Y.; Liu, S.; et al. Structural Basis for Difunctional Mechanism of M-AMSA against African Swine Fever Virus pP1192R. Nucleic Acids Res. 2024, 52, 11301–11316. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An Updated Comprehensive Online ADMET Prediction Platform Enhanced with Broader Coverage, Improved Performance, API Functionality and Decision Support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chembridge ID | XP GScore | ΔG Binding (kcal/mol) | ΔEvdW (kcal/mol) | ΔEelec (kcal/mol) | ΔGpol (kcal/mol) |
|---|---|---|---|---|---|
| 10012949 | −10.552 | −70 ± 5.64 | −43.55 ± 2.76 | −14.12 ± 4.49 | 20.79 ± 2.94 |
| 40242484 | −9.511 | −73.88 ± 5.78 | −43.39 ± 2.72 | −12.37 ± 3.73 | 22.78 ± 2.27 |
| 46712145 | −11.929 | −74.43 ± 5.67 | −42.25 ± 2.42 | −18.77 ± 3.8 | 25.02 ± 2.52 |
| 15880207 | −9.394 | −65.25 ± 7.07 | −43.28 ± 3.24 | −19.67 ± 4.59 | 27.71 ± 2.42 |
| 33688815 | −10.222 | −63.05 ± 7.08 | −42.6 ± 3.12 | −16.41 ± 6.3 | 25.38 ± 2.48 |
| Chembridge ID | MW | TPSA | logS | logD | logP | caco2 | BBB | cl-plasma |
|---|---|---|---|---|---|---|---|---|
| 10012949 | 263.09 | 89.37 | −2.06 | 0.69 | 0.26 | −5.17 | 0.94 | 4.12 |
| 40242484 | 268.07 | 46.01 | −3.57 | 3.03 | 2.84 | −4.88 | 0.65 | 4.94 |
| 46712145 | 237.09 | 61.54 | −3.14 | 2.21 | 2.31 | −5.15 | 0.21 | 6.61 |
| 15880207 | 277.08 | 79.36 | −3.00 | 1.27 | 1.06 | −5.52 | 0.56 | 5.72 |
| 33688815 | 295.06 | 70.98 | −3.52 | 1.94 | 1.53 | −5.64 | 0.07 | 4.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Hou, L.; Tesfagaber, W.; Song, L.; Zhang, Z.; Li, F.; Bu, Z.; Zhao, D. Virtual Screening and Molecular Dynamics Simulation Targeting the ATP Domain of African Swine Fever Virus Type II DNA Topoisomerase. Viruses 2025, 17, 681. https://doi.org/10.3390/v17050681
Zhao R, Hou L, Tesfagaber W, Song L, Zhang Z, Li F, Bu Z, Zhao D. Virtual Screening and Molecular Dynamics Simulation Targeting the ATP Domain of African Swine Fever Virus Type II DNA Topoisomerase. Viruses. 2025; 17(5):681. https://doi.org/10.3390/v17050681
Chicago/Turabian StyleZhao, Rui, Lezi Hou, Weldu Tesfagaber, Linfei Song, Zhenjiang Zhang, Fang Li, Zhigao Bu, and Dongming Zhao. 2025. "Virtual Screening and Molecular Dynamics Simulation Targeting the ATP Domain of African Swine Fever Virus Type II DNA Topoisomerase" Viruses 17, no. 5: 681. https://doi.org/10.3390/v17050681
APA StyleZhao, R., Hou, L., Tesfagaber, W., Song, L., Zhang, Z., Li, F., Bu, Z., & Zhao, D. (2025). Virtual Screening and Molecular Dynamics Simulation Targeting the ATP Domain of African Swine Fever Virus Type II DNA Topoisomerase. Viruses, 17(5), 681. https://doi.org/10.3390/v17050681