Correction: Moonga et al. Application of a Sensitive Capture Sequencing Approach to Reservoir Surveillance Detects Novel Viruses in Zambian Wild Rodents. Viruses 2024, 16, 1754

 , , ,
, , ,  , ,
, ,  and
and Text Correction
Error in Figure/Table
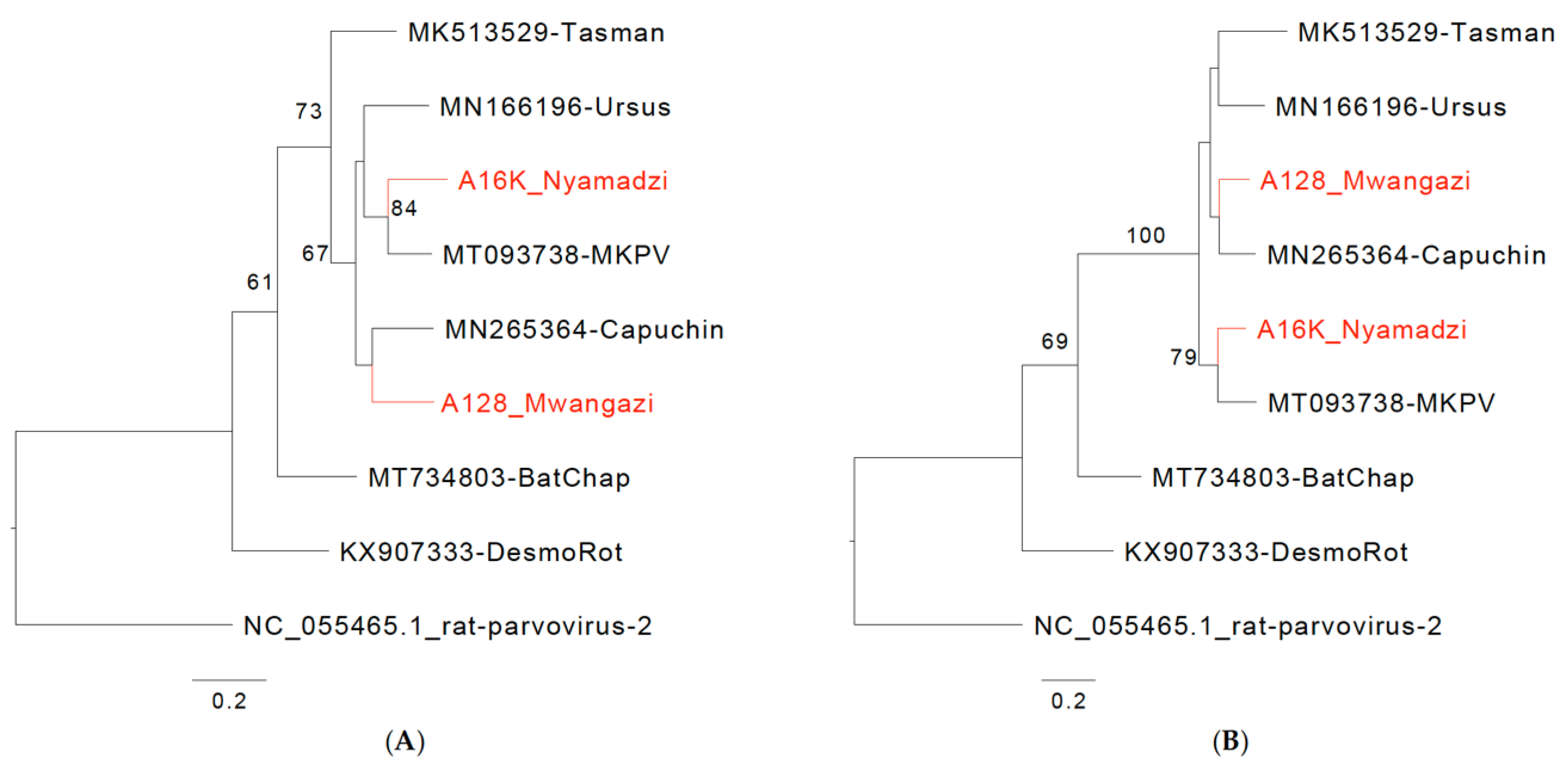


{kind=link}
{kind=link}
| Canonical donor consensus | mAG GTr | mAG GTr | |
| MKPV | D1 GAAGGAG GTGAGTCAG | D2 GCCGAAG GTAATTAAA | |
| CKPV | D1 GAAAGAG GTGAGTCGC | D2 CCTGAAG GTACTTATC | |
| Nyamadzi virus | D1 GGTGGAG GTGAGGGAG | D2 GCGGAAG GTACTTATT | |
| Mwangazi virus | D1 GCAGAAG GAGAATCGG | D2 CCAGAAG GTACTTATT | |
| D1a CCACAAG GTGCGAAAA | |||
| Canonical acceptor consensus | cAG Gk | cAG Gk | cAG Gk |
| MKPV | A1 CTTCTTACAG ATGTCTAT | A2 TTATTTGCAG AGCTAGTG | A3 TTATTTACAG AAACACTA |
| CKPV | A1 ATGCATGCAG ATGTCTAT | A2 TCTTTTGCAG AACTAGTG | A3 TTATTTACAG CAACAATA |
| Nyamadzi virus | A1 TAATTTACAG ATGTCTCT | A2 TTATTTGCAG AGCTAGTG | A3 TCATTTACAG AAACAATA |
| Mwangazi virus | A1 TCATCTACAG ATGTCTAT | A2 TTGTTTGCAG AATTAGTG | A3 TTATTTGCAG AACAAATA |
Reference
- Moonga, L.C.; Chipinga, J.; Collins, J.P.; Kapoor, V.; Saasa, N.; Nalubamba, K.S.; Hang’ombe, B.M.; Namangala, B.; Lundu, T.; Lu, X.-J.; et al. Application of a Sensitive Capture Sequencing Approach to Reservoir Surveillance Detects Novel Viruses in Zambian Wild Rodents. Viruses 2024, 16, 1754. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moonga, L.C.; Chipinga, J.; Collins, J.P.; Kapoor, V.; Saasa, N.; Nalubamba, K.S.; Hang’ombe, B.M.; Namangala, B.; Lundu, T.; Lu, X.-J.; et al. Correction: Moonga et al. Application of a Sensitive Capture Sequencing Approach to Reservoir Surveillance Detects Novel Viruses in Zambian Wild Rodents. Viruses 2024, 16, 1754. Viruses 2025, 17, 561. https://doi.org/10.3390/v17040561
Moonga LC, Chipinga J, Collins JP, Kapoor V, Saasa N, Nalubamba KS, Hang’ombe BM, Namangala B, Lundu T, Lu X-J, et al. Correction: Moonga et al. Application of a Sensitive Capture Sequencing Approach to Reservoir Surveillance Detects Novel Viruses in Zambian Wild Rodents. Viruses 2024, 16, 1754. Viruses. 2025; 17(4):561. https://doi.org/10.3390/v17040561
Chicago/Turabian StyleMoonga, Lavel C., Jones Chipinga, John P. Collins, Vishal Kapoor, Ngonda Saasa, King S. Nalubamba, Bernard M. Hang’ombe, Boniface Namangala, Tapiwa Lundu, Xiang-Jun Lu, and et al. 2025. "Correction: Moonga et al. Application of a Sensitive Capture Sequencing Approach to Reservoir Surveillance Detects Novel Viruses in Zambian Wild Rodents. Viruses 2024, 16, 1754" Viruses 17, no. 4: 561. https://doi.org/10.3390/v17040561
APA StyleMoonga, L. C., Chipinga, J., Collins, J. P., Kapoor, V., Saasa, N., Nalubamba, K. S., Hang’ombe, B. M., Namangala, B., Lundu, T., Lu, X.-J., Yingst, S., Wickiser, J. K., & Briese, T. (2025). Correction: Moonga et al. Application of a Sensitive Capture Sequencing Approach to Reservoir Surveillance Detects Novel Viruses in Zambian Wild Rodents. Viruses 2024, 16, 1754. Viruses, 17(4), 561. https://doi.org/10.3390/v17040561