
The Chikungunya Virus nsP3 Macro Domain Inhibits Activation of the NF-κB Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Stocks and Infection
2.3. Plaque Assays
2.4. Plasmid Constructs
2.5. Luciferase Assays
2.6. Immunofluorescence
2.7. Western Blotting
3. Results
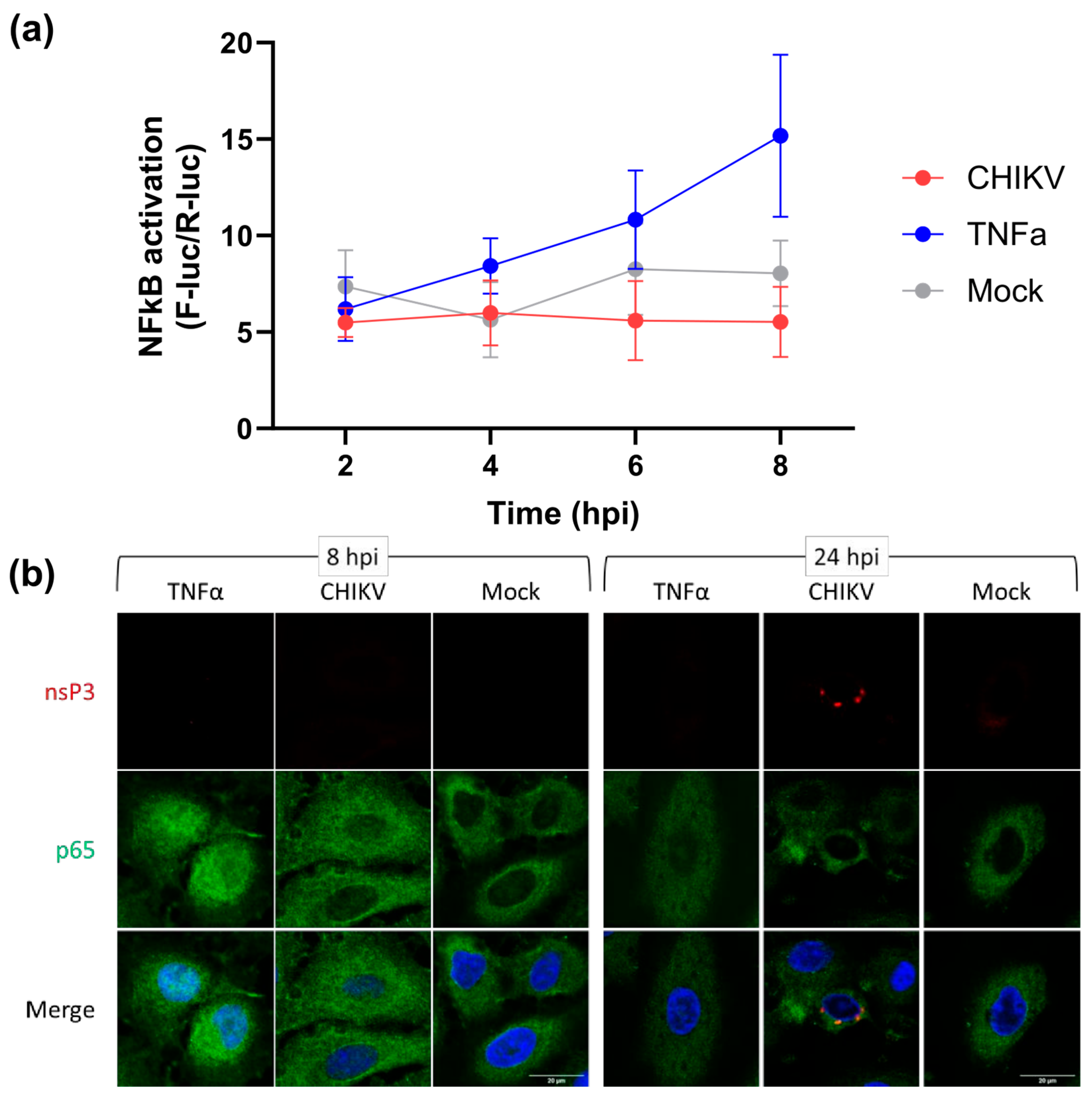
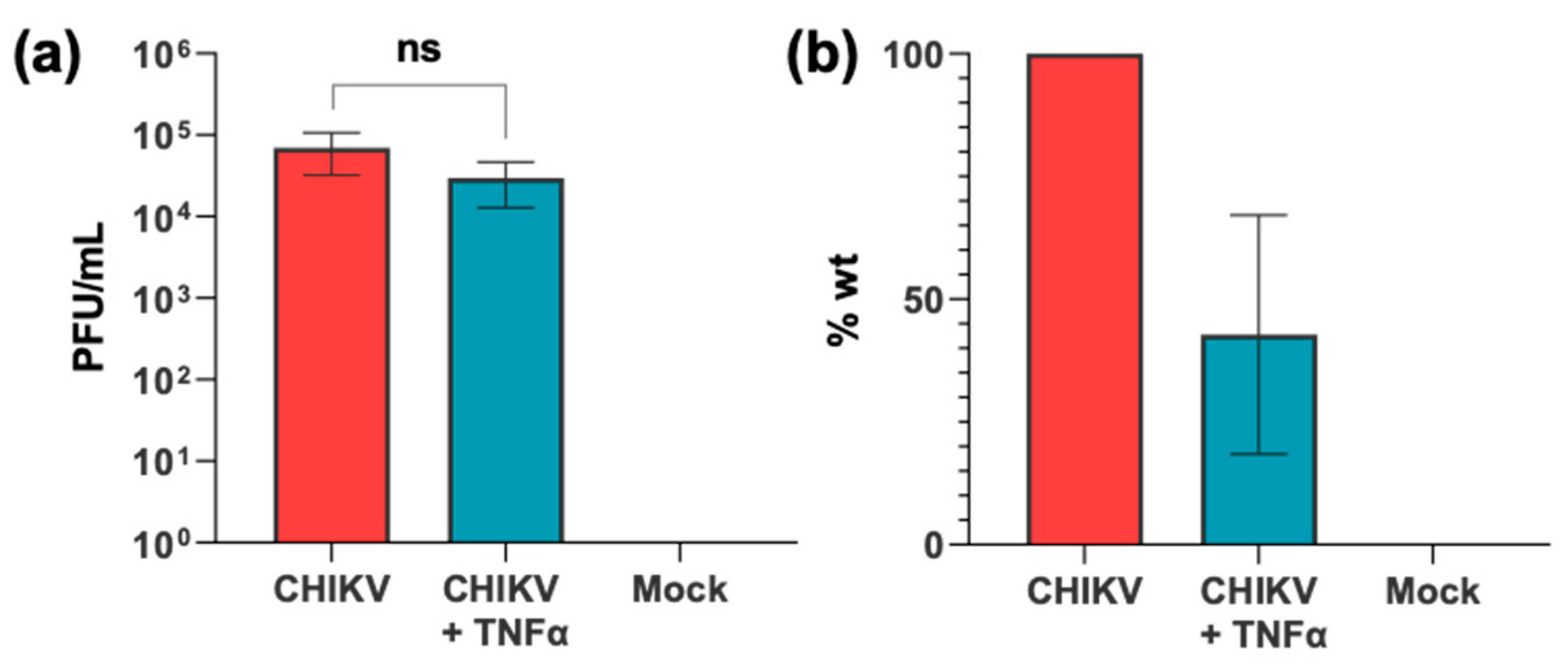
3.1. CHIKV Infection Does Not Activate the NF-κB Pathway
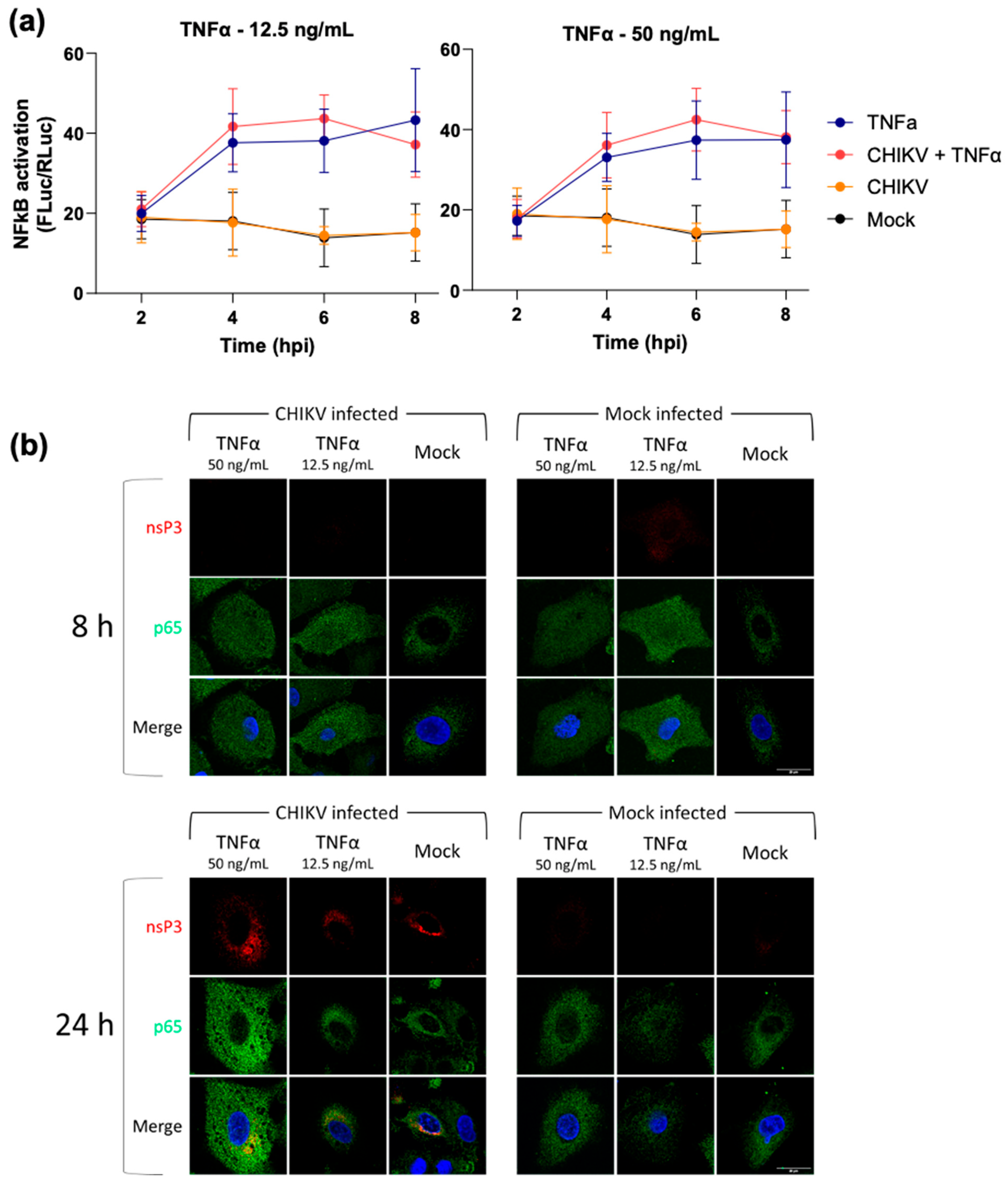
3.2. CHIKV Infection Does Not Inhibit Exogenous Activation of the NF-κB Pathway
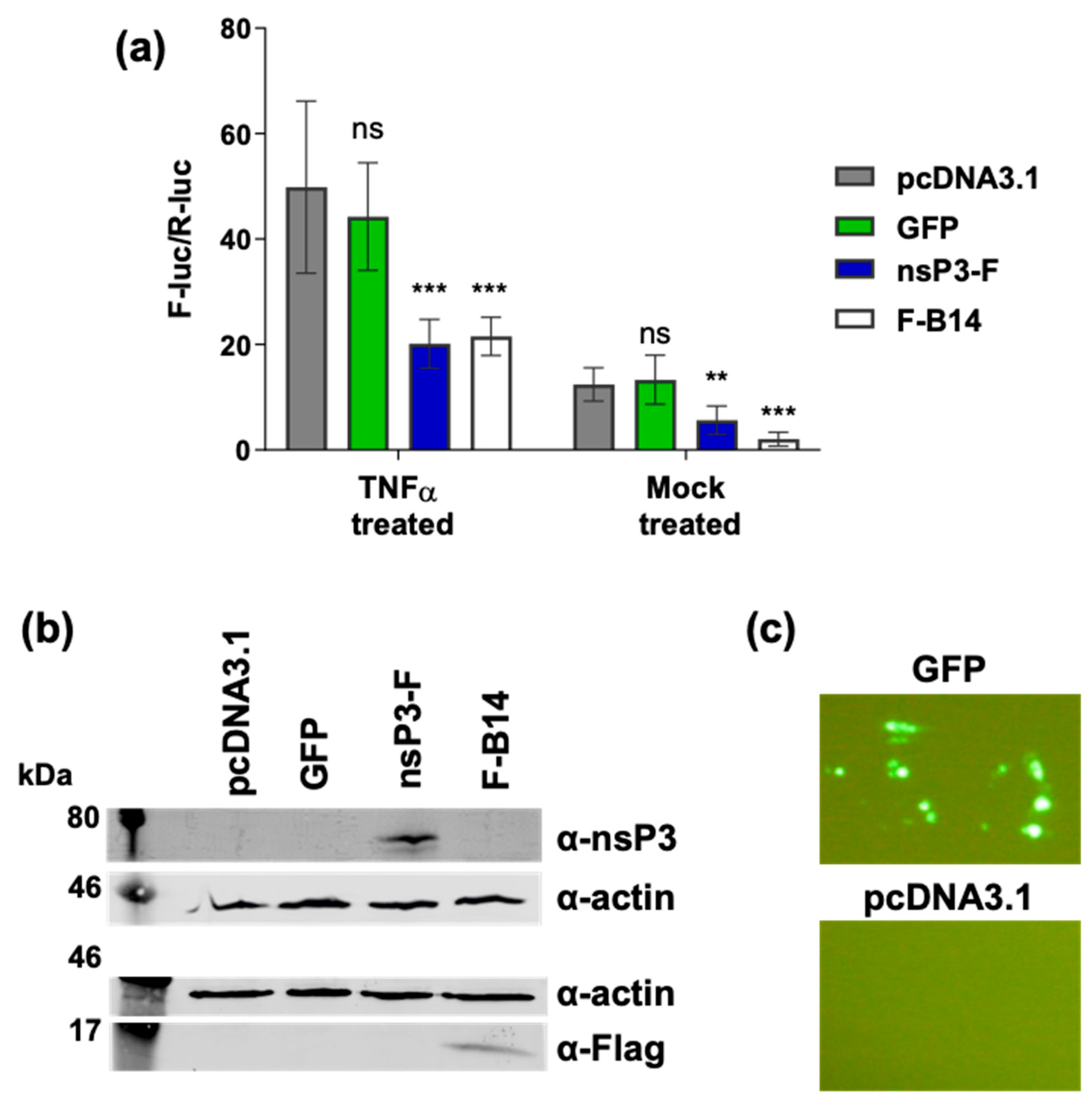
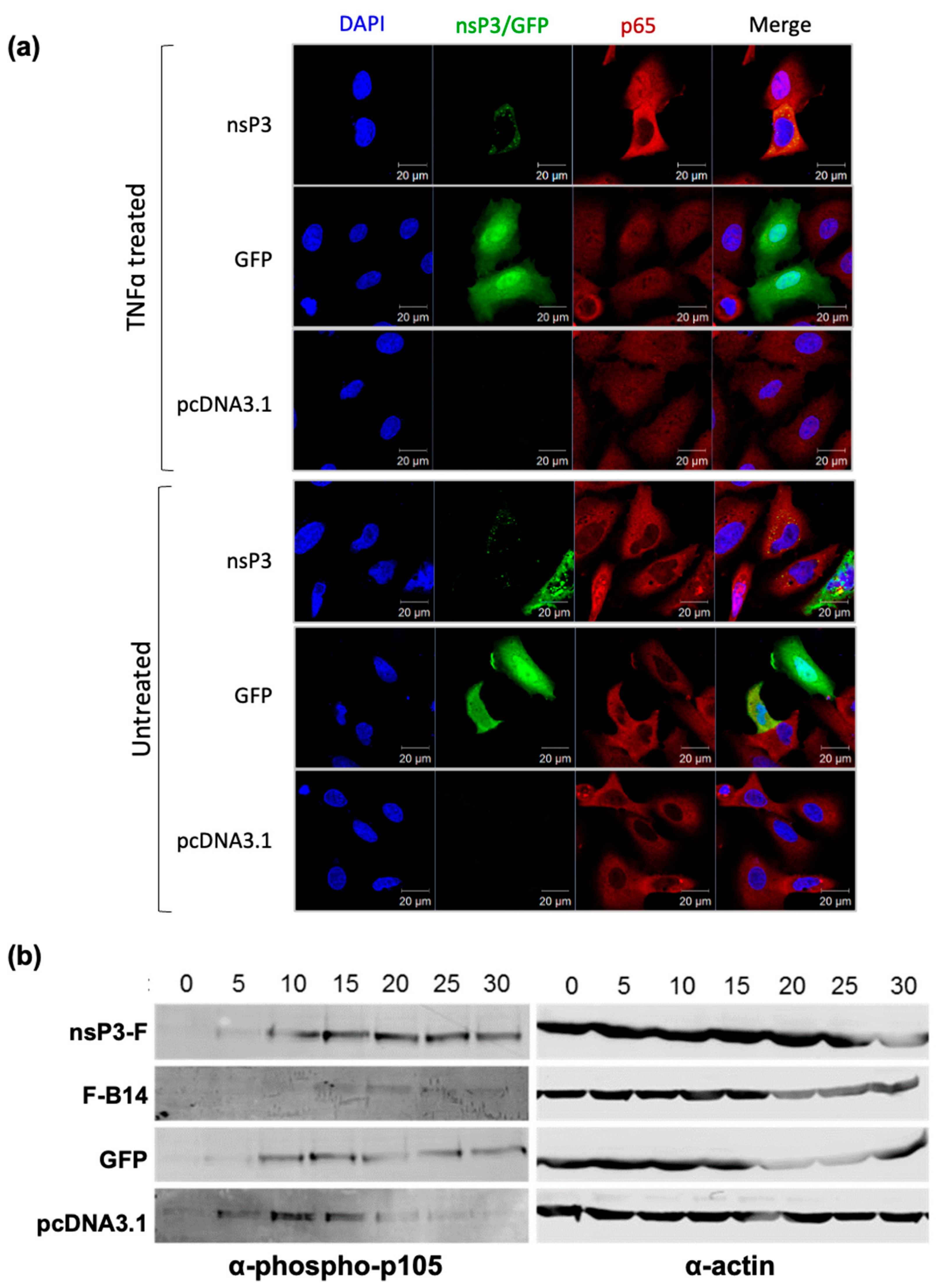
3.3. Ectopic Expression of nsP3 in the Absence of Other CHIKV Proteins Inhibits the NF-κB Pathway
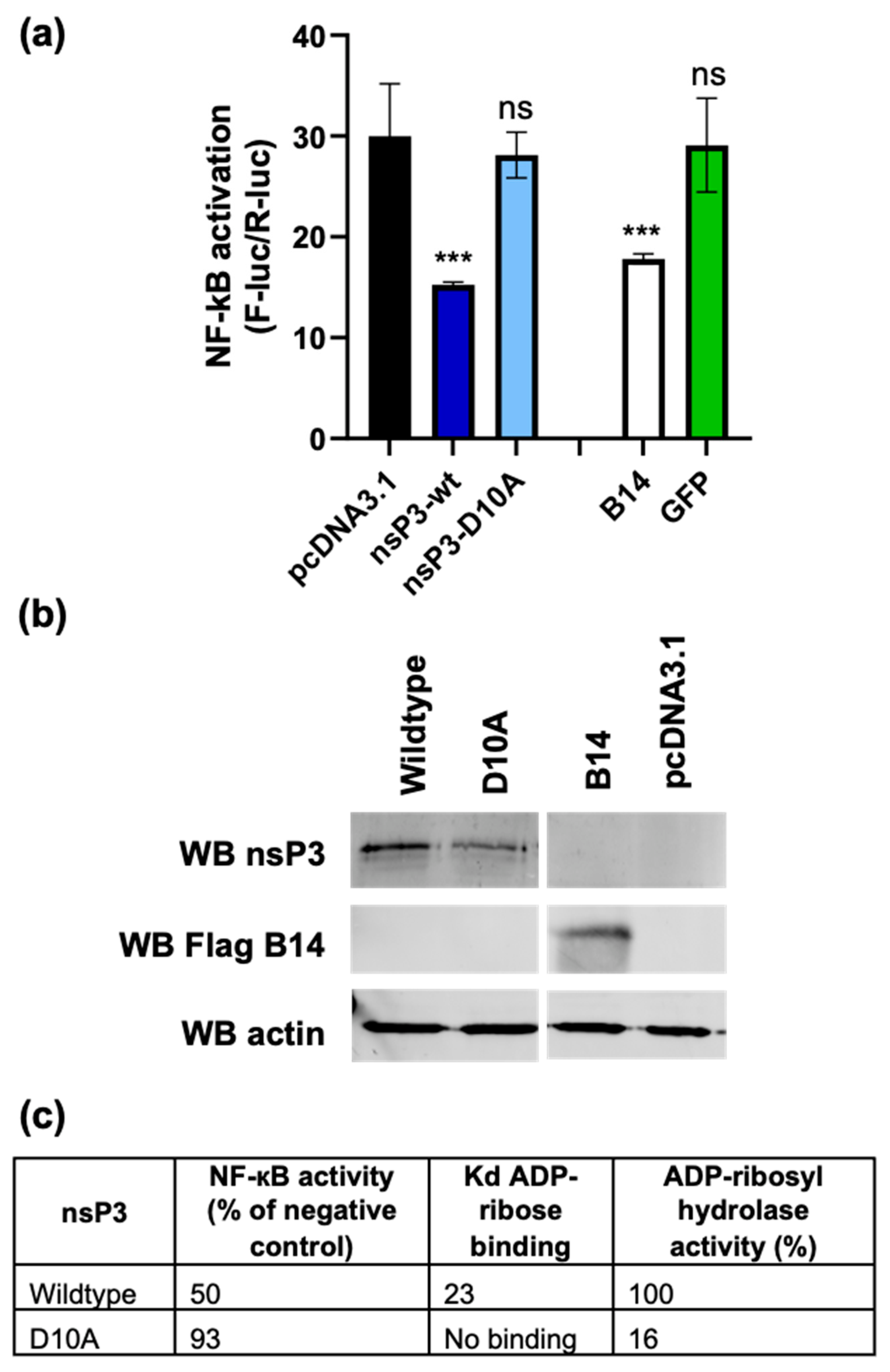
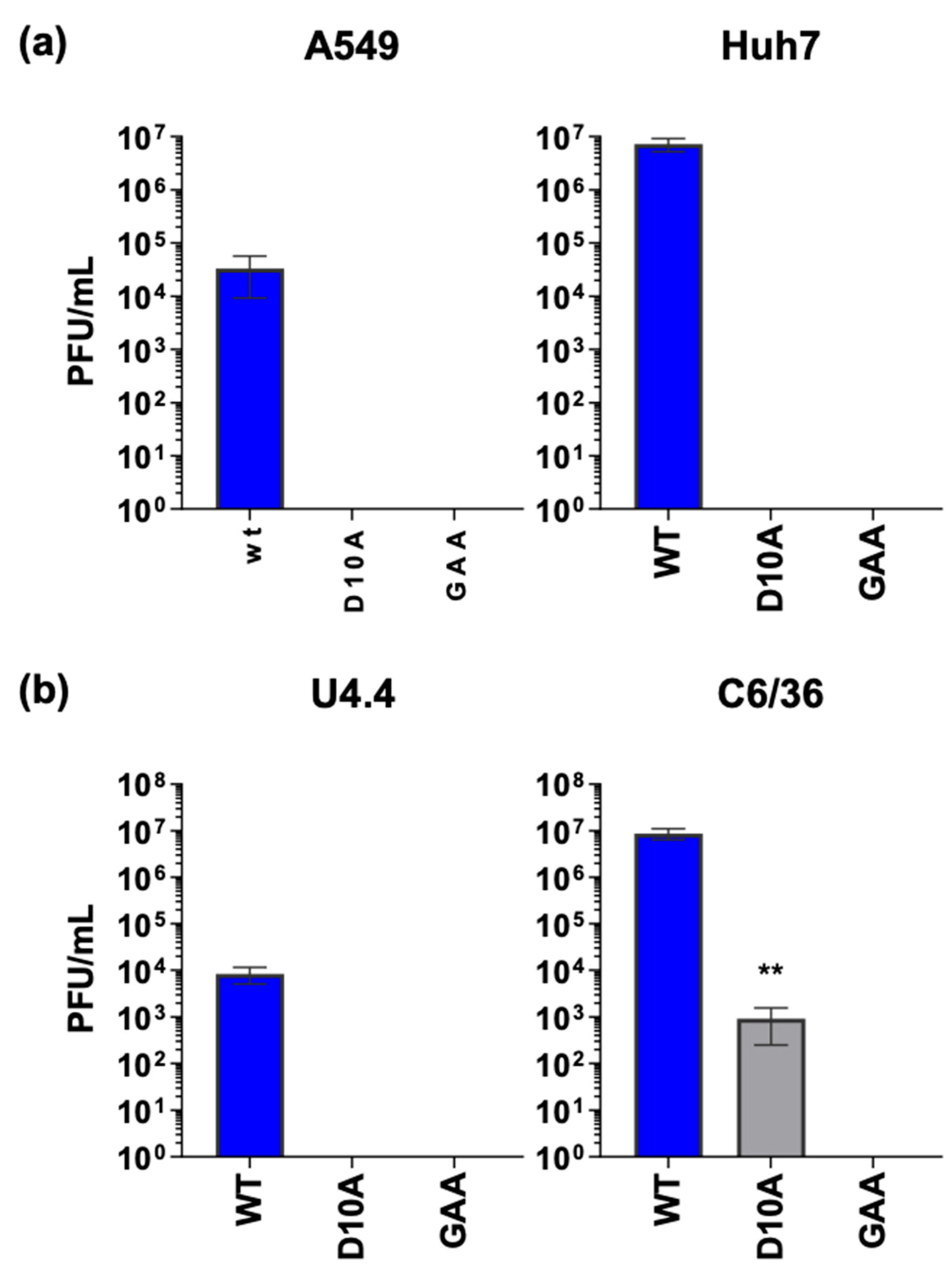
3.4. The nsP3 Macro Domain Contributes to Inhibition of NF-κB Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, R.; Mukhopadhyay, S.; Merits, A.; Bolling, B.; Nasar, F.; Coffey, L.L.; Powers, A.; Weaver, S.C.; Lefkowitz, E.J.; Davison, A.J.; et al. ICTV virus taxonomy profile: Togaviridae. J. Gen. Virol. 2018, 99, 761–762. [Google Scholar] [CrossRef] [PubMed]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Global expansion of chikungunya virus: Mapping the 64-year history. Int. J. Infect. Dis. 2017, 58, 69–76. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Heath, C.J.; Lowther, J.; Noël, T.P.; Mark-George, I.; Boothroyd, D.B.; Mitchell, G.; MacPherson, C.; LaBeaud, A.D. The identification of risk factors for chronic chikungunya arthralgia in Grenada, West Indies: A cross-sectional cohort study. Open Forum Infect. Dis. 2018, 5, ofx234. [Google Scholar] [CrossRef] [PubMed]
- Zaid, A.; Gérardin, P.; Taylor, A.; Mostafavi, H.; Malvy, D.; Mahalingam, S. Review: Chikungunya Arthritis: Implications of Acute and Chronic Inflammation Mechanisms on Disease Management. Arthritis Rheumatol. 2018, 70, 484–495. [Google Scholar] [CrossRef]
- Chang, A.Y.; Encinales, L.; Porras, A.; Pacheco, N.; Reid, S.P.; Martins, K.A.O.; Pacheco, S.; Bravo, E.; Navarno, M.; Rico Mendoza, A.; et al. Frequency of Chronic Joint Pain Following Chikungunya Virus Infection. Arthritis Rheumatol. 2018, 70, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Economopoulou, A.; Dominguez, M.; Helynck, B.; Sissoko, D.; Wichmann, O.; Quenel, P.; Germonneau, P.; Quatresous, I. Atypical Chikungunya virus infections: Clinical manifestations, mortality and risk factors for severe disease during the 2005–2006 outbreak on Réunion. Epidemiol. Infect. 2009, 137, 534–541. [Google Scholar] [CrossRef]
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef]
- Lemm, J.A.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M.; Rümenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7517863 (accessed on 19 November 2024). [CrossRef] [PubMed]
- Singh, A.; Kumar, A.; Yadav, R.; Uversky, V.N.; Giri, R. Deciphering the dark proteome of Chikungunya virus. Sci. Rep. 2018, 8, 5822. [Google Scholar] [CrossRef]
- Law, M.C.Y.; Zhang, K.; Tan, Y.B.; Nguyen, T.M.; Luo, D. Chikungunya virus nonstructural protein 1 is a versatile RNA capping and decapping enzyme. J. Biol. Chem. 2023, 299, 105415. [Google Scholar] [CrossRef]
- Zhang, K.; Law, Y.S.; Law, M.C.Y.; Tan, Y.B.; Wirawan, M.; Luo, D. Structural insights into viral RNA capping and plasma membrane targeting by Chikungunya virus nonstructural protein 1. Cell Host Microbe 2021, 29, 757–764.e3. [Google Scholar] [CrossRef]
- Rikkonen, M. Functional Significance of the Nuclear-Targeting and NTP-Binding Motifs of Semliki Forest Virus Nonstructural Protein nsP2. Virology 1996, 218, 352–361. [Google Scholar] [CrossRef]
- Russo, A.T.; White, M.A.; Watowich, S.J. The Crystal Structure of the Venezuelan Equine Encephalitis Alphavirus nsP2 Protease. Structure 2006, 14, 1449–1458. [Google Scholar] [CrossRef]
- Chen, M.W.; Tan, Y.B.; Zheng, J.; Zhao, Y.; Lim, B.T.; Cornvik, T.; Lescar, J.; Ng, L.F.P.; Luo, D. Chikungunya virus nsP4 RNA-dependent RNA polymerase core domain displays detergent-sensitive primer extension and terminal adenylyltransferase activities. Antivir. Res. 2017, 143, 38–47. [Google Scholar] [CrossRef]
- Götte, B.; Liu, L.; McInerney, G.M. The Enigmatic Alphavirus Non-Structural Protein 3 (nsP3) Revealing Its Secrets at Last. Viruses 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.L.; McPherson, R.L.; Griffin, D.E. Macrodomain ADP-ribosylhydrolase and the pathogenesis of infectious diseases. PLoS Pathog. 2018, 14, e1006864. [Google Scholar] [CrossRef] [PubMed]
- Malet, H.; Coutard, B.; Jamal, S.; Dutartre, H.; Papageorgiou, N.; Neuvonen, M.; Ahola, T.; Forrester, N.; Gould, E.A.; Lafitte, D.; et al. The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol. 2009, 83, 6534–6545. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Debing, Y.; Jankevicius, G.; Neyts, J.; Ahel, I.; Coutard, B.; Canard, B. Viral Macro Domains Reverse Protein ADP-Ribosylation. J. Virol. 2016, 90, 8478–8486. [Google Scholar] [CrossRef]
- McPherson, R.L.; Abraham, R.; Sreekumar, E.; Ong, S.-E.; Cheng, S.-J.; Baxter, V.K.; Kistemaker, H.A.V.; Filippov, D.V.; Griffin, D.E.; Leung, A.K.L. ADP-ribosylhydrolase activity of Chikungunya virus macrodomain is critical for virus replication and virulence. Proc. Natl. Acad. Sci. USA 2017, 114, 1666–1671. [Google Scholar] [CrossRef]
- Krieg, S.; Pott, F.; Potthoff, L.; Verheirstraeten, M.; Bütepage, M.; Golzmann, A.; Lippok, B.; Goffinet, C.; Lüscher, B.; Korn, P. Mono-ADP-ribosylation by PARP10 inhibits Chikungunya virus nsP2 proteolytic activity and viral replication. Cell. Mol. Life Sci. 2023, 80, 72. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Goonawardane, N.; Ward, J.; Tuplin, A.; Harris, M. Multiple roles of the non-structural protein 3 (nsP3) alphavirus unique domain (AUD) during Chikungunya virus genome replication and transcription. PLoS Pathog. 2019, 15, e1007239. [Google Scholar] [CrossRef]
- Abdullah, N.; Ahemad, N.; Aliazis, K.; Khairat, J.E.; Lee, T.C.; Abdul Ahmad, S.A.; Adnan NA, A.; Macha, N.O.; Hassan, S.S. The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence. Viruses 2021, 13, 1021. [Google Scholar] [CrossRef]
- Vihinen, H.; Ahola, T.; Tuittila, M.; Merits, A.; Kääriäinen, L. Elimination of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J. Biol. Chem. 2001, 276, 5745–5752. [Google Scholar] [CrossRef]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Selvamani, S.P.; Mishra, R.; Singh, S.K. Chikungunya Virus Exploits miR-146a to Regulate NF-κB Pathway in Human Synovial Fibroblasts. PLoS ONE 2014, 9, e103624. [Google Scholar] [CrossRef]
- Bae, S.; Lee, J.Y.; Myoung, J. Chikungunya Virus nsP2 Impairs MDA5/RIG-I-Mediated Induction of NF-κB Promoter Activation: A Potential Target for Virus-Specific Therapeutics. J. Microbiol. Biotechnol. 2020, 30, 1801–1809. [Google Scholar] [CrossRef]
- Gao, S.; Liu, X.; Han, B.; Wang, N.; Lv, X.; Guan, X.; Xu, G.; Huang, J.; Shi, W.; Liu, M. Salmonid alphavirus non-structural protein 2 is a key protein that activates the NF-κB signaling pathway to mediate inflammatory responses. Fish Shellfish. Immunol. 2022, 129, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; De La Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The role of IKKβ in Venezuelan equine encephalitis virus infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef]
- Bakovic, A.; Bhalla, N.; Kortchak, S.; Sun, C.; Zhou, W.; Ahmed, A.; Risner, K.; Klimstra, W.B.; Narayanan, A. Venezuelan Equine Encephalitis Virus nsP3 Phosphorylation Can Be Mediated by IKKβ Kinase Activity and Abrogation of Phosphorylation Inhibits Negative-Strand Synthesis. Viruses 2020, 12, 1021. [Google Scholar] [CrossRef]
- Yeh, J.X.; Park, E.; Schultz, K.L.W.; Griffin, D.E. NF-κB Activation Promotes Alphavirus Replication in Mature Neurons. J. Virol. 2019, 93, 24. [Google Scholar] [CrossRef] [PubMed]
- Verheugd, P.; Forst, A.H.; Milke, L.; Herzog, N.; Feijs, K.L.H.; Kremmer, E.; Kleine, H.; Lüscher, B. Regulation of NF-κB signalling by the mono-ADP-ribosyltransferase ARTD10. Nat. Commun. 2013, 4, 1683. [Google Scholar] [CrossRef] [PubMed]
- Pohjala, L.; Utt, A.; Varjak, M.; Lulla, A.; Merits, A.; Ahola, T.; Tammela, P. Inhibitors of Alphavirus Entry and Replication Identified with a Stable Chikungunya Replicon Cell Line and Virus-Based Assays. PLoS ONE 2011, 6, e28923. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.A.-J.; Ryzhakov, G.; Cooray, S.; Randow, F.; Smith, G.L. Inhibition of IκB Kinase by Vaccinia Virus Virulence Factor B14. PLoS Pathog. 2008, 4, e22. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen RA, J.; Whalen, D.M.; Abrescia NG, A.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia virus proteins A52 and B14 share a Bcl-2-like fold but have evolved to inhibit Nf-κB rather than apoptosis. PLoS Pathog. 2008, 4, e1000128. [Google Scholar] [CrossRef]
- Abdul-Sada, H.; Müller, M.; Mehta, R.; Toth, R.; Arthur, J.S.C.; Whitehouse, A.; Macdonald, A. The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-κB by merkel polyomavirus small tumour antigen. Oncotarget 2017, 8, 25418–25432. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zeng, Q.; Wang, M.; Cheng, A.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Yang, Q.; Wu, Y.; et al. Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism. Viruses 2018, 10, 409. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G. Viral interference with innate immunity by preventing NF-κB activity. Cell. Microbiol. 2012, 14, 168–181. [Google Scholar] [CrossRef]
- Roberts, G.C.; Zothner, C.; Remenyi, R.; Merits, A.; Stonehouse, N.J.; Harris, M. Evaluation of a range of mammalian and mosquito cell lines for use in Chikungunya virus research. Sci. Rep. 2017, 7, 14641. [Google Scholar] [CrossRef]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Truitt, R.; Tan, L.H.; Dong, B.; Alysandratos, K.D.; et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118. [Google Scholar] [CrossRef] [PubMed]
- Morazzani, E.M.; Wiley, M.R.; Murreddu, M.G.; Adelman, Z.N.; Myles, K.M. Production of Virus-Derived Ping-Pong-Dependent piRNA-like Small RNAs in the Mosquito Soma. PLoS Pathog. 2012, 8, e1002470. [Google Scholar] [CrossRef] [PubMed]
- Sumner, R.P.; Motes, C.M.; de Veyer, D.L.; Smith, G.L. Vaccinia Virus Inhibits NF-κB-Dependent Gene Expression Downstream of p65 Translocation. J. Virol. 2014, 88, 3092. [Google Scholar] [CrossRef] [PubMed]
- Eckei, L.; Krieg, S.; Bütepage, M.; Lehmann, A.; Gross, A.; Lippok, B.; Grimm, A.R.; Kümmerer, B.M.; Rossetti, G.; Lüscher, B.; et al. The conserved macrodomains of the non-structural proteins of Chikungunya virus and other pathogenic positive strand RNA viruses function as mono-ADP-ribosylhydrolases. Sci. Rep. 2017, 7, 41746. [Google Scholar] [CrossRef]
- Makrynitsa, G.I.; Ntonti, D.; Marousis, K.D.; Birkou, M.; Matsoukas, M.-T.; Asami, S.; Bentrop, D.; Papageorgiou, N.; Canard, B.; Coutard, B.; et al. Conformational plasticity of the VEEV macro domain is important for binding of ADP-ribose. J Struct Biol. 2019, 206, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Strauss, E.G.; Rice, C.M.; Strauss, J.H. Complete nucleotide sequence of the genomic RNA of Sindbis virus. Virology 1984, 133, 92–110. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, G.C.; Stonehouse, N.J.; Harris, M. The Chikungunya Virus nsP3 Macro Domain Inhibits Activation of the NF-κB Pathway. Viruses 2025, 17, 191. https://doi.org/10.3390/v17020191
Roberts GC, Stonehouse NJ, Harris M. The Chikungunya Virus nsP3 Macro Domain Inhibits Activation of the NF-κB Pathway. Viruses. 2025; 17(2):191. https://doi.org/10.3390/v17020191
Chicago/Turabian StyleRoberts, Grace C., Nicola J. Stonehouse, and Mark Harris. 2025. "The Chikungunya Virus nsP3 Macro Domain Inhibits Activation of the NF-κB Pathway" Viruses 17, no. 2: 191. https://doi.org/10.3390/v17020191
APA StyleRoberts, G. C., Stonehouse, N. J., & Harris, M. (2025). The Chikungunya Virus nsP3 Macro Domain Inhibits Activation of the NF-κB Pathway. Viruses, 17(2), 191. https://doi.org/10.3390/v17020191