
CIEVaD: A Lightweight Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection


Abstract
1. Introduction
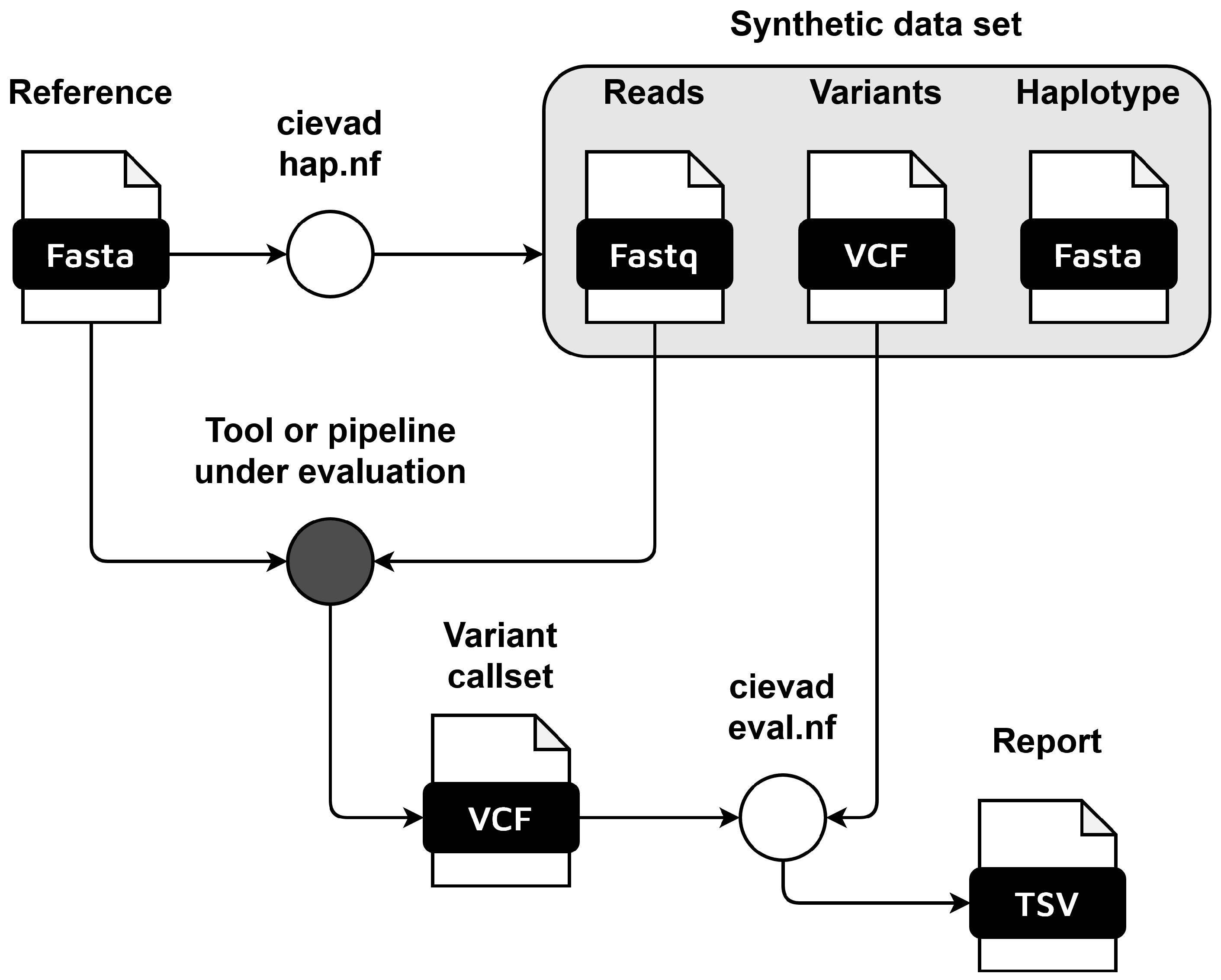
2. Materials and Methods
2.1. Haplotype Workflow
2.2. Evaluation Workflow
3. Results
3.1. Assessing Variant Detection from NGS Data as Part of SARS-CoV-2 Genome Reconstruction
- Install Conda and Nextflow;
- Download a reference genome;
- Run CIEVaD hap.nf;
- Run CoVpipe2;
- Prepare input for CIEVaD eval.nf;
- Run CIEVaD eval.nf;
- Check results.
3.2. Assessing Variant Detection from Long-Read Data as Part of Nanopore Sequencing Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
| Type | Total in Truthset | Total in Query | TP | FP | FN | Recall | Precision | F1-Score |
|---|---|---|---|---|---|---|---|---|
| indels | 158.67 | 134.33 | 133.67 | 0.67 | 25 | 0.84 | 0.996 | 0.91 |
| SNVs | 287.33 | 268.67 | 268.67 | 0 | 18.67 | 0.93 | 1 | 0.97 |
Appendix C

Appendix D
| Type | Total in Truthset | Total in Query | TP | FP | FN | Recall | Precision | F1-Score |
|---|---|---|---|---|---|---|---|---|
| indels | 158.67 | 166 | 119 | 47 | 39.67 | 0.75 | 0.72 | 0.73 |
| SNVs | 287.33 | 263.33 | 261 | 2.33 | 26.33 | 0.91 | 0.99 | 0.95 |
References
- Shastry, B.S. SNP alleles in human disease and evolution. J. Hum. Genet. 2002, 47, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Jiang, G.; Yang, W.; Jin, W.; Gong, J.; Xu, X.; Niu, X. Animal-SNPAtlas: A comprehensive SNP database for multiple animals. Nucleic Acid Res. 2023, 51, D816–D826. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017. [Google Scholar] [CrossRef]
- Majidian, S.; Agustinho, D.P.; Chin, C.S.; Sedlazeck, F.J.; Mahmoud, M. Genomic variant benchmark: If you cannot measure it, you cannot improve it. Genome Biol. 2023, 24, 221. [Google Scholar] [CrossRef] [PubMed]
- Holtgrewe, M. Mason—A Read Simulator for Second Generation Sequencing Data; Technical Report FU Berlin; Freie Universität Berlin: Berlin, Germany, 2010. [Google Scholar]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Picard Toolkit; Broad Institute: Cambridge, MA, USA, 2019; GitHub Repository; Available online: http://broadinstitute.github.io/picard (accessed on 3 September 2024).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Krusche, P.; Trigg, L.; Boutros, P.C.; Mason, C.E.; De La Vega, F.M.; Moore, B.L.; Gonzalez-Porta, M.; Eberle, M.A.; Tezak, Z.; Lababidi, S.; et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat. Biotechnol. 2019, 37, 555–560. [Google Scholar] [CrossRef]
- Olson, N.D.; Wagner, J.; McDaniel, J.; Stephens, S.H.; Westreich, S.T.; Prasanna, A.G.; Johanson, E.; Boja, E.; Maier, E.J.; Serang, O.; et al. PrecisionFDA Truth Challenge V2: Calling variants from short and long reads in difficult-to-map regions. Cell Genom. 2022, 2, 100129. [Google Scholar] [CrossRef]
- Dunn, T.; Narayanasamy, S. vcfdist: Accurately benchmarking phased small variant calls in human genomes. Nat. Commun. 2023, 14, 8149. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, F.; Gabernet, G.; Smith, N.H.; Mertes, C.; Neogi, A.G.; Brandhoff, L.; Ossowski, A.; Altmueller, J.; Becker, K.; Petzold, A.; et al. NCBench: Providing an open, reproducible, transparent, adaptable, and continuous benchmark approach for DNA-sequencing-based variant calling [version 1; peer review: 1 approved with reservations]. F1000Research 2023, 12, 1125. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Hamada, M.; Asai, K. PBSIM3: A simulator for all types of PacBio and ONT long reads. NAR Genom. Bioinform. 2022, 4, lqac092. [Google Scholar] [CrossRef] [PubMed]
- Lataretu, M.; Drechsel, O.; Kmiecinski, R.; Trappe, K.; Hölzer, M.; Fuchs, S. Lessons learned: Overcoming common challenges in reconstructing the SARS-CoV-2 genome from short-read sequencing data via CoVpipe2 [version 2; peer review: 2 approved]. F1000Research 2024, 12, 1091. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar] [CrossRef]
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Hölzer, M. Corrigendum: PoreCov—An Easy to Use, Fast, and Robust Workflow for SARS CoV-2 Genome Reconstruction via Nanopore Sequencing. Front. Genet. 2022, 13, 875644. [Google Scholar] [CrossRef] [PubMed]
- Köndgen, S.; Oh, D.Y.; Thürmer, A.; Sedaghatjoo, S.; Patrono, L.V.; Calvignac-Spencer, S.; Biere, B.; Wolff, T.; Dürrwald, R.; Fuchs, S.; et al. A Robust, Scalable, and Cost-Efficient Approach to Whole Genome Sequencing of RSV Directly from Clinical Samples. J. Clin. Microbiol. 2024, 62, e0111123, Erratum in J. Clin. Microbiol. 2024, 62, e0078424. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core Framework for Community-Curated Bioinformatics Pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; Rademakers, R. Nanopack2: Population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39, btad311. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krannich, T.; Ternovoj, D.; Paraskevopoulou, S.; Fuchs, S. CIEVaD: A Lightweight Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection. Viruses 2024, 16, 1444. https://doi.org/10.3390/v16091444
Krannich T, Ternovoj D, Paraskevopoulou S, Fuchs S. CIEVaD: A Lightweight Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection. Viruses. 2024; 16(9):1444. https://doi.org/10.3390/v16091444
Chicago/Turabian StyleKrannich, Thomas, Dimitri Ternovoj, Sofia Paraskevopoulou, and Stephan Fuchs. 2024. "CIEVaD: A Lightweight Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection" Viruses 16, no. 9: 1444. https://doi.org/10.3390/v16091444
APA StyleKrannich, T., Ternovoj, D., Paraskevopoulou, S., & Fuchs, S. (2024). CIEVaD: A Lightweight Workflow Collection for the Rapid and On-Demand Deployment of End-to-End Testing for Genomic Variant Detection. Viruses, 16(9), 1444. https://doi.org/10.3390/v16091444