
Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis


Abstract
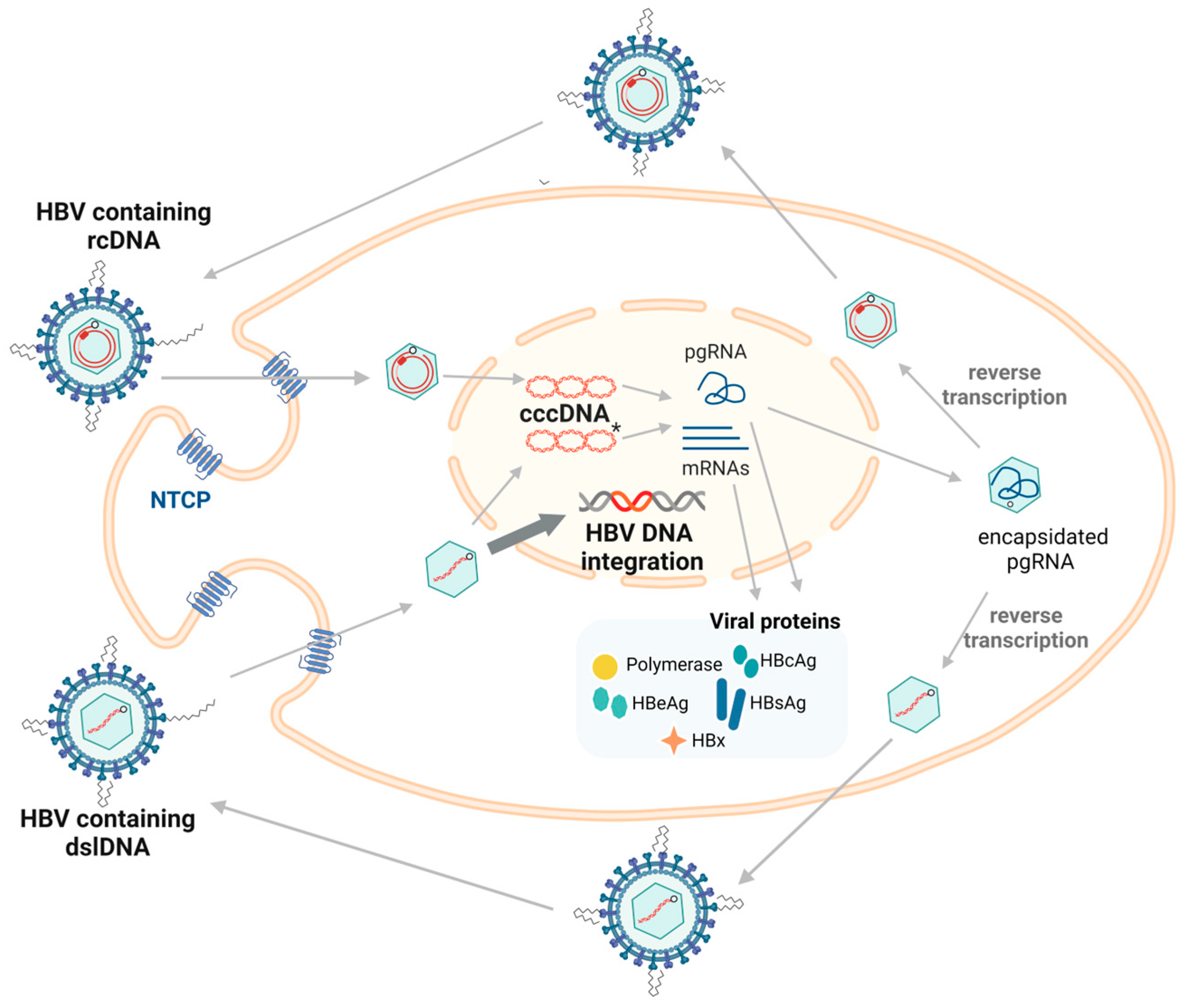
1. Introduction
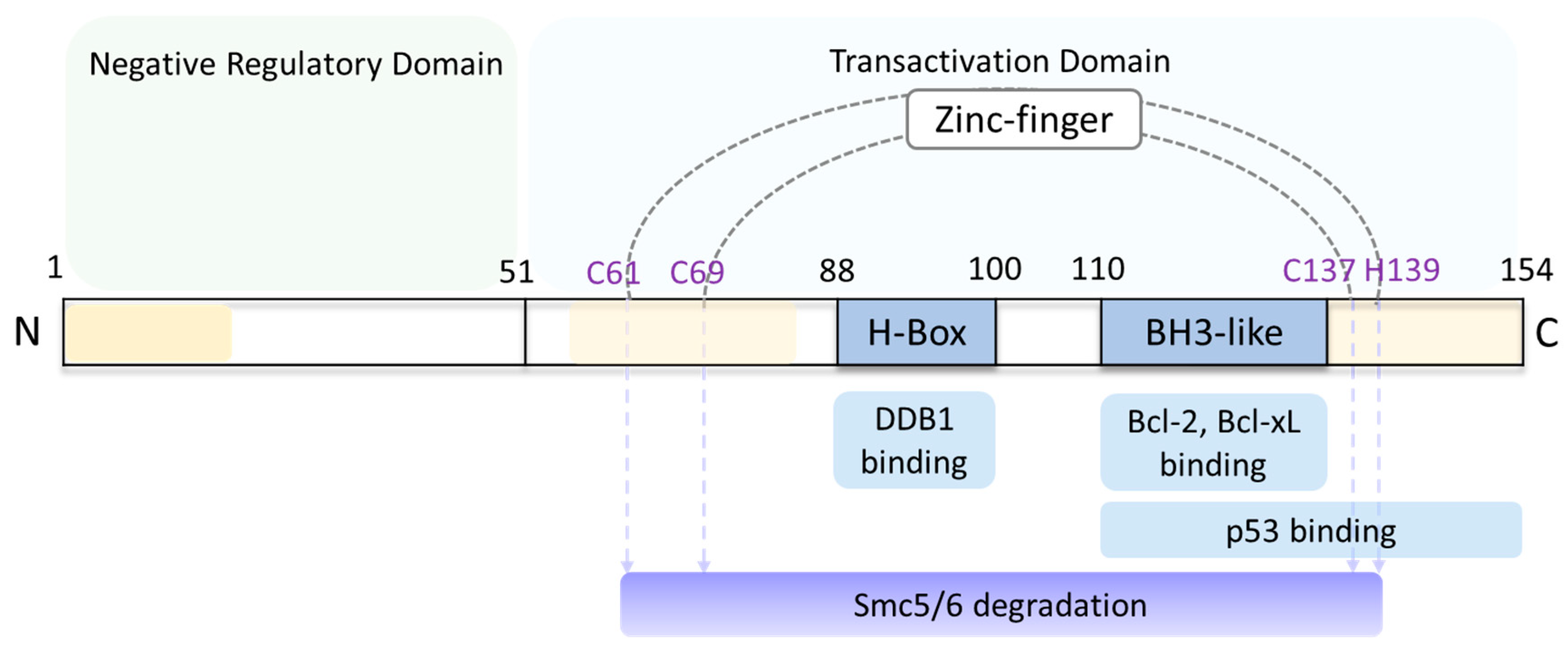
4. HBx
5. Role of HBx in HBV Replication
5.1. HBx and DDB1
5.2. HBx and Histone Modification
5.3. HBx and Cell Signaling Pathways
6. Antiviral Approaches by Therapeutically Targeting HBx Functions
7. HBx in Hepadnavirus-Associated Liver Cancer
7.1. HBx and Smc5/6 Complex Disruption
7.2. HBx and Cancer-Related Signaling Pathways
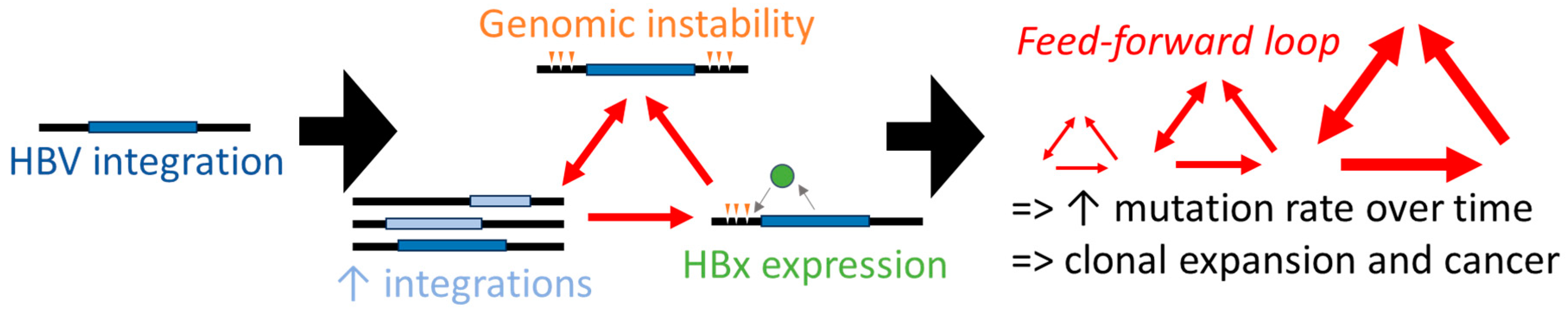
8. A Hypothetical Model of HBx-Mediated Carcinogenesis
9. Unanswered Questions in HBx Research
9.1. What Is the Structure of HBx in Various Cellular Contexts?
9.2. Is Transcriptional Regulation of Integrated HBV DNA Genomes Independent of HBx?
9.3. What Are the Most Appropriate Models to Test HBx Function and Anti-HBx Therapies?
9.4. Can Anti-HBx Therapies Prevent or Reduce HBV-Associated Liver Cancer?
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hsu, Y.-C.; Huang, D.Q.; Nguyen, M.H. Global burden of hepatitis B virus: Current status, missed opportunities and a call for action. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Global Hepatitis Report 2024: Action for Access in Low- and Middle-Income Countries; States News Service: Washington, DC, USA, 2024.
- Sherman, M. Hepatocellular Carcinoma: Epidemiology, Surveillance, and Diagnosis. Semin. Liver Dis. 2010, 30, 003–016. [Google Scholar] [CrossRef]
- Jin, Y.M.; Yun, C.; Park, C.; Wang, H.; Cho, H. Expression of hepatitis B virus X protein is closely correlated with the high periportal inflammatory activity of liver diseases. J. Viral Hepat. 2001, 8, 322–330. [Google Scholar] [CrossRef]
- Magnius, L.; Mason, W.S.; Taylor, J.; Kann, M.; Glebe, D.; Dény, P.; Sureau, C.; Norder, H.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Hepadnaviridae. J. Gen. Virol. 2020, 101, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49 (Suppl. S5), S13–S21. [Google Scholar] [CrossRef]
- Gavilanes, F.; Gonzalez-Ros, J.M.; Peterson, D.L. Structure of hepatitis B surface antigen. Characterization of the lipid components and their association with the viral proteins. J. Biol. Chem. 1982, 257, 7770–7777. [Google Scholar] [CrossRef]
- Hu, J.; Seeger, C. Hepadnavirus Genome Replication and Persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, W.H.; Robinson, W.S. Hepatitis B virus contains protein attached to the 5′ terminus of its complete DNA strand. Cell 1980, 21, 801–809. [Google Scholar] [CrossRef]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef]
- Hirsch, R.C.; Loeb, D.D.; Pollack, J.R.; Ganem, D. cis-acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol. 1991, 65, 3309–3316. [Google Scholar] [CrossRef]
- Selzer, L.; Zlotnick, A. Assembly and Release of Hepatitis B Virus. Cold Spring Harb. Perspect. Med. 2015, 5, a021394. [Google Scholar] [CrossRef]
- Chen, M.T.; Billaud, J.-N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A Function of the Hepatitis B Virus Precore Protein Is to Regulate the Immune Response to the Core Antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef] [PubMed]
- Wettengel, J.M.; Burwitz, B.J. Innovative HBV Animal Models Based on the Entry Receptor NTCP. Viruses 2020, 12, 828. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, R.J.; Bagga, S.; Bouchard, M.J. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res. 2016, 2, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-C.; Huang, E.-Y.; Su, P.-Y.; Wu, S.-Y.; Yang, C.-C.; Lin, Y.-S.; Chang, W.-C.; Shih, C. Nuclear Export and Import of Human Hepatitis B Virus Capsid Protein and Particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef]
- Urban, S.; Schulze, A.; Dandri, M.; Petersen, J. The replication cycle of hepatitis B virus. J. Hepatol. 2010, 52, 282–284. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Zhang, B.-H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-Cell Analysis of Covalently Closed Circular DNA Copy Numbers in a Hepadnavirus-Infected Liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [CrossRef]
- Tu, T.; Jilbert, A.R. Detection of Hepatocyte Clones Containing Integrated Hepatitis B Virus DNA Using Inverse Nested PCR. In Hepatitis B Virus; Humana Press: New York, NY, USA, 2017; pp. 97–118. [Google Scholar]
- Tu, T.; Zhang, H.; Urban, S. Hepatitis B Virus DNA Integration: In Vitro Models for Investigating Viral Pathogenesis and Persistence. Viruses 2021, 13, 180. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Salpini, R.; D’anna, S.; Benedetti, L.; Piermatteo, L.; Gill, U.; Svicher, V.; Kennedy, P.T.F. Hepatitis B virus DNA integration as a novel biomarker of hepatitis B virus-mediated pathogenetic properties and a barrier to the current strategies for hepatitis B virus cure. Front. Microbiol. 2022, 13, 972687. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef]
- Bill, C.A.; Summers, J. Genomic DNA Double-Strand Breaks Are Targets for Hepadnaviral DNA Integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef]
- Mason, W.S.; Low, H.-C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of Clonally Expanded Hepatocytes in Chimpanzees with Chronic Hepatitis B Virus Infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte Turnover during Resolution of a Transient Hepadnaviral Infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.J.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal Expansion of Hepatocytes during Chronic Woodchuck Hepatitis Virus Infection. Proc. Natl. Acad. Sci. USA 2005, 102, 1139–1144. [Google Scholar] [CrossRef]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal Expansion of Normal-Appearing Human Hepatocytes during Chronic Hepatitis B Virus Infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef] [PubMed]
- Podlaha, O.; Wu, G.; Downie, B.; Ramamurthy, R.; Gaggar, A.; Subramanian, M.; Ye, Z.; Jiang, Z. Genomic modeling of hepatitis B virus integration frequency in the human genome. PLoS ONE 2019, 14, e0220376. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Yuen, M.-F.; Chan, H.L.-Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhang, Y.; Gu, W.; Wang, Z.; Li, D.; Zhang, F.; Qiu, G.; Xie, K. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: A key to the cell cycle and apoptosis. Int. J. Oncol. 2005, 26, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Shaul, Y. Enhancer I Predominance in Hepatitis B Virus Gene Expression. Mol. Cell. Biol. 2004, 24, 1799–1808. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef]
- Kumar, V.; Jayasuryan, N.; Kumar, R. A Truncated Mutant (Residues 58-140) of the Hepatitis B Virus X Protein Retains Transactivation Function. Proc. Natl. Acad. Sci. USA 1996, 93, 5647–5652. [Google Scholar] [CrossRef]
- Kidd-Ljunggren, K.; Oberg, M.; Kidd, A.H. The hepatitis B virus X gene: Analysis of functional domain variation and gene phylogeny using multiple sequences. J. Gen. Virol. 1995, 76, 2119–2130. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Hildt, E.; Chang, S.-F.; Terradillos, O.; Pollicino, T.; Lanford, R.; Gerlich, W.H.; Will, H.; Schaefer, S. Conserved transactivating and pro-apoptotic functions of hepadnaviral X protein in ortho- and avihepadnaviruses. Oncogene 2002, 21, 6606–6613. [Google Scholar] [CrossRef]
- Prescott, N.A.; Bram, Y.; Schwartz, R.E.; David, Y. Targeting Hepatitis B Virus Covalently Closed Circular DNA and Hepatitis B Virus X Protein: Recent Advances and New Approaches. ACS Infect. Dis. 2019, 5, 1657–1667. [Google Scholar] [CrossRef]
- Rui, E.; de Moura, P.R.; Gonçalves, K.d.A.; Kobarg, J. Expression and spectroscopic analysis of a mutant hepatitis B virus onco-protein HBx without cysteine residues. J. Virol. Methods 2005, 126, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.-Y.; Chen, H.-Y.; Cao, J.-L.; Xiong, H.-L.; Mo, X.-B.; Li, T.-L.; Kang, X.-Z.; Zhao, J.-H.; Yin, B.; Zhao, X.; et al. Structural and functional analyses of hepatitis B virus X protein BH3-like domain and Bcl-xL interaction. Nat. Commun. 2019, 10, 3192. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, D.; Xing, W.; Beran, R.K.; Chemuru, S.; Rohrs, H.; Niedziela-Majka, A.; Marchand, B.; Mehra, U.; Zábranský, A.; Doležal, M.; et al. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, M.; Wu, J.; Shi, Y. Structural and biochemical analysis of Bcl-2 interaction with the hepatitis B virus protein HBx. Proc. Natl. Acad. Sci. USA 2016, 113, 2074–2079. [Google Scholar] [CrossRef]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous α-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Elmore, L.W.; Hancock, A.R.; Chang, S.-F.; Wang, X.W.; Chang, S.; Callahan, C.P.; Geller, D.A.; Will, H.; Harris, C.C. Hepatitis B Virus X Protein and p53 Tumor Suppressor Interactions in the Modulation of Apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 14707–14712. [Google Scholar] [CrossRef]
- Liu, W.; Yao, Q.; Su, X.; Deng, Y.; Yang, M.; Peng, B.; Zhao, F.; Du, C.; Zhang, X.; Zhu, J.; et al. Molecular insights into Spindlin1-HBx interplay and its impact on HBV transcription from cccDNA minichromosome. Nat. Commun. 2023, 14, 4663. [Google Scholar] [CrossRef] [PubMed]
- Schollmeier, A.; Glitscher, M.; Hildt, E. Relevance of HBx for Hepatitis B Virus-Associated Pathogenesis. Int. J. Mol. Sci. 2023, 24, 4964. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vilchez, S.; Lara-Pezzi, E.; Trapero-Marugán, M.; Moreno-Otero, R.; Sanz-Cameno, P. The molecular and pathophysiological implications of hepatitis B X antigen in chronic hepatitis B virus infection. Rev. Med. Virol. 2011, 21, 315–329. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Ryu, D.-K.; Jung, H.-S.; Chang, H.-E.; Ryu, W.-S. Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 2009, 90, 978–986. [Google Scholar] [CrossRef]
- Henkler, F.; Hoare, J.; Waseem, N.; Goldin, R.D.; McGarvey, M.J.; Koshy, R.; King, I.A. Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 2001, 82, 871–882. [Google Scholar] [CrossRef]
- Ueda, C.; Langton, M.; Chen, J.; Pandelia, M.-E. The HBx protein from hepatitis B virus coordinates a redox-active Fe-S cluster. J. Biol. Chem. 2022, 298, 101698. [Google Scholar] [CrossRef]
- Zoulim, F.; Saputelli, J.; Seeger, C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 1994, 68, 2026–2030. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Kaneko, S.; Girones, R.; Anderson, R.W.; Hornbuckle, W.E.; Tennant, B.C.; Cote, P.J.; Gerin, J.L.; Purcell, R.H.; Miller, R.H. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol. 1993, 67, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Hepatitis B virus HBx protein localized to the nucleus restores HBx-deficient virus replication in HepG2 cells and in vivo in hydrodynamically-injected mice. Virology 2009, 390, 122–129. [Google Scholar] [CrossRef]
- Lin-Marq, N.; Bontron, S.; Leupin, O.; Strubin, M. Hepatitis B Virus X Protein Interferes with Cell Viability through Interaction with the p127-kDa UV-Damaged DNA-Binding Protein. Virology 2001, 287, 266–274. [Google Scholar] [CrossRef]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B Virus X Protein Stimulates Viral Genome Replication via a DDB1-Dependent Pathway Distinct from That Leading to Cell Death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, A.J.; Hyser, J.M.; Keasler, V.V.; Cang, Y.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 2012, 426, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 Complex Restricts HBV when Localized to ND10 without Inducing an Innate Immune Response and Is Counteracted by the HBV X Protein Shortly after Infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef]
- Rivière, L.; Quioc-Salomon, B.; Fallot, G.; Halgand, B.; Féray, C.; Buendia, M.A.; Neuveut, C. Hepatitis B virus replicating in hepatocellular carcinoma encodes HBx variants with preserved ability to antagonize restriction by Smc5/6. Antivir. Res. 2019, 172, 104618. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.-Y.; Liu, F.; Fung, J.; Seto, W.-K.; Lai, K.K.-Y.; Lai, C.-L.; Yuen, M.-F.; et al. HBV X protein mutations affect HBV transcription and association of histone-modifying enzymes with covalently closed circular DNA. Sci. Rep. 2020, 10, 802. [Google Scholar] [CrossRef]
- Benhenda, S.; Ducroux, A.; Rivière, L.; Sobhian, B.; Ward, M.D.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.-L.; Benkirane, M.; et al. Methyltransferase PRMT1 Is a Binding Partner of HBx and a Negative Regulator of Hepatitis B Virus Transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef]
- Ducroux, A.; Benhenda, S.; Rivière, L.; Semmes, O.J.; Benkirane, M.; Neuveut, C. The Tudor Domain Protein Spindlin1 Is Involved in Intrinsic Antiviral Defense against Incoming Hepatitis B Virus and Herpes Simplex Virus Type 1. PLoS Pathog. 2014, 10, e1004343. [Google Scholar] [CrossRef]
- Rivière, L.; Gerossier, L.; Ducroux, A.; Dion, S.; Deng, Q.; Michel, M.-L.; Buendia, M.-A.; Hantz, O.; Neuveut, C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J. Hepatol. 2015, 63, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, V.; Hernández, S.; Rubio, L.; Alvarez, F.; Flores, Y.; Varas-Godoy, M.; De Ferrari, G.V.; Kann, M.; Villanueva, R.A.; Loyola, A. The enzymes LSD1 and Set1A cooperate with the viral protein HBx to establish an active hepatitis B viral chromatin state. Sci. Rep. 2016, 6, 25901. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Wang, L.-H.; Schneider, R.J. Calcium Signaling by HBx Protein in Hepatitis B Virus DNA Replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef] [PubMed]
- McClain, S.L.; Clippinger, A.J.; Lizzano, R.; Bouchard, M.J. Hepatitis B Virus Replication Is Associated with an HBx-Dependent Mitochondrion-Regulated Increase in Cytosolic Calcium Levels. J. Virol. 2007, 81, 12061–12065. [Google Scholar] [CrossRef]
- Choi, Y.; Park, S.G.; Yoo, J.-H.; Jung, G. Calcium ions affect the hepatitis B virus core assembly. Virology 2005, 332, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91 Pt 7, 1854–1864. [Google Scholar] [CrossRef]
- Sitterlin, D.; Lee, T.H.; Prigent, S.; Tiollais, P.; Butel, J.S.; Transy, C. Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J. Virol. 1997, 71, 6194–6199. [Google Scholar] [CrossRef]
- Kanno, T.; Berta, D.G.; Sjögren, C. The Smc5/6 Complex Is an ATP-Dependent Intermolecular DNA Linker. Cell Rep. 2015, 12, 1471–1482. [Google Scholar] [CrossRef]
- Choonnasard, A.; Shofa, M.; Okabayashi, T.; Saito, A. Conserved Functions of Orthohepadnavirus X Proteins to Inhibit Type-I Interferon Signaling. Int. J. Mol. Sci. 2024, 25, 3753. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Al-Anazi, M.R.; Nazir, N.; Ghai, R.; Abdo, A.A.; Sanai, F.M.; Al-Hamoudi, W.K.; Alswat, K.A.; Al-Ashgar, H.I.; Khan, M.Q.; et al. Hepatitis B virus (HBV) X gene mutations and their association with liver disease progression in HBV-infected patients. Oncotarget 2017, 8, 105115–105125. [Google Scholar] [CrossRef]
- Abdul, F.; Filleton, F.; Gerossier, L.; Paturel, A.; Hall, J.; Strubin, M.; Etienne, L. Smc5/6 Antagonism by HBx Is an Evolutionarily Conserved Function of Hepatitis B Virus Infection in Mammals. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B Virus Replication Is Regulated by the Acetylation Status of Hepatitis B Virus cccDNA-Bound H3 and H4 Histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Diao, J.; Garces, R.; Richardson, C.D. X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev. 2001, 12, 189–205. [Google Scholar] [CrossRef]
- Agustiningsih, A.; Rasyak, M.R.; Turyadi; Jayanti, S.; Sukowati, C. The oncogenic role of hepatitis B virus X gene in hepatocarcinogenesis: Recent updates. Explor. Target. Anti-Tumor Ther. 2024, 5, 120–134. [Google Scholar] [CrossRef]
- Slagle, B.L.; Andrisani, O.M.; Bouchard, M.J.; Lee, C.G.; Ou, J.J.; Siddiqui, A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015, 61, 1416–1424. [Google Scholar] [CrossRef]
- Allweiss, L.; Giersch, K.; Pirosu, A.; Volz, T.; Muench, R.C.; Beran, R.K.; Urban, S.; Javanbakht, H.; Fletcher, S.P.; Lütgehetmann, M.; et al. Therapeutic shutdown of HBV transcripts promotes reappearance of the SMC5/6 complex and silencing of the viral genome in vivo. Gut 2022, 71, 372–381. [Google Scholar] [CrossRef]
- Han, Q.; Hou, Z.; Yin, C.; Zhang, C.; Zhang, J. 5′-triphosphate siRNA targeting HBx elicits a potent anti-HBV immune response in pAAV-HBV transfected mice. Antivir. Res. 2019, 161, 36–45. [Google Scholar] [CrossRef]
- Rossignol, J.F.; Bréchot, C. A Pilot Clinical Trial of Nitazoxanide in the Treatment of Chronic Hepatitis B. Hepatol. Commun. 2019, 3, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-T.; Hu, J.-L.; Ren, J.-H.; Yu, H.-B.; Zhong, S.; Wong, V.K.W.; Law, B.Y.K.; Chen, W.-X.; Xu, H.-M.; Zhang, Z.-Z.; et al. Dicoumarol, an NQO1 inhibitor, blocks cccDNA transcription by promoting degradation of HBx. J. Hepatol. 2021, 74, 522–534. [Google Scholar] [CrossRef]
- Tan, G.; Yi, Z.; Song, H.; Xu, F.; Li, F.; Aliyari, R.; Zhang, H.; Du, P.; Ding, Y.; Niu, J.; et al. Type-I-IFN-Stimulated Gene TRIM5γ Inhibits HBV Replication by Promoting HBx Degradation. Cell Rep. 2019, 29, 3551–3563.e3. [Google Scholar] [CrossRef]
- Bergametti, F.; Sitterlin, D.; Transy, C. Turnover of Hepatitis B Virus X Protein Is Regulated by Damaged DNA-Binding Complex. J. Virol. 2002, 76, 6495–6501. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.S.; Klote, L.; Aoki, N. Hepadnaviruses in cirrhotic liver and hepatocellular carcinoma. J. Med. Virol. 1990, 31, 18–32. [Google Scholar] [CrossRef]
- Gerin, J.L.; Cote, P.J.; Korba, B.E.; Tennant, B.C. Hepadnavirus-induced liver cancer in woodchucks. Cancer Detect. Prev. 1989, 14, 227–229. [Google Scholar]
- Pesavento, P.A.; Jackson, K.; Scase, T.; Tse, T.; Hampson, B.; Munday, J.S.; Barrs, V.R.; Beatty, J.A. A Novel Hepadnavirus is Associated with Chronic Hepatitis and Hepatocellular Carcinoma in Cats. Viruses 2019, 11, 969. [Google Scholar] [CrossRef]
- Yang, C.; Ruan, P.; Ou, C.; Su, J.; Cao, J.; Luo, C.; Tang, Y.; Wang, Q.; Qin, H.; Sun, W.; et al. Chronic hepatitis B virus infection and occurrence of hepatocellular carcinoma in tree shrews (Tupaia belangeri chinensis). Virol. J. 2015, 12, 26. [Google Scholar] [CrossRef]
- Marion, P.L.; Van Davelaar, M.J.; Knight, S.S.; Salazar, F.H.; Garcia, G.; Popper, H.; Robinson, W.S. Hepatocellular Carcinoma in Ground Squirrels Persistently Infected with Ground Squirrel Hepatitis Virus. Proc. Natl. Acad. Sci. USA 1986, 83, 4543–4546. [Google Scholar] [CrossRef]
- Sivasudhan, E.; Blake, N.; Lu, Z.; Meng, J.; Rong, R. Hepatitis B Viral Protein HBx and the Molecular Mechanisms Modulating the Hallmarks of Hepatocellular Carcinoma: A Comprehensive Review. Cells 2022, 11, 741. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Volz, T.; Giersch, K.; Kah, J.; Raffa, G.; Petersen, J.; Lohse, A.W.; Beninati, C.; Pollicino, T.; Urban, S.; et al. Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut 2018, 67, 542–552. [Google Scholar] [CrossRef]
- Tu, T.; Zehnder, B.; Wettengel, J.M.; Zhang, H.; Coulter, S.; Ho, V.; Douglas, M.W.; Protzer, U.; George, J.; Urban, S. Mitosis of hepatitis B virus-infected cells in vitro results in uninfected daughter cells. JHEP Rep. 2022, 4, 100514. [Google Scholar] [CrossRef]
- Yan, Y.; Allweiss, L.; Yang, D.; Kang, J.; Wang, J.; Qian, X.; Zhang, T.; Liu, H.; Wang, L.; Liu, S.; et al. Down-regulation of cell membrane localized NTCP expression in proliferating hepatocytes prevents hepatitis B virus infection. Emerg. Microbes Infect. 2019, 8, 879–894. [Google Scholar] [CrossRef]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E., IV; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy: cccDNA levels in chronic hepatitis B patients. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef] [PubMed]
- Bréchot, C.; Gozuacik, D.; Murakami, Y.; Paterlini-Bréchot, P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2000, 10, 211–231. [Google Scholar] [CrossRef]
- Hayashi, S.; Isogawa, M.; Kawashima, K.; Ito, K.; Chuaypen, N.; Morine, Y.; Shimada, M.; Higashi-Kuwata, N.; Watanabe, T.; Tangkijvanich, P.; et al. Droplet digital PCR assay provides intrahepatic HBV cccDNA quantification tool for clinical application. Sci. Rep. 2022, 12, 2133. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Deng, H.; Zhen, X.; Xiong, J.; Hu, Y. Quantification of intrahepatic cccDNA in HBV associated hepatocellular carcinoma by improved ddPCR method. J. Virol. Methods 2022, 299, 114334. [Google Scholar] [CrossRef]
- Lizzano, R.A.; Yang, B.; Clippinger, A.J.; Bouchard, M.J. The C-terminal region of the hepatitis B virus X protein is essential for its stability and function. Virus Res. 2011, 155, 231–239. [Google Scholar] [CrossRef]
- Ma, N.-F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.-C.; Fung, J.; Bai, X.; et al. COOH-Terminal Truncated HBV X Protein Plays Key Role in Hepatocarcinogenesis. Clin. Cancer Res. 2008, 14, 5061–5068. [Google Scholar] [CrossRef]
- Sachs, A. The role of poly(A) in the translation and stability of mRNA. Curr. Opin. Cell Biol. 1990, 2, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Russnak, R.; Ganem, D. Sequences 5’ to the polyadenylation signal mediate differential poly(A) site use in hepatitis B viruses. Genes Dev. 1990, 4, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, Q.; Gong, L.; Xu, H.; Liu, B.; Fang, X.; Yu, D.; Li, L.; Wei, T.; Wang, Y.; et al. C-terminal truncated HBx initiates hepatocarcinogenesis by downregulating TXNIP and reprogramming glucose metabolism. Oncogene 2021, 40, 1147–1161. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.-Y.; Chai, S.; Tong, M.; Guan, X.-Y.; Lin, C.-H.; Ching, Y.P.; Xie, D.; Cheng, A.S.L.; Ma, S.K.Y. C-terminal truncated hepatitis B virus X protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 2016, 7, 24005–24017. [Google Scholar] [CrossRef]
- Sze, K.M.F.; Chu, G.K.; Lee, J.M.; Ng, I.O. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by c-jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef]
- Ching, R.H.H.; Sze, K.M.F.; Lau, E.Y.T.; Chiu, Y.-T.; Lee, J.M.F.; Ng, I.O.L.; Lee, T.K.W. C-terminal truncated hepatitis B virus X protein regulates tumorigenicity, self-renewal and drug resistance via STAT3/Nanog signaling pathway. Oncotarget 2017, 8, 23507–23516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, W.; Liu, Q.; Zhang, X.; Lv, N.; Ye, L.; Zhang, X. A Mutant of Hepatitis B Virus X Protein (HBxΔ127) Promotes Cell Growth through A Positive Feedback Loop Involving 5-Lipoxygenase and Fatty Acid Synthase. Neoplasia 2010, 12, 103–115, IN1–IN3. [Google Scholar] [CrossRef]
- Mao, X.; Tey, S.K.; Ko, F.C.; Kwong, E.M.; Gao, Y.; Ng, I.O.; Cheung, S.T.; Guan, X.Y.; Yam, J.W. C-terminal truncated HBx protein activates caveolin-1/LRP6/β-catenin/FRMD5 axis in promoting hepatocarcinogenesis. Cancer Lett. 2019, 444, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, M.; Liao, D.; Lu, X.; Gu, X.; Zhang, Q.; Zhang, Z.; Li, H. Carboxyl-terminal truncated HBx contributes to invasion and metastasis via deregulating metastasis suppressors in hepatocellular carcinoma. Oncotarget 2016, 7, 55110–55127. [Google Scholar] [CrossRef]
- Montalbano, R.; Honrath, B.; Wissniowski, T.T.; Elxnat, M.; Roth, S.; Ocker, M.; Quint, K.; Churin, Y.; Roederfeld, M.; Schroeder, D.; et al. Exogenous hepatitis B virus envelope proteins induce endoplasmic reticulum stress: Involvement of cannabinoid axis in liver cancer cells. Oncotarget 2016, 7, 20312–20323. [Google Scholar] [CrossRef]
- Jung, S.-Y.; Kim, Y.-J. C-terminal region of HBx is crucial for mitochondrial DNA damage. Cancer Lett. 2013, 331, 76–83. [Google Scholar] [CrossRef]
- Wu, X.; Ni, Z.; Song, T.; Lv, W.; Chen, Y.; Huang, D.; Xie, Y.; Huang, W.; Niu, Y. C-Terminal Truncated HBx Facilitates Oncogenesis by Modulating Cell Cycle and Glucose Metabolism in FXR-Deficient Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 5174. [Google Scholar] [CrossRef]
- Becker, S.A.; Lee, T.-H.; Butel, J.S.; Slagle, B.L. Hepatitis B Virus X Protein Interferes with Cellular DNA Repair. J. Virol. 1998, 72, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.M.; Murray, J.M. Smc5/6: A link between DNA repair and unidirectional replication? Nature reviews. Mol. Cell Biol. 2008, 9, 177–182. [Google Scholar]
- Atkins, A.; Xu, M.J.; Li, M.; Rogers, N.P.; Pryzhkova, M.V.; Jordan, P.W. SMC5/6 is required for replication fork stability and faithful chromosome segregation during neurogenesis. eLife 2020, 9, e61171. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Jordan, P.W. SMC5/6 Promotes Replication Fork Stability via Negative Regulation of the COP9 Signalosome. Int. J. Mol. Sci. 2024, 25, 952. [Google Scholar] [CrossRef]
- Moradi-Fard, S.; Sarthi, J.; Tittel-Elmer, M.; Lalonde, M.; Cusanelli, E.; Chartrand, P.; Cobb, J.A. Smc5/6 Is a Telomere-Associated Complex that Regulates Sir4 Binding and TPE. PLoS Genet. 2016, 12, e1006268. [Google Scholar] [CrossRef]
- Sekiba, K.; Otsuka, M.; Funato, K.; Miyakawa, Y.; Tanaka, E.; Seimiya, T.; Yamagami, M.; Tsutsumi, T.; Okushin, K.; Miyakawa, K.; et al. HBx-induced degradation of Smc5/6 complex impairs homologous recombination-mediated repair of damaged DNA. J. Hepatol. 2022, 76, 53–62. [Google Scholar] [CrossRef]
- Lee, T.H.; Finegold, M.J.; Shen, R.F.; DeMayo, J.L.; Woo, S.L.; Butel, J.S. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J. Virol. 1990, 64, 5939–5947. [Google Scholar] [CrossRef] [PubMed]
- Jacome, A.; Gutierrez-Martinez, P.; Schiavoni, F.; Tenaglia, E.; Martinez, P.; Rodríguez-Acebes, S.; Lecona, E.; Murga, M.; Méndez, J.; Blasco, M.A.; et al. NSMCE2 suppresses cancer and aging in mice independently of its SUMO ligase activity. EMBO J. 2015, 34, 2604–2619. [Google Scholar] [CrossRef]
- Groisman, I.J.; Koshy, R.; Henkler, F.; Groopman, J.D.; Alaoui-Jamali, M.A. Downregulation of DNA excision repair by the hepatitis B virus-x protein occurs in p53-proficient and p53-deficient cells. Carcinogenesis 1999, 20, 479–483. [Google Scholar] [CrossRef]
- Arzumanyan, A.; Reis, H.M.G.P.V.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-M.; Lee, S.-Y.; Kim, B.-J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef]
- Yip, K.M.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Atomic-resolution protein structure determination by cryo-EM. Nature 2020, 587, 157–161. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Trabut, J.-B.; Delpuech, O.; Carnot, F.; Colombo, M.; Kremsdorf, D.; Bréchot, C.; Thiers, V. Characterisation of hepatitis B virus X protein mutants in tumour and non-tumour liver cells using laser capture microdissection. J. Hepatol. 2003, 39, 253–261. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HBx Activity | Experimental Models | Year | References | |
|---|---|---|---|---|
| HBx and DDB1 | Interferes with cell viability | In vitro primary REFs and HeLa cells transfected with HBx | 2001 | Lin-Marq et al. [62] |
| Stimulates viral genome replication distinct from leading to cell death | In vitro HepG2 and Huh-7 cells transfected with HBx | 2005 | Leupin et al. [63] | |
| Anchors viral hijackers and substrate receptors to the Cul4–DDB1 ubiquitin ligase machinery through a promiscuous α-helical motif | In vitro transfection experiments with HeLa and HepG2 cells | 2010 | Li et al. [51] | |
| HBx-DDB1 is required for maximal HBV replication | In vitro HepG2 cells transfected with pHBV1.3 and point mutant HBx; in vivo HBx-transgenic mice | 2012 | Hodgson et al. [64] | |
| Identified the Smc5/6 complex as a host restriction factor | In vitro transfection of human hepatoma cells and HepaRG cells; in vitro infection of PHH; in vivo infection of human liver chimeric uPA-SCID mice | 2016 | Decorsière et al. [65] | |
| Promotes degradation of Smc 5/6 to enhance HBV replication | In vitro wild type and mutant HBx-expressing HepG2, HepAD38, HepG2-NTCP cell lines | 2016 | Murphy et al. [66] | |
| Smc5/6 complex restricts HBx-DDB1 when localized to ND10 without inducing an innate immune response | In vitro PHH infection with wild type and mutant HBx HBV; in vivo infection of human liver chimeric uPA-SCID mice | 2017 | Niu et al. [67] | |
| HBV replicating in HCC encodes HBx variants with preserved ability to antagonize restriction by Smc5/6 | In vitro dHepaRG cells infected with wild type and mutant HBx-expressing HepG2; HeLa cells transfected with HBx | 2019 | Rivière et al. [68] | |
| HBx and histone modification | HBx is recruited onto the cccDNA with a kinetic paralleling HBV replication | In vitro transiently transfected HepG2 cells with full-length WT and HBx mutant HBV | 2009 | Belloni et al. [69] |
| Epigenetic regulation of HBV transcription from cccDNA | In vitro HBV infection of primary human hepatocytes (PHH) and HepaRG cells with wild type and mutant HBx-expressing HepG2 cells | 2011 | Lucifora et al. [19] | |
| In vitro transiently transfected HepG2-NTCP cells with full-length WT and truncated-HBx plasmids | 2020 | Chong et al. [70] | ||
| Binding to PRMT1/Spindlin-1, blocks the inhibitory activity of PRMT1 on HBV transcription | In vitro transfection of HepG2 cells; in vitro infection of PHH; in vivo mouse model infected with AAV2/8-HBV or AAV2/8-empty virus vector (single tail vein injection) | 2013 | Benhenda et al. [71] | |
| In vitro infection of HepaRG cells with HBVwt and HBVX-; HEK293 cell transfection | 2014 | Ducroux et al. [72] | ||
| HBx relieves chromatin-mediated transcriptional repression of cccDNA involving SETDB1 histone methyltransferase | In vitro HBV infection of PHH and dHepaRG cells with HBVwt or HBVX- | 2015 | Rivière et al. [73] | |
| HBx regulates the recruitment of chromatin modifying enzymes(LSD1/Set1A) to an active cccDNA chromatin state | In vitro transient transfection of Huh7 and HepG2 cells | 2016 | Alarcon et al. [74] | |
| Epigenetic switch to an H3K4me3-marked active state; a conformational switch may occur in coordination with HBx-DDB1 | In vitro HBV infection models in HepG2-NTCP cells | 2023 | Liu et al. [53] | |
| HBx and cell signaling pathways | HBx targets mitochondrial calcium regulation, thereby activating Pyk2/Src and FAK pathways | In vitro HepG2 cells transfected with full-length HBx | 2001, 2007 | Bouchard et al., McClain et al. [75,76] |
| Enhances HBV core assembly | In vitro HepG2 cells transfected with pHBV1.2x (WT and mutant) | 2005 | Choi et al. [77] | |
| A truncated mutant (aa58–140) of HBx retains transactivation function | In vitro transiently transfected HepG2 cells | 1996 | Kumar et al. [41] | |
| HBx-deficient HBV genomes are compromised for HBV replication | In vitro transfection of HepG2 cells with pHBV1.2 and an HBx-deficient plasmid; in vivo infection mice model (in hydrodynamic injection) | 2009 | Keasler et al. [61] | |
| The stimulation of viral genome replication by HBx is linked to both nuclear and cytoplasmic HBx | in vitro transfection of HepG2 cells with pHBV1.2 Wt or HBx-null construct | 2009 | Cha et al. [56] | |
| HBx is indispensable for HBV replication | In vivo infection of human hepatocyte chimeric mice with WT (pHBV1.4) and HBx-def HBV | 2010 | Tsuge et al. [78] |
| Pathogenesis of C-Terminal Truncated HBx | Study Design | Year | References | |
|---|---|---|---|---|
| Cancer-related signalling pathways | Abrogates the antiproliferative and transactivation effects of HBx | In vitro HepG2 and MIHA cells transfected with full-length and mutant HBx, HCC patient samples | 2008 | Ma et al. [111] |
| Downregulates TXNIP protein to reprogram glucose metabolism | In vitro HBx-expressing MIHA and LO-2 cell lines; in vivo mice model and HCC patient samples | 2021 | Zhang et al. [114] | |
| Induces cancer and stem cell-like properties in HCC cell lines through overexpression | In vitro HCC cells, Huh7 and immortalized normal liver cells MIHA with or without HBx-ΔC mutants stably overexpressed | 2016 | Ng et al. [115] | |
| Enhances cell invasiveness and metastasis in HCC by activating MMP10 through C-Jun | In vitro full-length and C-truncated HBx-expressing human hepatoma cells and human HCC samples | 2013 | Sze et al. [116] | |
| Regulates tumorigenicity, self-renewal and drug resistance via STAT3/Nanog signaling pathway | In vitro full-length and C-truncated HBx-expressing human hepatoma cells | 2017 | Ching et al. [117] | |
| Upregulates transcription of FAS, mediated by 5-lipoxygenase (5-LOX) | In vitro HBx-expressing human hepatoma HepG2 and H7402 cells | 2010 | Wang et al. [118] | |
| Activates caveolin-1/LRP6/β-catenin/ FRMD5 axis in promoting hepatocarcinogenesis | In vitro full-length and C-truncated HBx-expressing human hepatoma cells; in vivo mice model; HCC clinical samples | 2019 | Mao et al. [119] | |
| Deregulates metastasis suppressors in hepatocellular carcinoma | In vitro full-length and C-truncated HBx-expressing human hepatoma cell lines and HCC samples | 2016 | Li et al. [120] | |
| Induces endoplasmic reticulum and mitochondrial stress responses | In vitro transfection in human HCC cells | 2016 | Montalbano et al. [121] | |
| Induces oxidative stress-associated tumor metastasis | In vitro transfection in Huh-7, HepG2, and Chang liver cells | 2013 | Jung et al. [122] | |
| Disrupts cell cycle regulation and glucose metabolis in FXR-deficient HCC | In vitro transfection with full-length and C-terminal truncated HBx in human Hep3B hepatocellular carcinoma cell line | 2023 | Wu et al. [123] | |
| Inducing genomic instability | Binds DDB proteins | In vitro wild-type or mutant HBx-expressing HepG2 cell lines | 1998 | Becker et al. [124] |
| Decreases HBx stability and HBV replication, impairs HBx activation of NF-κB and a minimal promoter | In vitro HepG2 cells transfected with full-length HBx and truncation mutants | 2011 | Lizzano et al. [110] | |
| Degradation of Smc5/6 | In vitro wild type and mutant HBx-expressing HepG2, HepAD38, and HepG2-NTCP cell lines | 2016 | Murphy et al. [66] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Hamadalnil, Y.; Tu, T. Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis. Viruses 2024, 16, 1361. https://doi.org/10.3390/v16091361
Li D, Hamadalnil Y, Tu T. Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis. Viruses. 2024; 16(9):1361. https://doi.org/10.3390/v16091361
Chicago/Turabian StyleLi, Dong, Yassir Hamadalnil, and Thomas Tu. 2024. "Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis" Viruses 16, no. 9: 1361. https://doi.org/10.3390/v16091361
APA StyleLi, D., Hamadalnil, Y., & Tu, T. (2024). Hepatitis B Viral Protein HBx: Roles in Viral Replication and Hepatocarcinogenesis. Viruses, 16(9), 1361. https://doi.org/10.3390/v16091361