

A Balancing Act: The Viral–Host Battle over RNA Binding Proteins

{kind=link}
{kind=link}
Abstract
1. Introduction
2. RNA Elements and Viral Infection
3. Host–Viral RBP Role during Viral Infections
4. RBP Modulation of RNA Stability in Subcellular Localizations during Viral Infections
4.1. Nuclear and Cytoplasmic Regulation
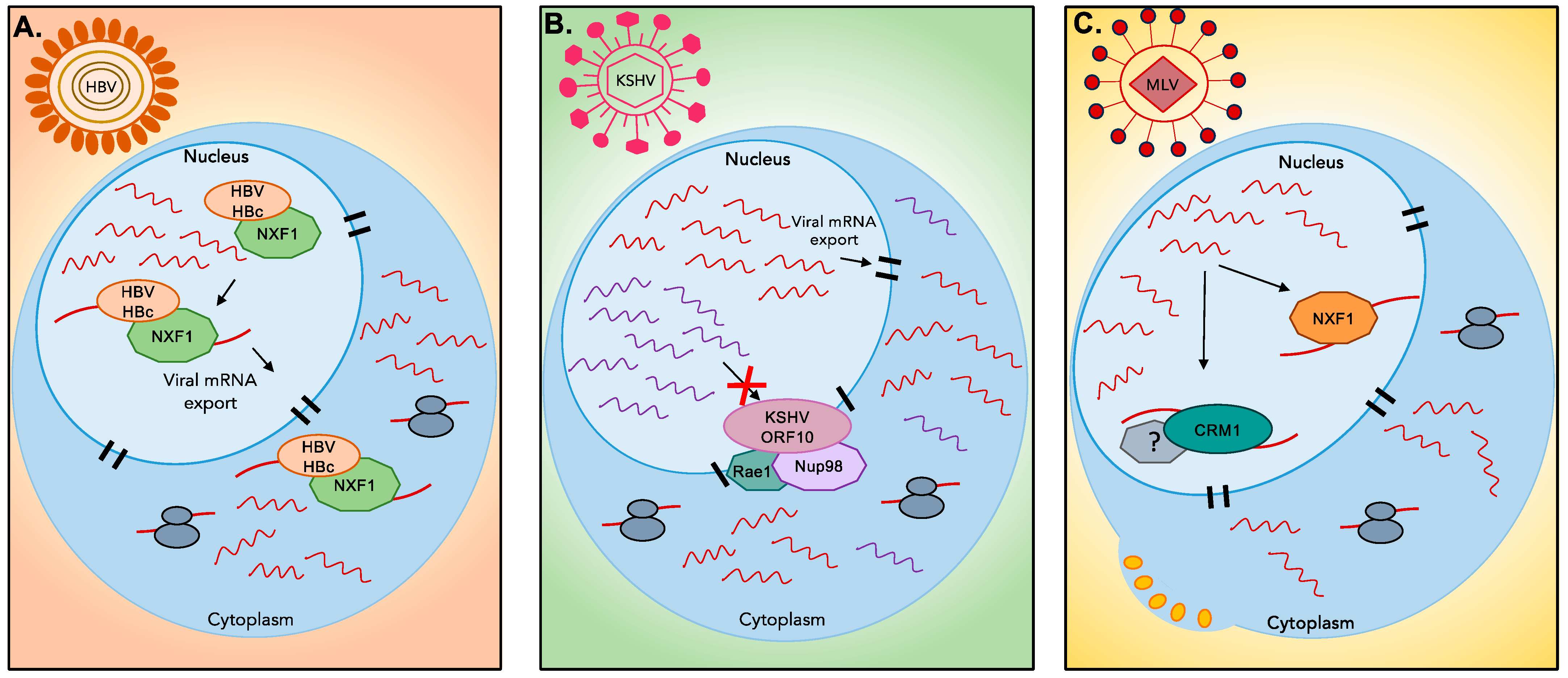
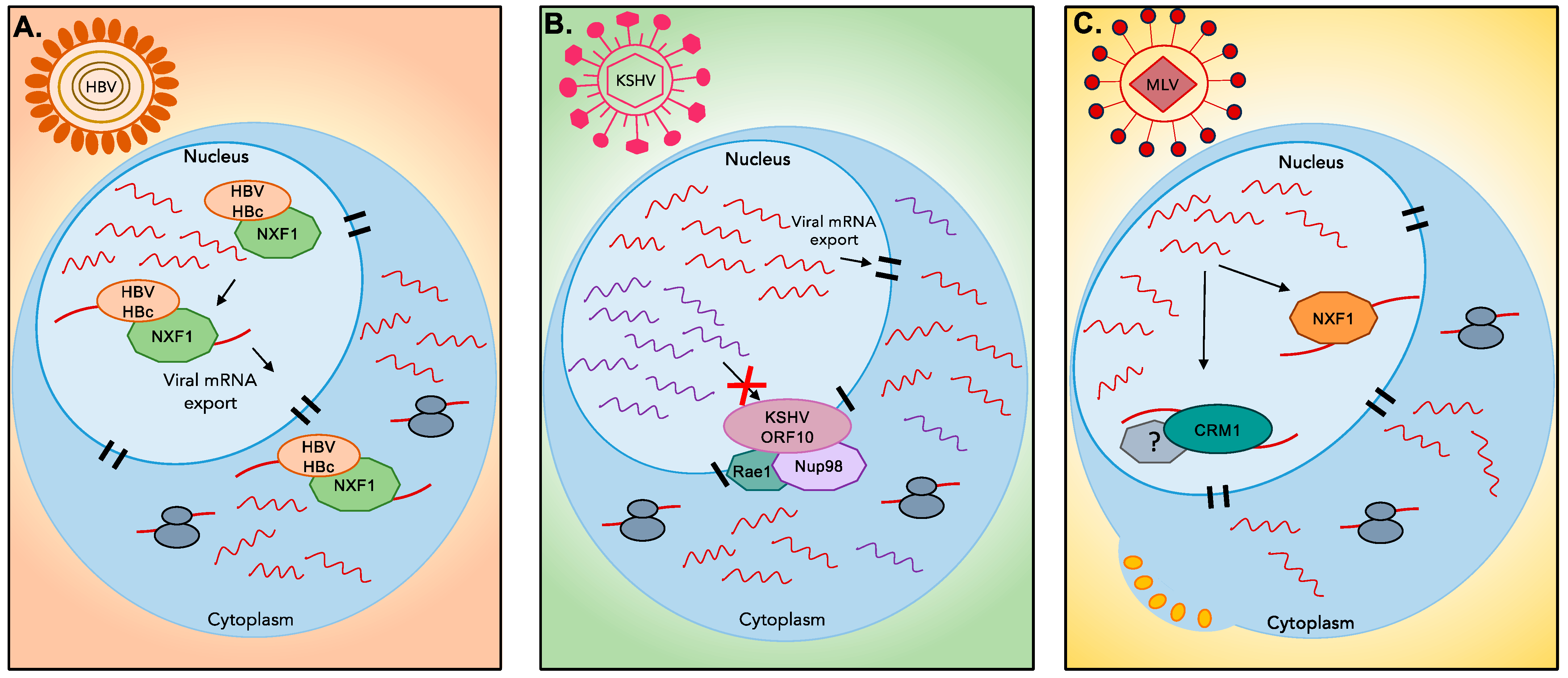
4.2. Nuclear Export
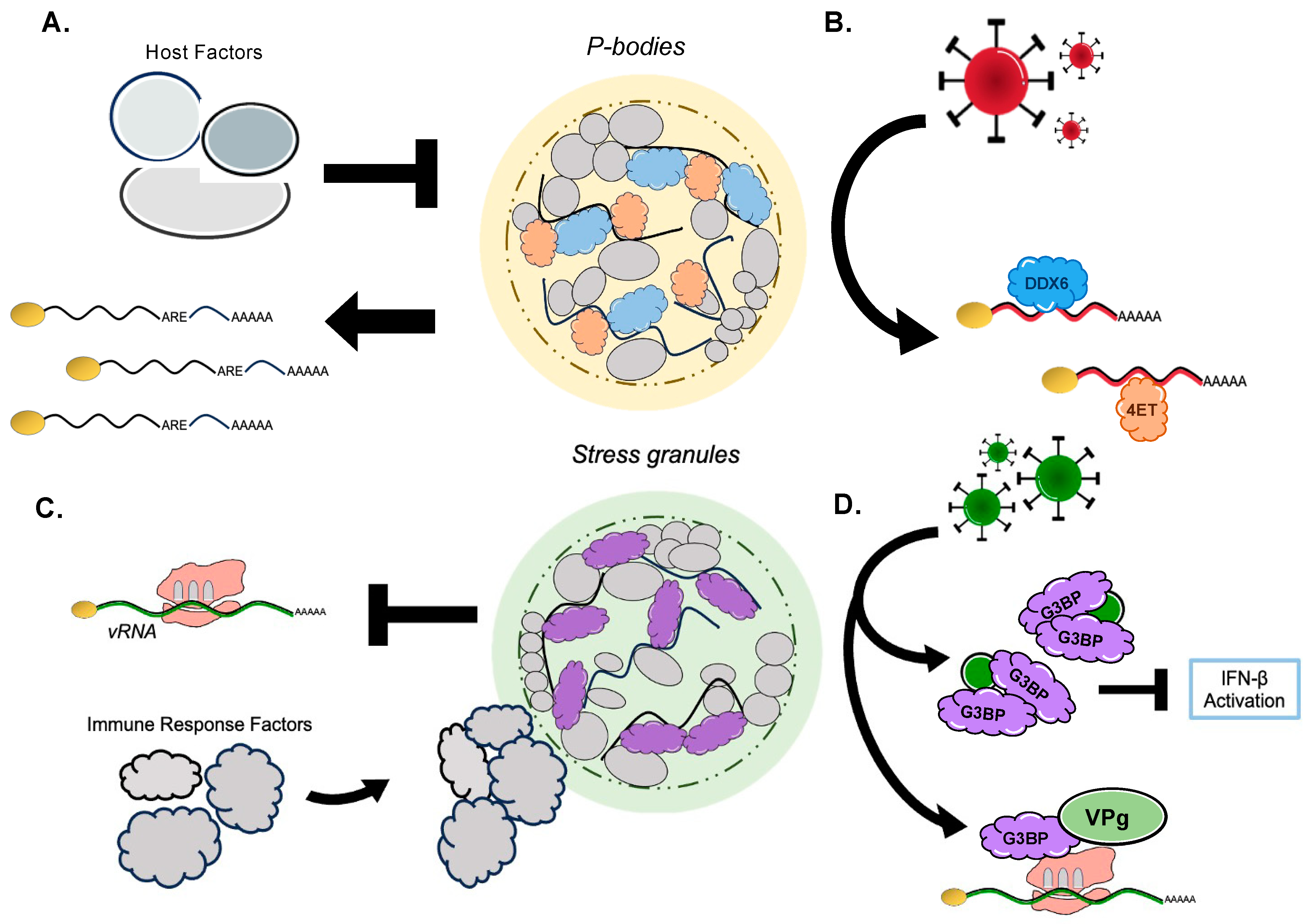
5. RNA Granules: A Nexus in the Viral–Host Struggle over RNA
5.1. Viral Factors Exercise Control over P-Body RBPs to Promote Viral Replication
5.2. Hosts Manipulate P-Body RBPs to Alter RNA Availability/Degradation to Combat Viral Infection
5.3. Viruses Influence Stress Granule RBPs to Suppress Host Immune Response Transcripts and Positively Regulate Viral RNA Fate
5.4. Host Agents Orchestrate Stress Granule RBPs to Effectively Quench Viral Replication
5.5. Granule Functionality/Effects Are Convoluted; Both Virus and Host Wield SG RBPs for Their Own Benefit
6. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Singh, G.; Pratt, G.; Yeo, G.W.; Moore, M.J. The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu. Rev. Biochem. 2015, 84, 325–354. [Google Scholar] [CrossRef]
- Mitchell, S.F.; Parker, R. Principles and properties of eukaryotic mRNPs. Mol. Cell 2014, 54, 547–558. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Lloyd, R.E. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip. Rev. RNA 2013, 4, 317–331. [Google Scholar] [CrossRef]
- Sagan, S.M.; Weber, S.C. Let’s phase it: Viruses are master architects of biomolecular condensates. Trends Biochem. Sci. 2023, 48, 229–243. [Google Scholar] [CrossRef]
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195. [Google Scholar] [CrossRef]
- Chahar, H.S.; Chen, S.; Manjunath, N. P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 2013, 436, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Iseni, F.; Garcin, D.; Nishio, M.; Kedersha, N.; Anderson, P.; Kolakofsky, D. Sendai virus trailer RNA binds TIAR, a cellular protein involved in virus-induced apoptosis. EMBO J. 2002, 21, 5141–5150. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Liu, S.; Li, Y.; Yang, G.; Luo, Y.; Li, S.; Du, H.; Zhao, Y.; Wang, D.; Chen, J.; et al. The Nuclear Matrix Protein SAFA Surveils Viral RNA and Facilitates Immunity by Activating Antiviral Enhancers and Super-enhancers. Cell Host Microbe 2019, 26, doi. [Google Scholar] [CrossRef]
- Rodriguez, W.; Mehrmann, T.; Hatfield, D.; Muller, M. Shiftless Restricts Viral Gene Expression and Influences RNA Granule Formation during Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2022, 96, e0146922. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcriptome-Wide Cleavage Site Mapping on Cellular mRNAs Reveals Features Underlying Sequence-Specific Cleavage by the Viral Ribonuclease SOX. PLoS Pathog. 2015, 11, e1005305. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.S.; Vogt, C.; Bohne, J.; Glaunsinger, B.A. Site specific target binding controls RNA cleavage efficiency by the Kaposi’s sarcoma-associated herpesvirus endonuclease SOX. Nucleic Acids Res. 2018, 46, 11968–11979. [Google Scholar] [CrossRef]
- Gaucherand, L.; Iyer, A.; Gilabert, I.; Rycroft, C.H.; Gaglia, M.M. Cut site preference allows influenza A virus PA-X to discriminate between host and viral mRNAs. Nat. Microbiol. 2023, 8, 1304–1317. [Google Scholar] [CrossRef]
- Gaucherand, L.; Porter, B.K.; Levene, R.E.; Price, E.L.; Schmaling, S.K.; Rycroft, C.H.; Kevorkian, Y.; McCormick, C.; Khaperskyy, D.A.; Gaglia, M.M. The Influenza A Virus Endoribonuclease PA-X Usurps Host mRNA Processing Machinery to Limit Host Gene Expression. Cell Rep. 2019, 27, 776–792.e7. [Google Scholar] [CrossRef] [PubMed]
- Hutin, S.; Lee, Y.; Glaunsinger, B.A. An RNA element in human interleukin 6 confers escape from degradation by the gammaherpesvirus SOX protein. J. Virol. 2013, 87, 4672–4682. [Google Scholar] [CrossRef]
- Muller, M.; Hutin, S.; Marigold, O.; Li, K.H.; Burlingame, A.; Glaunsinger, B.A. A ribonucleoprotein complex protects the interleukin-6 mRNA from degradation by distinct herpesviral endonucleases. PLoS Pathog. 2015, 11, e1004899. [Google Scholar] [CrossRef]
- Muller, M.; Glaunsinger, B.A. Nuclease escape elements protect messenger RNA against cleavage by multiple viral endonucleases. PLoS Pathog. 2017, 13, e1006593. [Google Scholar] [CrossRef]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of evolution and host gene mimicry in influenza and other RNA viruses. PLoS Pathog. 2008, 4, e1000079. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Mukerji, R.; Smith, G.J. RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion. PLoS Pathog. 2015, 11, e1004902. [Google Scholar] [CrossRef]
- Smyth, R.P.; Negroni, M.; Lever, A.M.; Mak, J.; Kenyon, J.C. RNA Structure-A Neglected Puppet Master for the Evolution of Virus and Host Immunity. Front. Immunol. 2018, 9, 2097. [Google Scholar] [CrossRef]
- Garcia-Moreno, M.; Jarvelin, A.I.; Castello, A. Unconventional RNA-binding proteins step into the virus-host battlefront. Wiley Interdiscip. Rev. RNA 2018, 9, e1498. [Google Scholar] [CrossRef] [PubMed]
- Lisy, S.; Rothamel, K.; Ascano, M. RNA Binding Proteins as Pioneer Determinants of Infection: Protective, Proviral, or Both? Viruses 2021, 13, 2172. [Google Scholar] [CrossRef] [PubMed]
- Brugier, A.; Hafirrassou, M.L.; Pourcelot, M.; Baldaccini, M.; Kril, V.; Couture, L.; Kummerer, B.M.; Gallois-Montbrun, S.; Bonnet-Madin, L.; Vidalain, P.O.; et al. RACK1 Associates with RNA-Binding Proteins Vigilin and SERBP1 to Facilitate Dengue Virus Replication. J. Virol. 2022, 96, e0196221. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Baum, S.; Bittins, M.; Frey, S.; Seedorf, M. Asc1p, a WD40-domain containing adaptor protein, is required for the interaction of the RNA-binding protein Scp160p with polysomes. Biochem. J. 2004, 380, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, E.; Leonard, M.; Mak, R.; Liao, J.; Fulzele, A.; Bennett, E.J. ZNF598 and RACK1 Regulate Mammalian Ribosome-Associated Quality Control Function by Mediating Regulatory 40S Ribosomal Ubiquitylation. Mol. Cell 2017, 65, 751–760.e4. [Google Scholar] [CrossRef] [PubMed]
- Ceci, M.; Gaviraghi, C.; Gorrini, C.; Sala, L.A.; Offenhauser, N.; Marchisio, P.C.; Biffo, S. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature 2003, 426, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Noerenberg, M.; Ni, S.; Jarvelin, A.I.; Gonzalez-Almela, E.; Lenz, C.E.; Bach-Pages, M.; Cox, V.; Avolio, R.; Davis, T.; et al. System-wide Profiling of RNA-Binding Proteins Uncovers Key Regulators of Virus Infection. Mol. Cell 2019, 74, 196–211.e11. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Krahling, V.; Kolesnikova, L.; Halwe, S.; Lier, C.; Baumeister, S.; Noda, T.; Biedenkopf, N.; Becker, S. Serine-Arginine Protein Kinase 1 Regulates Ebola Virus Transcription. mBio 2020, 11, e02565-19. [Google Scholar] [CrossRef]
- Merino, V.F.; Yan, Y.; Ordonez, A.A.; Bullen, C.K.; Lee, A.; Saeki, H.; Ray, K.; Huang, T.; Jain, S.K.; Pomper, M.G. Nucleolin mediates SARS-CoV-2 replication and viral-induced apoptosis of host cells. Antivir. Res. 2023, 211, 105550. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 2019, 176, 625–635.e14. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, W.; Muller, M. Shiftless, a Critical Piece of the Innate Immune Response to Viral Infection. Viruses 2022, 14, 1338. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Kong, N.; Zhang, Y.; Wang, C.; Dong, S.; Zhai, H.; Zhai, X.; Yang, X.; Ye, C.; Ye, M.; et al. PTBP1 suppresses porcine epidemic diarrhea virus replication via inducing protein degradation and IFN production. J. Biol. Chem. 2023, 299, 104987. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.H.; Verschueren, E.; Jang, G.M.; Kleffman, K.; Johnson, J.R.; Park, J.; Von Dollen, J.; Maher, M.C.; Johnson, T.; Newton, W.; et al. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 2015, 57, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403.e19. [Google Scholar] [CrossRef]
- Chen, T.; Yang, H.; Liu, P.; Hamiti, M.; Zhang, X.; Xu, Y.; Quan, W.; Zhang, Y.; Yu, W.; Jiao, L.; et al. Splicing factor SF3B3, a NS5-binding protein, restricts ZIKV infection by targeting GCH1. Virol. Sin. 2023, 38, 222–232. [Google Scholar] [CrossRef]
- Schneider-Lunitz, V.; Ruiz-Orera, J.; Hubner, N.; van Heesch, S. Multifunctional RNA-binding proteins influence mRNA abundance and translational efficiency of distinct sets of target genes. PLoS Comput. Biol. 2021, 17, e1009658. [Google Scholar] [CrossRef]
- Mayya, V.K.; Duchaine, T.F. Ciphers and Executioners: How 3′-Untranslated Regions Determine the Fate of Messenger RNAs. Front. Genet. 2019, 10, 6. [Google Scholar] [CrossRef]
- Timmers, H.T.M.; Tora, L. Transcript Buffering: A Balancing Act between mRNA Synthesis and mRNA Degradation. Mol. Cell 2018, 72, 10–17. [Google Scholar] [CrossRef]
- Chin, A.; Lecuyer, E. RNA localization: Making its way to the center stage. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2956–2970. [Google Scholar] [CrossRef]
- Gilbertson, S.; Federspiel, J.D.; Hartenian, E.; Cristea, I.M.; Glaunsinger, B. Changes in mRNA abundance drive shuttling of RNA binding proteins, linking cytoplasmic RNA degradation to transcription. eLife 2018, 7, e37663. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Glaunsinger, B.A. Aberrant herpesvirus-induced polyadenylation correlates with cellular messenger RNA destruction. PLoS Biol. 2009, 7, e1000107. [Google Scholar] [CrossRef]
- Park, R.; El-Guindy, A.; Heston, L.; Lin, S.F.; Yu, K.P.; Nagy, M.; Borah, S.; Delecluse, H.J.; Steitz, J.; Miller, G. Nuclear translocation and regulation of intranuclear distribution of cytoplasmic poly(A)-binding protein are distinct processes mediated by two Epstein Barr virus proteins. PLoS ONE 2014, 9, e92593. [Google Scholar] [CrossRef] [PubMed]
- Richner, J.M.; Clyde, K.; Pezda, A.C.; Cheng, B.Y.; Wang, T.; Kumar, G.R.; Covarrubias, S.; Coscoy, L.; Glaunsinger, B. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog. 2011, 7, e1002150. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Glaunsinger, B.A. Feedback to the central dogma: Cytoplasmic mRNA decay and transcription are interdependent processes. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 385–398. [Google Scholar] [CrossRef]
- Sandri-Goldin, R.M. Nuclear export of herpes virus RNA. Curr. Top. Microbiol. Immunol. 2001, 259, 2–23. [Google Scholar] [PubMed]
- Carmody, S.R.; Wente, S.R. mRNA nuclear export at a glance. J. Cell Sci. 2009, 122, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef]
- Guo, J.; Zhu, Y.; Ma, X.; Shang, G.; Liu, B.; Zhang, K. Virus Infection and mRNA Nuclear Export. Int. J. Mol. Sci. 2023, 24, 12593. [Google Scholar] [CrossRef]
- Stewart, M. Polyadenylation and nuclear export of mRNAs. J. Biol. Chem. 2019, 294, 2977–2987. [Google Scholar] [CrossRef]
- Vogt, C.; Bohne, J. The KSHV RNA regulator ORF57: Target specificity and its role in the viral life cycle. Wiley Interdiscip. Rev. RNA 2016, 7, 173–185. [Google Scholar] [CrossRef]
- Gales, J.P.; Kubina, J.; Geldreich, A.; Dimitrova, M. Strength in Diversity: Nuclear Export of Viral RNAs. Viruses 2020, 12, 1014. [Google Scholar] [CrossRef]
- Gong, D.; Kim, Y.H.; Xiao, Y.; Du, Y.; Xie, Y.; Lee, K.K.; Feng, J.; Farhat, N.; Zhao, D.; Shu, S.; et al. A Herpesvirus Protein Selectively Inhibits Cellular mRNA Nuclear Export. Cell Host Microbe 2016, 20, 642–653. [Google Scholar] [CrossRef]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef]
- Pardamean, C.I.; Wu, T.T. Inhibition of Host Gene Expression by KSHV: Sabotaging mRNA Stability and Nuclear Export. Front. Cell Infect. Microbiol. 2021, 11, 648055. [Google Scholar] [CrossRef] [PubMed]
- Mougel, M.; Akkawi, C.; Chamontin, C.; Feuillard, J.; Pessel-Vivares, L.; Socol, M.; Laine, S. NXF1 and CRM1 nuclear export pathways orchestrate nuclear export, translation and packaging of murine leukaemia retrovirus unspliced RNA. RNA Biol. 2020, 17, 528–538. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Dai, T.; Qin, Z.; Lu, H.; Zhang, L.; Zhou, F. Liquid-liquid phase separation in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 290. [Google Scholar] [CrossRef] [PubMed]
- Dundr, M.; Hebert, M.D.; Karpova, T.S.; Stanek, D.; Xu, H.; Shpargel, K.B.; Meier, U.T.; Neugebauer, K.M.; Matera, A.G.; Misteli, T. In vivo kinetics of Cajal body components. J. Cell Biol. 2004, 164, 831–842. [Google Scholar] [CrossRef]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Pappu, R.V. A conceptual framework for understanding phase separation and addressing open questions and challenges. Mol. Cell 2022, 82, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Alshareedah, I.; Moosa, M.M.; Pham, M.; Potoyan, D.A.; Banerjee, P.R. Programmable viscoelasticity in protein-RNA condensates with disordered sticker-spacer polypeptides. Nat. Commun. 2021, 12, 6620. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Sandoval, L.P.; Kumar, S.; Heris, H.K.; Chang, C.L.A.; Cornell, C.E.; Keller, S.L.; Francois, P.; Hendricks, A.G.; Ehrlicher, A.J.; Pappu, R.V.; et al. Endocytic proteins with prion-like domains form viscoelastic condensates that enable membrane remodeling. Proc. Natl. Acad. Sci. USA 2021, 118, e2113789118. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Kota, D.; Zhou, H.X. Shear relaxation governs fusion dynamics of biomolecular condensates. Nat. Commun. 2021, 12, 5995. [Google Scholar] [CrossRef] [PubMed]
- Jawerth, L.; Fischer-Friedrich, E.; Saha, S.; Wang, J.; Franzmann, T.; Zhang, X.; Sachweh, J.; Ruer, M.; Ijavi, M.; Saha, S.; et al. Protein condensates as aging Maxwell fluids. Science 2020, 370, 1317–1323. [Google Scholar] [CrossRef]
- Shen, Y.; Ruggeri, F.S.; Vigolo, D.; Kamada, A.; Qamar, S.; Levin, A.; Iserman, C.; Alberti, S.; George-Hyslop, P.S.; Knowles, T.P.J. Biomolecular condensates undergo a generic shear-mediated liquid-to-solid transition. Nat. Nanotechnol. 2020, 15, 841–847. [Google Scholar] [CrossRef]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032813. [Google Scholar] [CrossRef] [PubMed]
- Andrei, M.A.; Ingelfinger, D.; Heintzmann, R.; Achsel, T.; Rivera-Pomar, R.; Luhrmann, R. A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 2005, 11, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Yang, W.H.; Gulick, T.; Bloch, K.D.; Bloch, D.B. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA 2005, 11, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Yu, J.H.; Gulick, T.; Bloch, K.D.; Bloch, D.B. RNA-associated protein 55 (RAP55) localizes to mRNA processing bodies and stress granules. RNA 2006, 12, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Aigueperse, C.; Kress, M.; Dautry, F.; Weil, D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J. Cell Sci. 2005, 118, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, S.; Chekulaeva, M.; Stoecklin, G. Human Pat1b connects deadenylation with mRNA decapping and controls the assembly of processing bodies. Mol. Cell Biol. 2010, 30, 4308–4323. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Maldonado, M.; Bugaut, A.; Balasubramanian, S.; Kress, M.; Weil, D.; Standart, N. Distinct functions of maternal and somatic Pat1 protein paralogs. RNA 2010, 16, 2094–2107. [Google Scholar] [CrossRef]
- Ayache, J.; Benard, M.; Ernoult-Lange, M.; Minshall, N.; Standart, N.; Kress, M.; Weil, D. P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Mol. Biol. Cell 2015, 26, 2579–2595. [Google Scholar] [CrossRef]
- Eulalio, A.; Behm-Ansmant, I.; Schweizer, D.; Izaurralde, E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol. Cell Biol. 2007, 27, 3970–3981. [Google Scholar] [CrossRef]
- Davidson, A.; Parton, R.M.; Rabouille, C.; Weil, T.T.; Davis, I. Localized Translation of gurken/TGF-alpha mRNA during Axis Specification Is Controlled by Access to Orb/CPEB on Processing Bodies. Cell Rep. 2016, 14, 2451–2462. [Google Scholar] [CrossRef]
- Fan, S.J.; Marchand, V.; Ephrussi, A. Drosophila Ge-1 promotes P body formation and oskar mRNA localization. PLoS ONE 2011, 6, e20612. [Google Scholar] [CrossRef]
- Eystathioy, T.; Jakymiw, A.; Chan, E.K.; Seraphin, B.; Cougot, N.; Fritzler, M.J. The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA 2003, 9, 1171–1173. [Google Scholar] [CrossRef]
- Sharma, N.R.; Zheng, Z.M. RNA Granules in Antiviral Innate Immunity: A Kaposi’s Sarcoma-Associated Herpesvirus Journey. Front. Microbiol. 2021, 12, 794431. [Google Scholar] [CrossRef]
- Buchan, J.R.; Muhlrad, D.; Parker, R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 2008, 183, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Xu, Y.; Wang, B.; David, M.D.; Schubert, P.; Kennedy, D.; Schrader, J.W. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell Biol. 2007, 27, 2324–2342. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Chen, S.; Gilks, N.; Li, W.; Miller, I.J.; Stahl, J.; Anderson, P. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol. Biol. Cell 2002, 13, 195–210. [Google Scholar] [CrossRef]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Kubica, N.; Crozier, S.J.; Williamson, D.L.; Farrell, P.A.; Kimball, S.R.; Jefferson, L.S. Immediate response of mammalian target of rapamycin (mTOR)-mediated signalling following acute resistance exercise in rat skeletal muscle. J. Physiol. 2003, 553, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Ninomiya, K.; Nakagawa, S.; Yamazaki, T. A guide to membraneless organelles and their various roles in gene regulation. Nat. Rev. Mol. Cell Biol. 2023, 24, 288–304. [Google Scholar] [CrossRef]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 2000, 151, 1257–1268. [Google Scholar] [CrossRef]
- Robinson, C.A.; Singh, G.K.; Kleer, M.; Katsademas, T.; Castle, E.L.; Boudreau, B.Q.; Corcoran, J.A. Kaposi’s sarcoma-associated herpesvirus (KSHV) utilizes the NDP52/CALCOCO2 selective autophagy receptor to disassemble processing bodies. PLoS Pathog. 2023, 19, e1011080. [Google Scholar] [CrossRef]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Yu, L.; Kang, J.G.; Yang, A.; Gu, S.; Fritzler, M.J.; Zheng, Z.M. KSHV RNA-binding protein ORF57 inhibits P-body formation to promote viral multiplication by interaction with Ago2 and GW182. Nucleic Acids Res. 2019, 47, 9368–9385. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Kushima, Y.; Osugi, K.; Hijikata, M.; Maki, M.; Ikeda, M.; Kato, N. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J. Virol. 2011, 85, 6882–6892. [Google Scholar] [CrossRef]
- Pager, C.T.; Schutz, S.; Abraham, T.M.; Luo, G.; Sarnow, P. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 2013, 435, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vilaro, G.; Scheller, N.; Saludes, V.; Diez, J. Hepatitis C virus infection alters P-body composition but is independent of P-body granules. J. Virol. 2012, 86, 8740–8749. [Google Scholar] [CrossRef]
- Ariumi, Y.; Kuroki, M.; Abe, K.; Dansako, H.; Ikeda, M.; Wakita, T.; Kato, N. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 2007, 81, 13922–13926. [Google Scholar] [CrossRef]
- Jangra, R.K.; Yi, M.; Lemon, S.M. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J. Virol. 2010, 84, 6810–6824. [Google Scholar] [CrossRef]
- Scheller, N.; Mina, L.B.; Galao, R.P.; Chari, A.; Gimenez-Barcons, M.; Noueiry, A.; Fischer, U.; Meyerhans, A.; Diez, J. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc. Natl. Acad. Sci. USA 2009, 106, 13517–13522. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Xu, Z.; Liu, P.; Qin, Y.; Chen, M. Enterovirus 71 2A Protease Inhibits P-Body Formation To Promote Viral RNA Synthesis. J. Virol. 2021, 95, e0092221. [Google Scholar] [CrossRef]
- Corcoran, J.A.; Johnston, B.P.; McCormick, C. Viral activation of MK2-hsp27-p115RhoGEF-RhoA signaling axis causes cytoskeletal rearrangements, p-body disruption and ARE-mRNA stabilization. PLoS Pathog. 2015, 11, e1004597. [Google Scholar] [CrossRef]
- Blanco, F.F.; Sanduja, S.; Deane, N.G.; Blackshear, P.J.; Dixon, D.A. Transforming growth factor beta regulates P-body formation through induction of the mRNA decay factor tristetraprolin. Mol. Cell Biol. 2014, 34, 180–195. [Google Scholar] [CrossRef]
- Franks, T.M.; Lykke-Andersen, J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes. Dev. 2007, 21, 719–735. [Google Scholar] [CrossRef]
- Vindry, C.; Marnef, A.; Broomhead, H.; Twyffels, L.; Ozgur, S.; Stoecklin, G.; Llorian, M.; Smith, C.W.; Mata, J.; Weil, D.; et al. Dual RNA Processing Roles of Pat1b via Cytoplasmic Lsm1-7 and Nuclear Lsm2-8 Complexes. Cell Rep. 2017, 20, 1187–1200. [Google Scholar] [CrossRef]
- Hubstenberger, A.; Courel, M.; Benard, M.; Souquere, S.; Ernoult-Lange, M.; Chouaib, R.; Yi, Z.; Morlot, J.B.; Munier, A.; Fradet, M.; et al. P-Body Purification Reveals the Condensation of Repressed mRNA Regulons. Mol. Cell 2017, 68, 144–157.e5. [Google Scholar] [CrossRef]
- Corcoran, J.A.; Khaperskyy, D.A.; Johnston, B.P.; King, C.A.; Cyr, D.P.; Olsthoorn, A.V.; McCormick, C. Kaposi’s sarcoma-associated herpesvirus G-protein-coupled receptor prevents AU-rich-element-mediated mRNA decay. J. Virol. 2012, 86, 8859–8871. [Google Scholar] [CrossRef]
- Bakheet, T.; Williams, B.R.; Khabar, K.S. ARED 3.0: The large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006, 34, D111–D114. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Contreras, D.; Irudayam, J.I.; Ali, A.; Yang, O.O.; Arumugaswami, V. C19ORF66 is an interferon-stimulated gene (ISG) which Inhibits human immunodeficiency virus-1. BioRxiv 2016, 050310. [Google Scholar] [CrossRef]
- Suzuki, Y.; Chin, W.X.; Han, Q.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016, 12, e1005357. [Google Scholar] [CrossRef]
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Bruggemann, Y.; Friesland, M.; et al. C19orf66 is an interferon-induced inhibitor of HCV replication that restricts formation of the viral replication organelle. J. Hepatol. 2020, 73, 549–558. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Yao, Z.; Dong, X.; Zhang, D.; Hu, Y.; Zhang, S.; Lin, J.; Chen, J.; An, S.; et al. C19orf66 interrupts Zika virus replication by inducing lysosomal degradation of viral NS3. PLoS Negl. Trop. Dis. 2020, 14, e0008083. [Google Scholar] [CrossRef]
- Hanners, N.W.; Mar, K.B.; Boys, I.N.; Eitson, J.L.; De La Cruz-Rivera, P.C.; Richardson, R.B.; Fan, W.; Wight-Carter, M.; Schoggins, J.W. Shiftless inhibits flavivirus replication in vitro and is neuroprotective in a mouse model of Zika virus pathogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2111266118. [Google Scholar] [CrossRef]
- Yu, D.; Zhao, Y.; Pan, J.; Yang, X.; Liang, Z.; Xie, S.; Cao, R. C19orf66 Inhibits Japanese Encephalitis Virus Replication by Targeting -1 PRF and the NS3 Protein. Virol. Sin. 2021, 36, 1443–1455. [Google Scholar] [CrossRef]
- Balinsky, C.A.; Schmeisser, H.; Wells, A.I.; Ganesan, S.; Jin, T.; Singh, K.; Zoon, K.C. IRAV (FLJ11286), an Interferon-Stimulated Gene with Antiviral Activity against Dengue Virus, Interacts with MOV10. J. Virol. 2017, 91, e01606-16. [Google Scholar] [CrossRef]
- Luo, Y.; Na, Z.; Slavoff, S.A. P-Bodies: Composition, Properties, and Functions. Biochemistry 2018, 57, 2424–2431. [Google Scholar] [CrossRef]
- Na, Z.; Luo, Y.; Schofield, J.A.; Smelyansky, S.; Khitun, A.; Muthukumar, S.; Valkov, E.; Simon, M.D.; Slavoff, S.A. The NBDY Microprotein Regulates Cellular RNA Decapping. Biochemistry 2020, 59, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Na, Z.; Luo, Y.; Cui, D.S.; Khitun, A.; Smelyansky, S.; Loria, J.P.; Slavoff, S.A. Phosphorylation of a Human Microprotein Promotes Dissociation of Biomolecular Condensates. J. Am. Chem. Soc. 2021, 143, 12675–12687. [Google Scholar] [CrossRef] [PubMed]
- Bloch, D.B.; Sinow, C.O.; Sauer, A.J.; Corman, B.H.P. Assembly and regulation of the mammalian mRNA processing body. PLoS ONE 2023, 18, e0282496. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Okamoto, T.; Fukuhara, T.; Kambara, H.; Morita, E.; Mori, Y.; Kamitani, W.; Matsuura, Y. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with Caprin-1 and facilitates viral propagation. J. Virol. 2013, 87, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika Virus Subverts Stress Granules To Promote and Restrict Viral Gene Expression. J. Virol. 2019, 93, e00520-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bai, Y.; Zhang, X.; Gao, T.; Liu, Y.; Li, E.; Wang, X.; Cao, Z.; Zhu, L.; Dong, Q.; et al. SARS-CoV-2 N Protein Antagonizes Stress Granule Assembly and IFN Production by Interacting with G3BPs to Facilitate Viral Replication. J. Virol. 2022, 96, e0041222. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ru, Y.; Ren, J.; Bai, J.; Wei, J.; Fu, S.; Liu, X.; Li, D.; Zheng, H. G3BP1 inhibits RNA virus replication by positively regulating RIG-I-mediated cellular antiviral response. Cell Death Dis. 2019, 10, 946. [Google Scholar] [CrossRef]
- Hosmillo, M.; Lu, J.; McAllaster, M.R.; Eaglesham, J.B.; Wang, X.; Emmott, E.; Domingues, P.; Chaudhry, Y.; Fitzmaurice, T.J.; Tung, M.K.; et al. Noroviruses subvert the core stress granule component G3BP1 to promote viral VPg-dependent translation. eLife 2019, 8, e46681. [Google Scholar] [CrossRef]
- Sun, L.; Chen, H.; Ming, X.; Bo, Z.; Shin, H.J.; Jung, Y.S.; Qian, Y. Porcine Epidemic Diarrhea Virus Infection Induces Caspase-8-Mediated G3BP1 Cleavage and Subverts Stress Granules To Promote Viral Replication. J. Virol. 2021, 95, e02344-20. [Google Scholar] [CrossRef]
- Ramnani, B.; Powell, S.; Shetty, A.G.; Manivannan, P.; Hibbard, B.R.; Leaman, D.W.; Malathi, K. Viral Hemorrhagic Septicemia Virus Activates Integrated Stress Response Pathway and Induces Stress Granules to Regulate Virus Replication. Viruses 2023, 15, 466. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermudez, Y.; Hatfield, D.; Muller, M. A Balancing Act: The Viral–Host Battle over RNA Binding Proteins. Viruses 2024, 16, 474. https://doi.org/10.3390/v16030474
Bermudez Y, Hatfield D, Muller M. A Balancing Act: The Viral–Host Battle over RNA Binding Proteins. Viruses. 2024; 16(3):474. https://doi.org/10.3390/v16030474
Chicago/Turabian StyleBermudez, Yahaira, David Hatfield, and Mandy Muller. 2024. "A Balancing Act: The Viral–Host Battle over RNA Binding Proteins" Viruses 16, no. 3: 474. https://doi.org/10.3390/v16030474
APA StyleBermudez, Y., Hatfield, D., & Muller, M. (2024). A Balancing Act: The Viral–Host Battle over RNA Binding Proteins. Viruses, 16(3), 474. https://doi.org/10.3390/v16030474