
Molecular Profile of Variants Potentially Associated with Severe Forms of COVID-19 in Amazonian Indigenous Populations

 , , ,
, , ,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Consent and Ethics
2.2. Study Population
2.3. DNA Extraction and Preparation of the Exome Library
2.4. Bioinformatics Analysis
2.5. Statistical Analyses
2.6. Selection of Variants
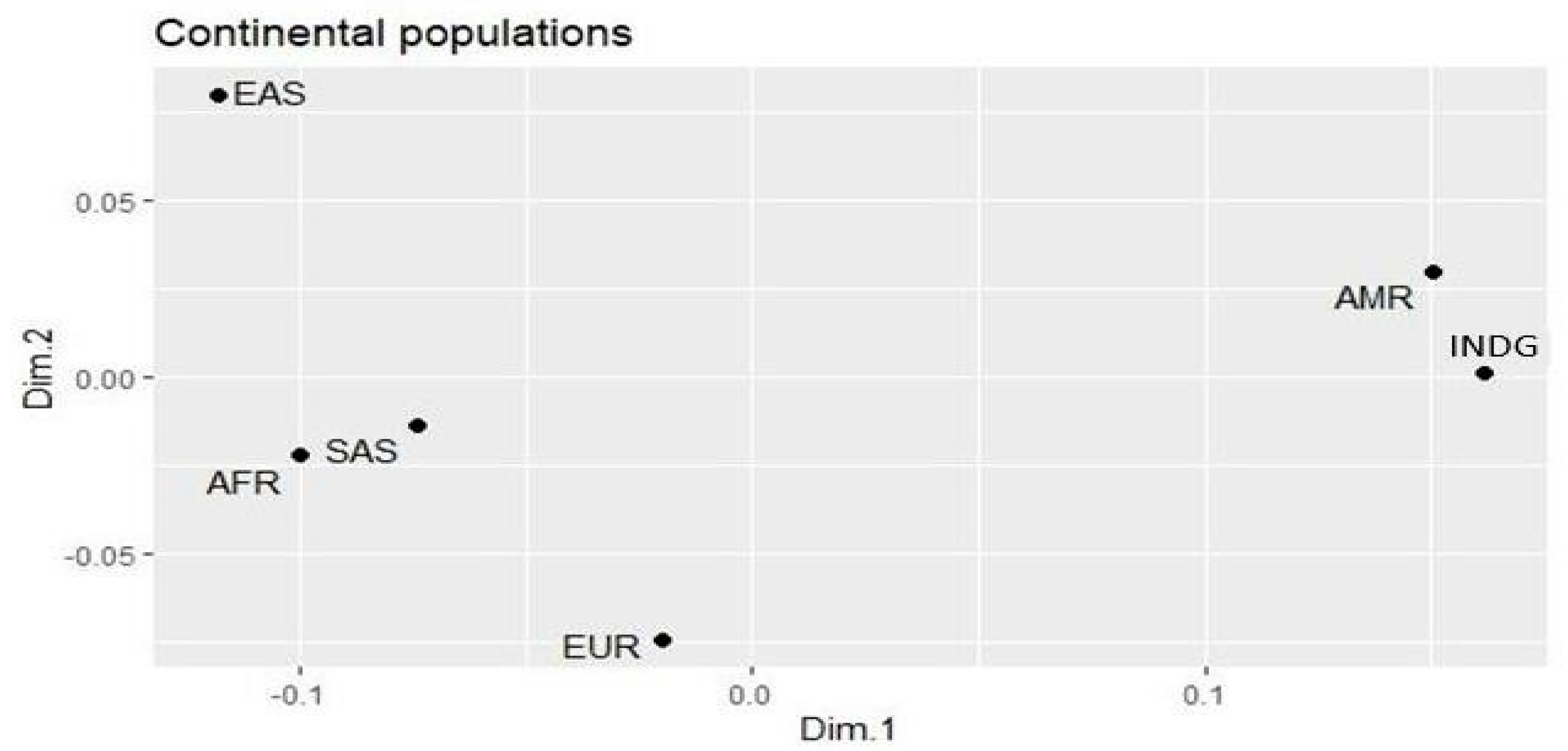
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, H.; Stratton, C.W.; Tang, Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020, 92, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 15, 497–506. [Google Scholar] [CrossRef]
- Dhama, K.; Patel, S.K.; Pathak, M.; Yatoo, M.I.; Tiwari, R.; Malik, Y.S.; Singh, R.; Sah, R.; Rabaan, A.A.; Bonilla-Aldana, D.K.; et al. An update on SARS-CoV-2/COVID-19 with particular reference to its clinical pathology, pathogenesis, immunopathology and mitigation strategies. Travel. Med. Infect. Dis. 2020, 37, 101755. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [PubMed]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 30 April 2023).
- da Silva Torres, M.K.; Bichara, C.D.A.; de Almeida, M.N.D.S.; Vallinoto, M.C.; Queiroz, M.A.F.; Vallinoto, I.M.V.C.; Dos Santos, E.J.M.; de Carvalho, C.A.M.; Vallinoto, A.C.R. The Complexity of SARS-CoV-2 Infection and the COVID-19 Pandemic. Front. Microbiol. 2022, 13, 789882. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; Aslibekyan, S.; Auton, A.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef]
- Cruz, R.; Almeida, S.D.; Heredia, M.L.; Quintela, I.; Ceballos, F.C.; Pita, G.; Lorenzo-Salazar, J.M.; González-Montelongo, R.; Gago-Domínguez, M.; Porras, M.S.; et al. Novel genes and sex differences in COVID-19 severity. Hum. Mol. Genet. 2022, 16, ddac132. [Google Scholar] [CrossRef]
- Pastana, L.F.; Silva, T.A. The Genomic Profile Associated with Risk of Severe Forms of COVID-19 in Amazonian Native American Populations. J. Pers. Med. 2022, 12, 554. [Google Scholar] [CrossRef]
- Leal, D.F.D.V.B.; Da Silva, M.N.S.; Fernandes, D.C.R.D.O.; Rodrigues, J.C.G.; Barros, M.C.D.C.; Pinto, P.D.D.C.; Pastana, L.F.; Da Silva, C.A.; Fernandes, M.R.; De Assumpção, P.P.; et al. Amerindian genetic ancestry as a risk factor for tuberculosis in an amazonian population. PLoS ONE 2020, 15, e0236033. [Google Scholar] [CrossRef]
- De Ramos, B.R.A.; D’Elia, M.P.B.; Amador, M.A.T.; Santos, N.P.C.; Santos, S.E.B.; da Castelli, E.C.; Witkin, S.S.; Miot, H.A.; Miot, L.D.B.; da Silva, M.G. Neither self-reported ethnicity nor declared family origin are reliable indicators of genomic ancestry. Genetica 2016, 144, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.C.G.; de Souza, T.P.; Pastana, L.F.; Ribeiro dos Santos, A.M.; Fernandes, M.R.; Pinto, P.; Wanderley, A.V.; De Souza, S.J.; Kroll, J.E.; Pereira, A.L.; et al. Identification of NUDT15 gene variants in Amazonian Amerindians and admixed individuals from northern Brazil. PLoS ONE 2020, 15, e0231651. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-dos-Santos, A.M.; Vidal, A.F.; Vinasco-Sandoval, T.; Guerreiro, J.; Santos, S.; Ribeiro-dos-Santos, Â. Exome Sequencing of Native Populations from the Amazon Reveals Patterns on the Peopling of South America. Front. Genet. 2020, 11, 548507. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.; Salgado, C.; Santos, N.P.C.; Santos, S.; Ribeiro-dos-Santos, Â. Influence of Genetic Ancestry on INDEL Markers of NFKβ1, CASP8, PAR1, IL4 and CYP19A1 Genes in Leprosy Patients. PLoS Negl. Trop. Dis. 2015, 9, e0004050. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Walker, R.S.; Sattenspiel, L.; Hill, K.R. Mortality from contact-related epidemics among indigenous populations in Greater Amazonia. Sci. Rep. 2015, 5, 14032. [Google Scholar] [CrossRef]
- Salzano, F.M. Fatores determinísticos e estocásticos no processo microevolucionário humano. Actas V Congr. Latinoam. Genet. 1982, 1, 81–89. [Google Scholar]
- De Brito Vargas, L.; Beltrame, M.H.; Ho, B.; Marin, W.M.; Dandekar, R.; Montero-Martín, G.; Fernández-Viña, M.A.; Hurtado, A.M.; Hill, K.R.; Tsuneto, L.T.; et al. Remarkably Low KIR and HLA Diversity in Amerindians Reveals Signatures of Strong Purifying Selection Shaping the Centromeric KIR Region. Mol. Biol. Evol. 2022, 39, msab298. [Google Scholar] [CrossRef]
- Verkman, A.S.; Hara-Chikuma, M.; Papadopoulos, M.C. Aquaporins–new players in cancer biology. J. Mol. Med. 2008, 86, 523–529. [Google Scholar] [CrossRef]
- Verkman, A.S.; Ruiz-Ederra, J.; Levin, M.H. Functions of aquaporins in the eye. Prog. Retin. Eye Res. 2008, 27, 420–433. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Lodder, E.M.; Wilders, R. Aquaporin channels in the heart-physiology and pathophysiology. Int. J. Mol. Sci. 2019, 20, 2039. [Google Scholar] [CrossRef] [PubMed]
- Meli, R.; Pirozzi, C.; Pelagalli, A. New perspectives on the potential role of aquaporins (AQPs) in the physiology of inflammation. Front. Physiol. 2018, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.K.; Raihan, T.; Ahmed, J.; Hakim, A.; Emon, T.H.; Chowdhury, P.A. Human Aquaporins: Functional Diversity and Potential Roles in Infectious and Non-infectious Diseases. Front. Genet. 2021, 12, 654865. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Rawlik, K.; Bretherick, A.D.; Qi, T.; Wu, Y.; Nassiri, I.; McConkey, G.A.; Zechner, M.; Klaric, L.; Griffiths, F.; et al. GWAS and meta-analysis identifies 49 genetic variants underlying critical COVID-19. Nature 2023, 617, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Fricke-Galindo, I.; Martínez-Morales, A.; Chávez-Galán, L.; Ocaña-Guzmán, R.; Buendía-Roldán, I.; Pérez-Rubio, G.; Hernández-Zenteno, R.J.; Verónica-Aguilar, A.; Alarcón-Dionet, A.; Aguilar-Duran, H.; et al. IFNAR2 relevance in the clinical outcome of individuals with severe COVID-19. Front. Immunol. 2022, 29, 949413. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2020, 591, 92–98. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. A second update on mapping the human genetic architecture of COVID-19. Nature 2023, 621, E7–E26. [Google Scholar] [CrossRef]
- Banday, A.R.; Stanifer, M.L.; Florez-Vargas, O.; Onabajo, O.O.; Zahoor, M.A.; Papenberg, B.W.; Ring, T.J.; Lee, C.H.; Andreakos, E.; Arons, E.; et al. Genetic regulation of OAS1 nonsense-mediated decay underlies association with risk of severe COVID-19. Nat. Genet. 2022, 54, 1103–1116. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, L.; Zhou, W.; Chen, Y.; Cheng, Y.; Dong, J. LIMD1 phosphorylation in mitosis is required for mitotic progression and its tumor-suppressing activity. FEBS J. 2019, 286, 963–974. [Google Scholar] [CrossRef]
- Sharp, T.V.; Munoz, F.; Bourboulia, D.; Presneau, N.; Darai, E.; Wang, H.W.; Cannon, M.; Butcher, D.N.; Nicholson, A.G.; Klein, G.; et al. LIM Domains-containing protein 1 (LIMD1), a tumor suppressor encoded at chromosome 3p21.3, binds pRB and represses E2F-driven transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 16531–16536. [Google Scholar] [CrossRef]
- Spendlove, I.; Al-Attar, A.; Watherstone, O.; Webb, T.M.; Ellis, I.O.; Longmore, G.D.; Sharp, T.V. Differential subcellular localisation of the tumour suppressor protein LIMD1 in breast cancer correlates with patient survival. Int. J. Cancer 2008, 123, 2247–2253. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Position | SNP ID | Ref a | Var b | Impact Predicted by SNPeff | Variant Allele Frequence | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| INDG | AFR | AMR | EAS | EUR | SAS | ||||||
| ARHGAP27 | 45404053 | rs201721078 | G | A | HIGH | 0.218 | 0.0009 | 0.0421 | - | 0.0001 | - |
| ARHGAP27 | 45429642 | rs2959953 | G | C | MODERATE | 0.375 | 0.6953 | 0.6537 | 0.520 | 0.717 | 0.673 |
| ARHGAP27 | 45429931 | rs12949256 | C | T | MODERATE | 0 | - | - | - | 0.5 | 0.166 |
| ARHGAP27 | 45395459 | rs117139057 | G | C | MODERATE | 0.125 | 0.089 | 0.2028 | 0.2768 | 0.1791 | 0.1189 |
| ARHGAP27 | 45430283 | rs7222444 | A | G | MODIFIER | 0.009 | 0.102 | 0.158 | 0.0005 | 0.243 | 0.071 |
| ARHGAP27 | 45395941 | rs62064597 | C | G | MODIFIER | 0.083 | 0.0424 | 0.132 | 0.0008 | 0.2141 | 0.073 |
| ARHGAP27 | 45429583 | rs115993362 | C | G | MODIFIER | 0.083 | 0.147 | 0.0062 | - | 0.0002 | 0.0006 |
| ARHGAP27 | 45410208 | rs35327136 | C | A | MODIFIER | 0.015 | 0.1308 | 0.1778 | - | 0.2233 | 0.0682 |
| ARHGAP27 | 45396860 | rs7213200 | G | A | MODIFIER | 0.016 | - | - | - | - | - |
| ARHGAP27 | 45410198 | rs142163608 | C | T | MODIFIER | 0.083 | 0.0162 | - | - | - | - |
| ARHGAP27 | 45404234 | rs184103721 | C | G | MODIFIER | 0.045 | 0.0004 | 0.048 | 0.010 | 0.0002 | 0.0004 |
| ARHGAP27 | 45410058 | rs35389313 | T | C | MODIFIER | 0.013 | - | - | - | - | - |
| AQP3 | 33442274 | rs2231235 | G | A | MODIFIER | 0 | 0.012 | 0.109 | 0.063 | 0.069 | 0.085 |
| AQP3 | 33442988 | rs2231231 | A | C | MODIFIER | 0.937 | 0.775 | 0.7272 | 0.721 | 0.639 | 0.586 |
| AQP3 | 33447549 | rs12555686 | G | T | MODIFIER | 0 | 0.0221 | 0.255 | 0.175 | 0.095 | 0.095 |
| IFNAR2 | 33241950 | rs1051393 | T | G | MODERATE | 0.789 | 0.193 | 0.487 | 0.583 | 0.336 | 0.476 |
| IFNAR2 | 33241945 | rs2229207 | T | C | MODERATE | 0.343 | 0.079 | 0.166 | 0.178 | 0.084 | 0.130 |
| IFNAR2 | 33262573 | rs1131668 | G | A | MODIFIER | 0.795 | 0.376 | 0.453 | 0.583 | 0.365 | 0.476 |
| IFNAR2 | 33262760 | rs9984273 | C | T | MODIFIER | 1 | 0.594 | 0.848 | 0.918 | 0.687 | 0.767 |
| IFNAR2 | 33245424 | rs2252639 | A | T | MODIFIER | 0.285 | 1 | 1 | 1 | 1 | 1 |
| IFNAR2 | 33244908 | rs2834158 | T | C | MODIFIER | 0.333 | 0.807 | 0.512 | 0.416 | 0.663 | 0.524 |
| IFNAR2 | 33230489 | rs17860118 | G | T | MODIFIER | 0.187 | 0.076 | 0.118 | 0.139 | 0.0864 | 0.120 |
| IFNAR2 | 33262748 | rs3216172 rs397789038 | G | GT | MODIFIER | 0.181 | 0.366 | 0.467 | 0.594 | 0.33 | 0.484 |
| IFNAR2 | 33230629 | rs12482556 | T | C | MODIFIER | 0.714 | - | - | - | - | - |
| IFNAR2 | 33252224 | rs56197608 | C | T | MODIFIER | 0 | - | 0.003 | 0.021 | 0.0002 | 0.0001 |
| ELF5 | 34489994 | rs2231825 | C | A | MODIFIER | 0.046 | 0.116 | 0.186 | 0.027 | 0.429 | 0.301 |
| ELF5 | 34505605 | rs2231821 | A | G | MODIFIER | 0.043 | 0.059 | 0.155 | 0.026 | 0.316 | 0.265 |
| ELF5 | 34493816 | rs556840829 | A | G | MODIFIER | 0.05 | - | - | - | - | - |
| ELF5 | 34511455 | rs28395819 | A | C | MODIFIER | 0.083 | - | - | - | - | - |
| ELF5 | 34493394 | rs737254 | T | G | MODIFIER | 0.040 | 0.286 | 0.327 | 0.051 | 0.467 | 0.339 |
| LIMD1 | 45595761 | rs267237 | C | T | MODERATE | 0.937 | 0.584 | 0.721 | 0.938 | 0.562 | 0.675 |
| LIMD1 | 45674275 | rs2742409 | G | A | MODIFIER | 0.083 | - | - | - | - | - |
| LIMD1 | 45674196 | rs2578679 | G | A | MODIFIER | 0.5 | - | - | - | - | - |
| OAS1 | 112919637 | rs2660 | G | A | MODERATE | 1 | 0.934 | 0.840 | 0.791 | 0.679 | 0.707 |
| OAS1 | 112911065 | rs1131454 | G | A | MODERATE | 0.645 | 0.228 | 0.703 | 0.614 | 0.575 | 0.572 |
| OAS1 | 112931839 | rs7968145 | C | T | MODIFIER | 1 | 0.989 | 0.955 | 1 | 0.924 | 0.955 |
| OAS1 | 112919404 | rs1131476 | G | A | MODIFIER | 1 | 0.934 | 0.8318 | 0.784 | 0.659 | 0.698 |
| OAS1 | 112919388 | rs10774671 | G | A | MODIFIER | 0.833 | 0.424 | 0.799 | 0.783 | 0.654 | 0.701 |
| OAS1 | 112931954 | rs7967461 | G | C | MODIFIER | 1 | 0.917 | 0.739 | 0.76 | 0.575 | 0.692 |
| OAS1 | 112931910 | rs11352835 | TA | T | MODIFIER | 1 | 0.916 | 0.738 | 0.769 | 0.59 | 0.691 |
| OAS1 | - | rs1051042 | G | C | MODIFIER | 1 | 0.934 | 0.831 | 0.784 | 0.657 | 0.693 |
| UPK1A | 35668466 | rs2267586 | T | G | MODERATE | 0.179 | 0.0715 | 0.1157 | 0.2241 | 0.1007 | 0.1026 |
| UPK1A | 35676195 | rs747589460 | CTTT | C | MODIFIER | 0.033 | 0.0227 | 0.037 | 0.0417 | 0.0587 | 0.0811 |
| UPK1A | 35673206 | rs75289222 | G | T | MODIFIER | 0.035 | 0.0653 | 0.115 | 0.1029 | 0.1016 | 0.0845 |
| UPK1A | 35678012 | rs2285421 | T | C | MODIFIER | 0.3 | 0.8012 | 0.5853 | 0.6216 | 0.5365 | 0.7368 |
| Gene | Position | SNP ID | Ref a | Var b | Allele Frequence | Impact Predicted by SNPeff |
|---|---|---|---|---|---|---|
| AQP3 | 33442882 | * | G | A | 8.3% | LOW |
| IFNAR2 | 33262799 | * | C | A | 8.3% | MODERATE |
| LIMD1 | 45676789 | * | G | T | 6.6% | MODIFIER |
| Gene | SNP Id | INDG vs. AFR * | INDG vs. AMR * | INDG vs. EAS * | INDG vs. EUR * | INDG vs. SAS * |
|---|---|---|---|---|---|---|
| ARHGAP27 | rs201721078 | 5.129 × 10−15 | 1.542 × 10−5 | NA | 1.381 × 10−14 | NA |
| AQP3 | rs2228332 | 0.00018 | 3.693 × 10−5 | 5.348 × 10−5 | 4.293 × 10−9 | 9.082 × 10−10 |
| AQP3 | rs591810 | 3.963 × 10−5 | 0.00075 | 1.031 × 10−5 | 3.214 × 10−7 | 1.087 × 10−8 |
| AQP3 | rs2231231 | 0.00117 | 0.00010 | 5.442 × 10−5 | 1.620 × 10−7 | 2.264 × 10−9 |
| UPK1A | rs2285421 | 3.016 × 10−16 | 3.180 × 10−5 | 1.341 × 10−6 | 0.00030 | 1.562 × 10−11 |
| OAS1 | rs2660 | 0.02583 | 7.618 × 10−5 | 1.374 × 10−6 | 1.525 × 10−10 | 2.482 × 10−9 |
| OAS1 | rs7967461 | 0.01055 | 3.650 × 10−8 | 1.260 × 10−7 | 1.118 × 10−14 | 6.043 × 10−10 |
| OAS1 | rs11352835 | 0.01087 | 3.432 × 10−8 | 2.366 × 10−7 | 3.623 × 10−14 | 6.043 × 10−10 |
| OAS1 | rs1051042 | 0.02583 | 4.458 × 10−5 | 7.665 × 10−7 | 3.220 × 10−11 | 5.950 × 10−10 |
| ARHGAP27 | rs2959953 | 6.052 × 10−7 | 4.116 × 10−5 | 0.03356 | 1.238 × 10−7 | 6.31360 × 10−6 |
| IFNAR2 | rs1051393 | 1.824 × 10−21 | 1.053 × 10−5 | 0.00258 | 1.215 × 10−11 | 3.942 × 10−6 |
| IFNAR2 | rs2229207 | 2.456 × 10−8 | 0.00186 | 0.003 | 1.035 × 10−7 | 6.205 × 10−5 |
| IFNAR2 | rs1131668 | 1.229 × 10−10 | 2.771 × 10−7 | 0.00098 | 7.147 × 10−11 | 9.955 × 10−7 |
| IFNAR2 | rs9984273 | 3.001 × 10−14 | 0.00013 | 0.00938 | 3.134 × 10−10 | 2.268 × 10−7 |
| IFNAR2 | rs2834158 | 5.32 × 10−15 | 0.00664 | 0.22382 | 3.818 × 10−7 | 0.00342 |
| IFNAR2 | rs149186597 rs79402470 | 8.004 × 10−5 | 0.00602 | 1.575 × 10−5 | 0.04188 | 0.04602 |
| IFNAR2 | rs3216172 rs397789038 | 0.00380 | 2.699 × 10−5 | 4.368 × 10−10 | 0.02185 | 3.997 × 10−6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, R.d.C.C.; Martins, C.L.e.L.P.; Pastana, L.F.; Rodrigues, J.C.G.; Aguiar, K.E.C.; Cohen-Paes, A.d.N.; Gellen, L.P.A.; Moraes, F.C.A.d.; Calderaro, M.C.L.; de Assunção, L.A.; et al. Molecular Profile of Variants Potentially Associated with Severe Forms of COVID-19 in Amazonian Indigenous Populations. Viruses 2024, 16, 359. https://doi.org/10.3390/v16030359
Coelho RdCC, Martins CLeLP, Pastana LF, Rodrigues JCG, Aguiar KEC, Cohen-Paes AdN, Gellen LPA, Moraes FCAd, Calderaro MCL, de Assunção LA, et al. Molecular Profile of Variants Potentially Associated with Severe Forms of COVID-19 in Amazonian Indigenous Populations. Viruses. 2024; 16(3):359. https://doi.org/10.3390/v16030359
Chicago/Turabian StyleCoelho, Rita de Cássia Calderaro, Carlliane Lima e Lins Pinto Martins, Lucas Favacho Pastana, Juliana Carla Gomes Rodrigues, Kaio Evandro Cardoso Aguiar, Amanda de Nazaré Cohen-Paes, Laura Patrícia Albarello Gellen, Francisco Cezar Aquino de Moraes, Maria Clara Leite Calderaro, Letícia Almeida de Assunção, and et al. 2024. "Molecular Profile of Variants Potentially Associated with Severe Forms of COVID-19 in Amazonian Indigenous Populations" Viruses 16, no. 3: 359. https://doi.org/10.3390/v16030359
APA StyleCoelho, R. d. C. C., Martins, C. L. e. L. P., Pastana, L. F., Rodrigues, J. C. G., Aguiar, K. E. C., Cohen-Paes, A. d. N., Gellen, L. P. A., Moraes, F. C. A. d., Calderaro, M. C. L., de Assunção, L. A., Monte, N., Pereira, E. E. B., Ribeiro-dos-Santos, A. M., Ribeiro-do-Santos, Â., Rodriguez Burbano, R. M., de Souza, S. J., Guerreiro, J. F., Assumpção, P. P. d., Santos, S. E. B. d., ... Santos, N. P. C. d. (2024). Molecular Profile of Variants Potentially Associated with Severe Forms of COVID-19 in Amazonian Indigenous Populations. Viruses, 16(3), 359. https://doi.org/10.3390/v16030359