
The Influenza A Virus Replication Cycle: A Comprehensive Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
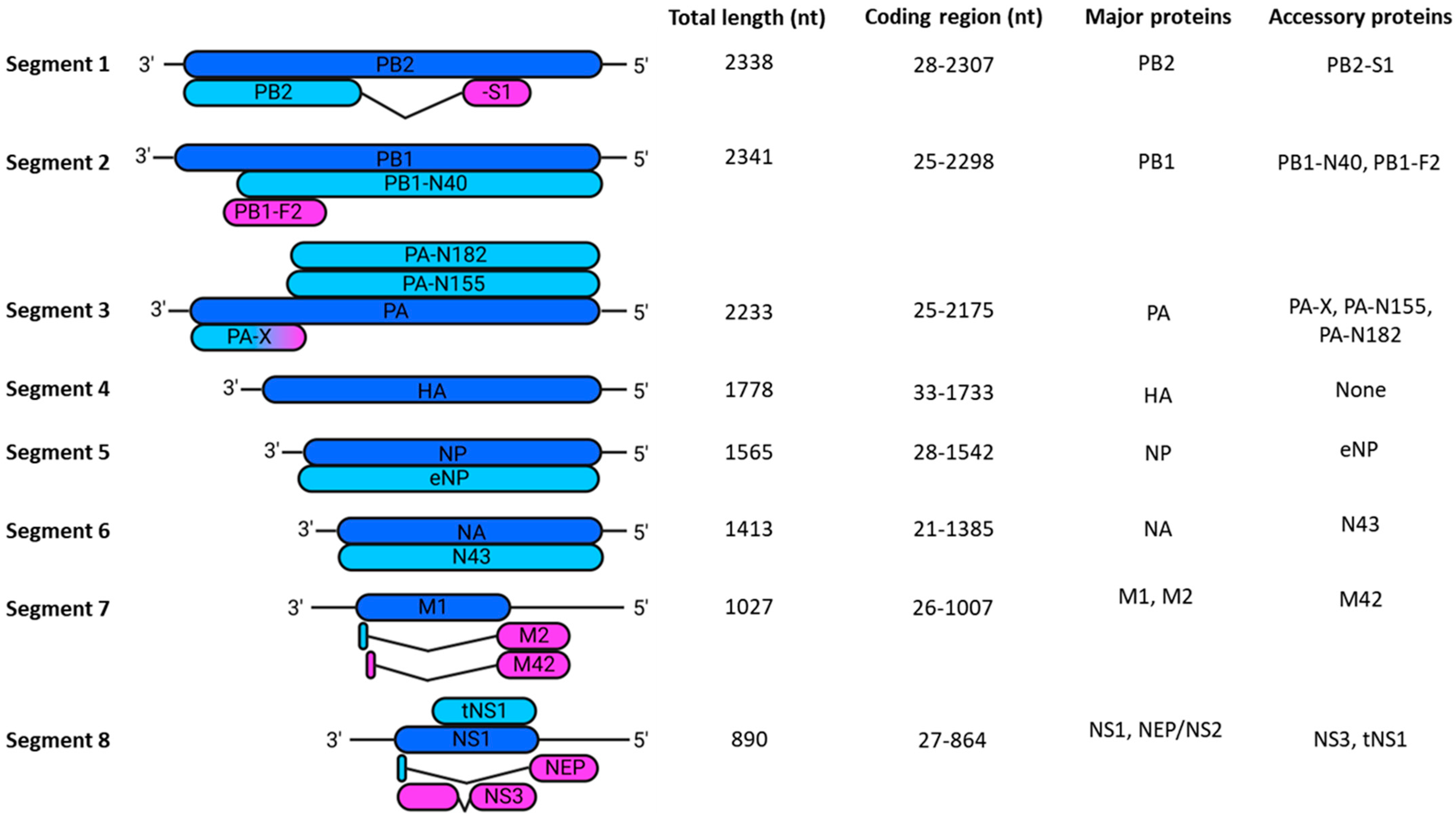
1.1. Overview of Essential IAV Proteins
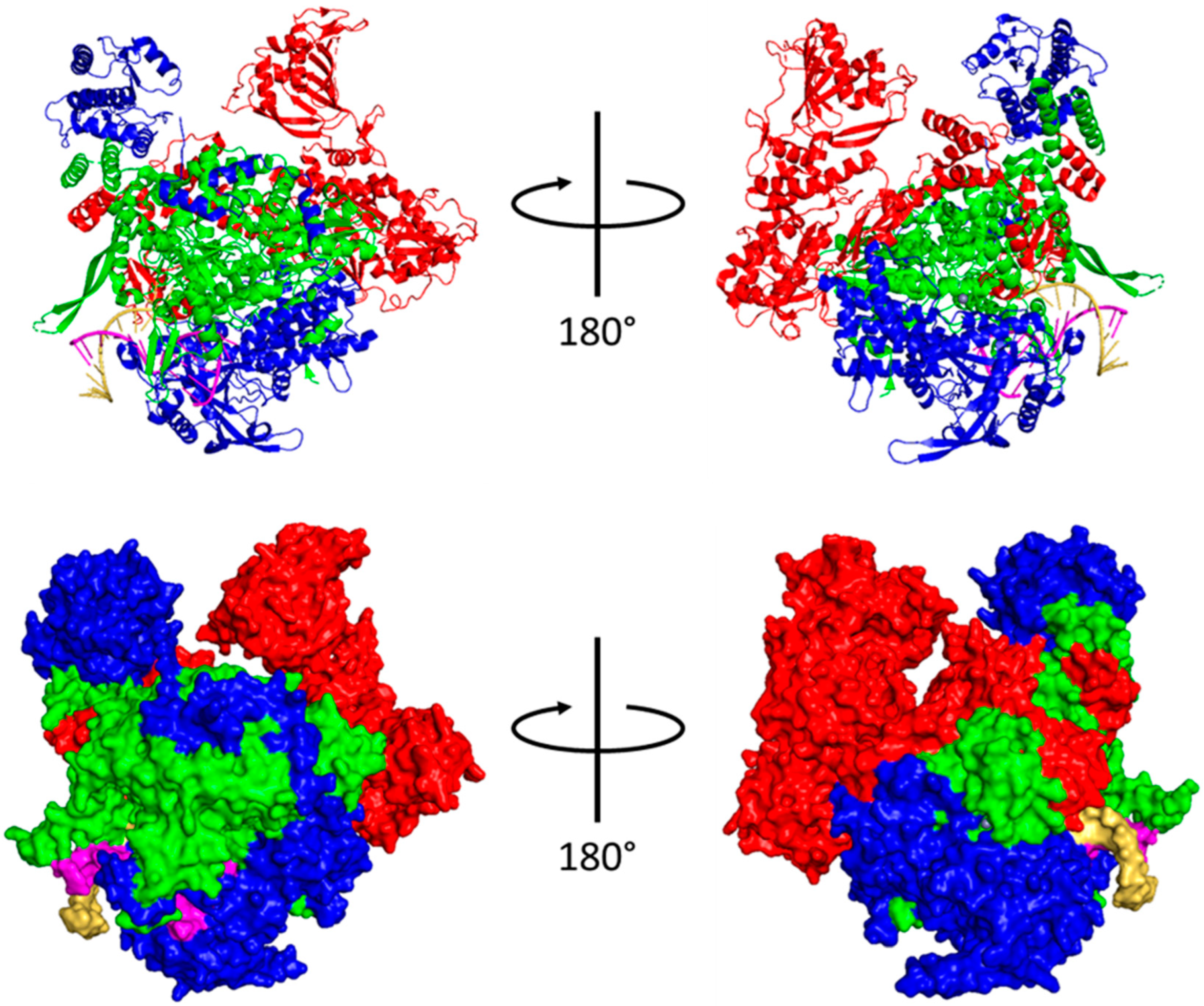
1.1.1. Polymerase
1.1.2. Polymerase Basic 2 (PB2)
1.1.3. Polymerase Basic 1 (PB1)
1.1.4. Polymerase Acidic (PA)
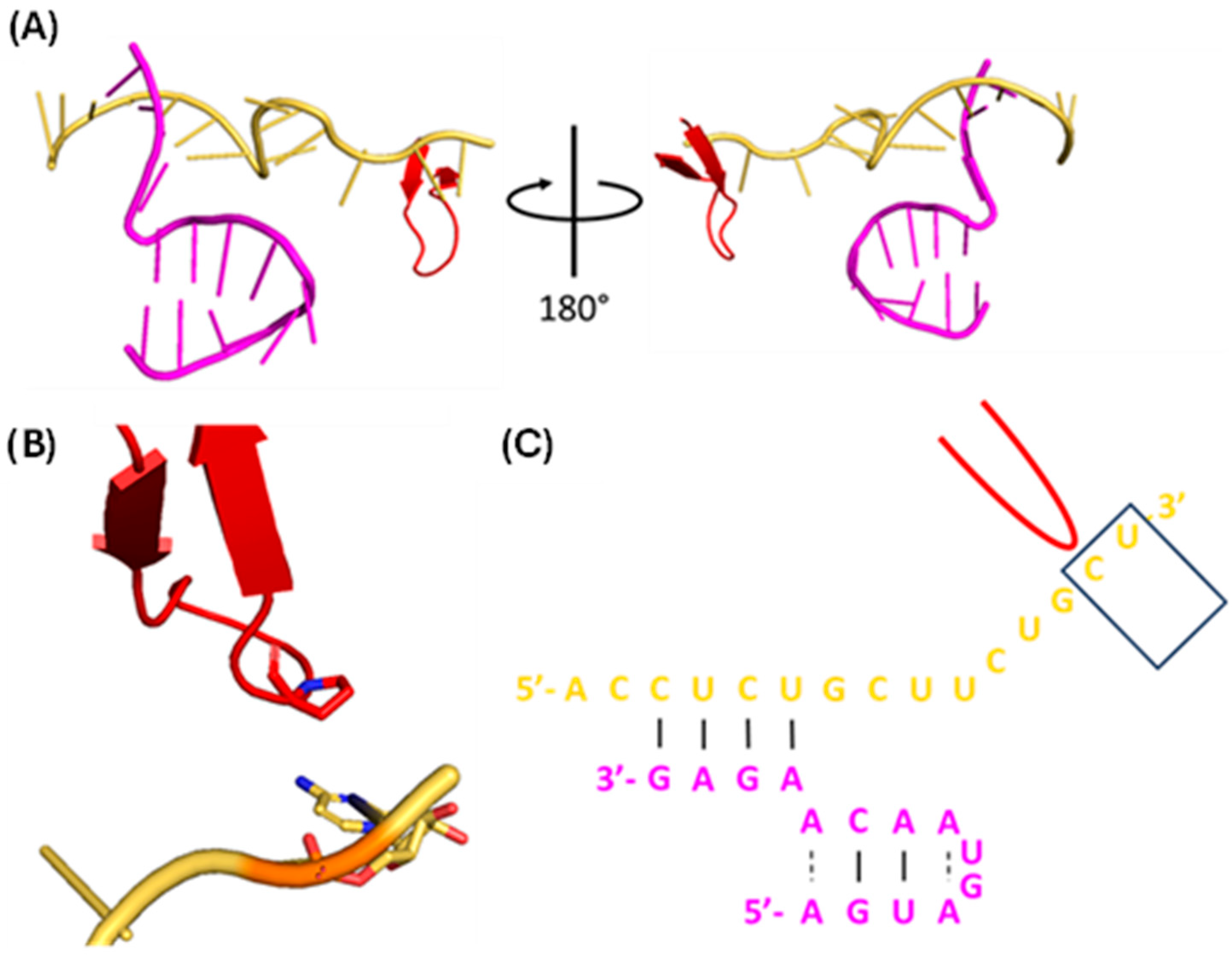
1.1.5. vRNA Promoter
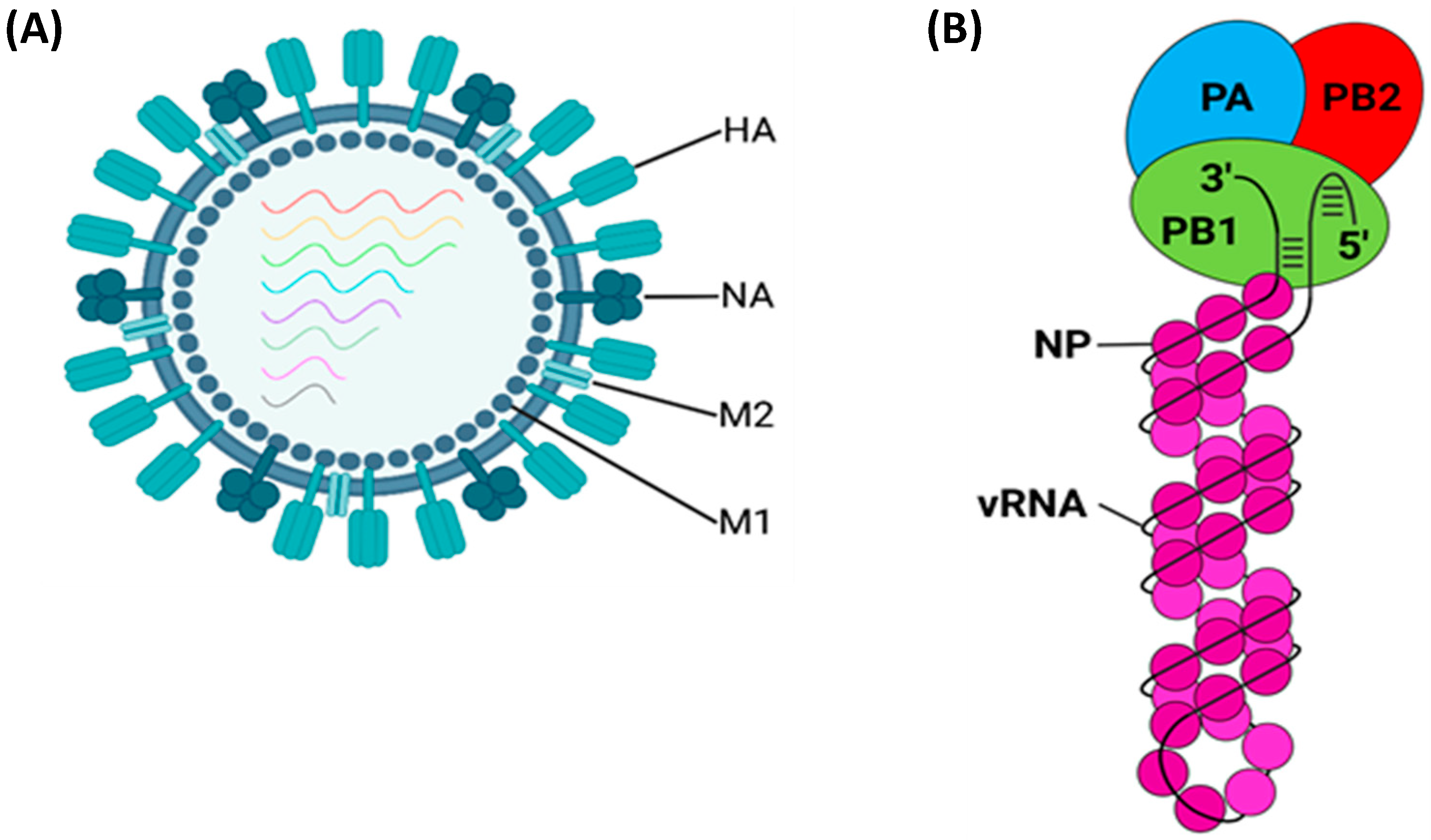
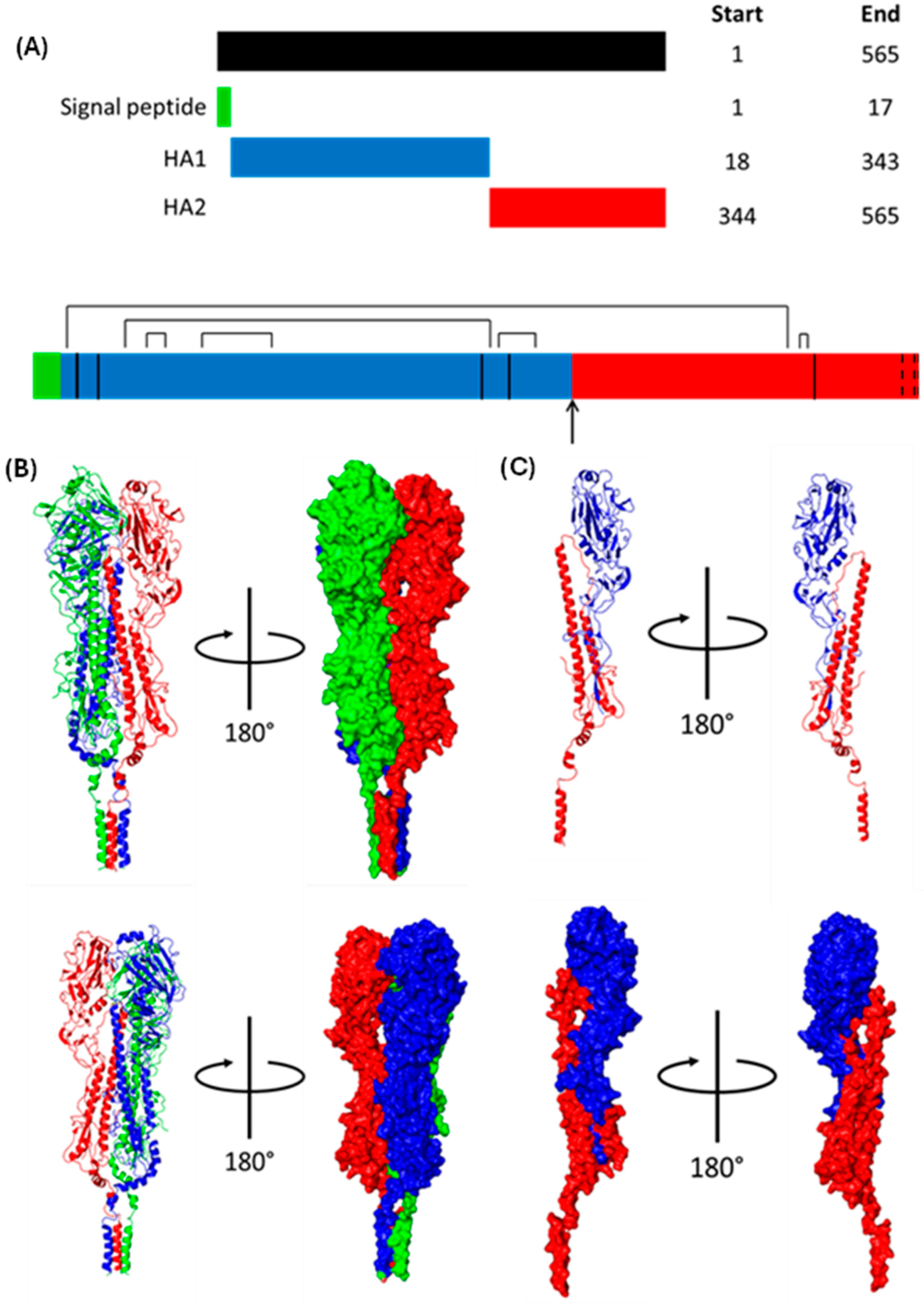
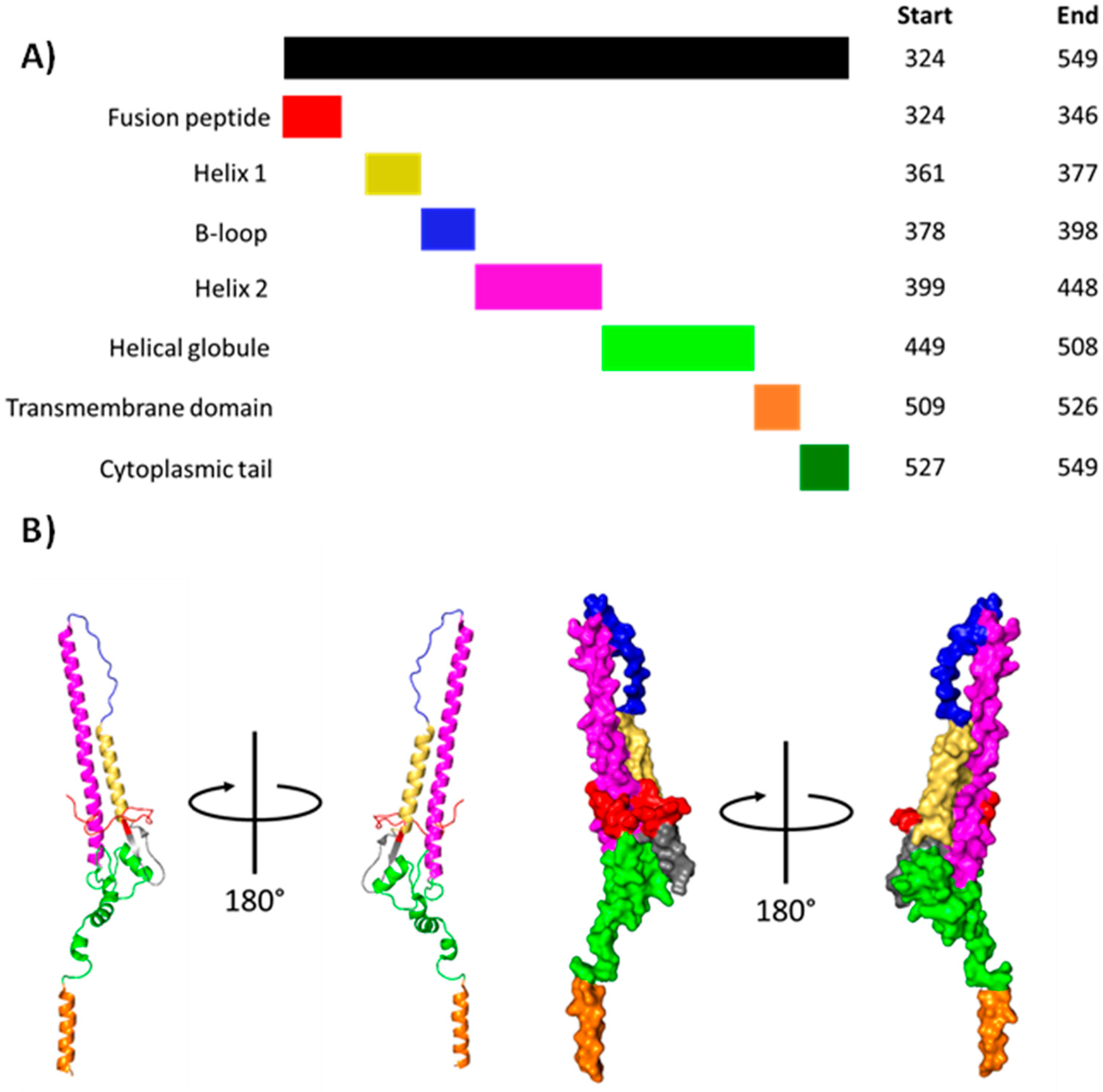
1.1.6. Haemagglutinin (HA)
1.1.7. Nucleoprotein (NP)
1.1.8. Neuraminidase (NA)
1.1.9. Matrix Protein 1 (M1)
1.1.10. Matrix Protein 2 (M2)
1.1.11. Nuclear Export Protein (NEP)
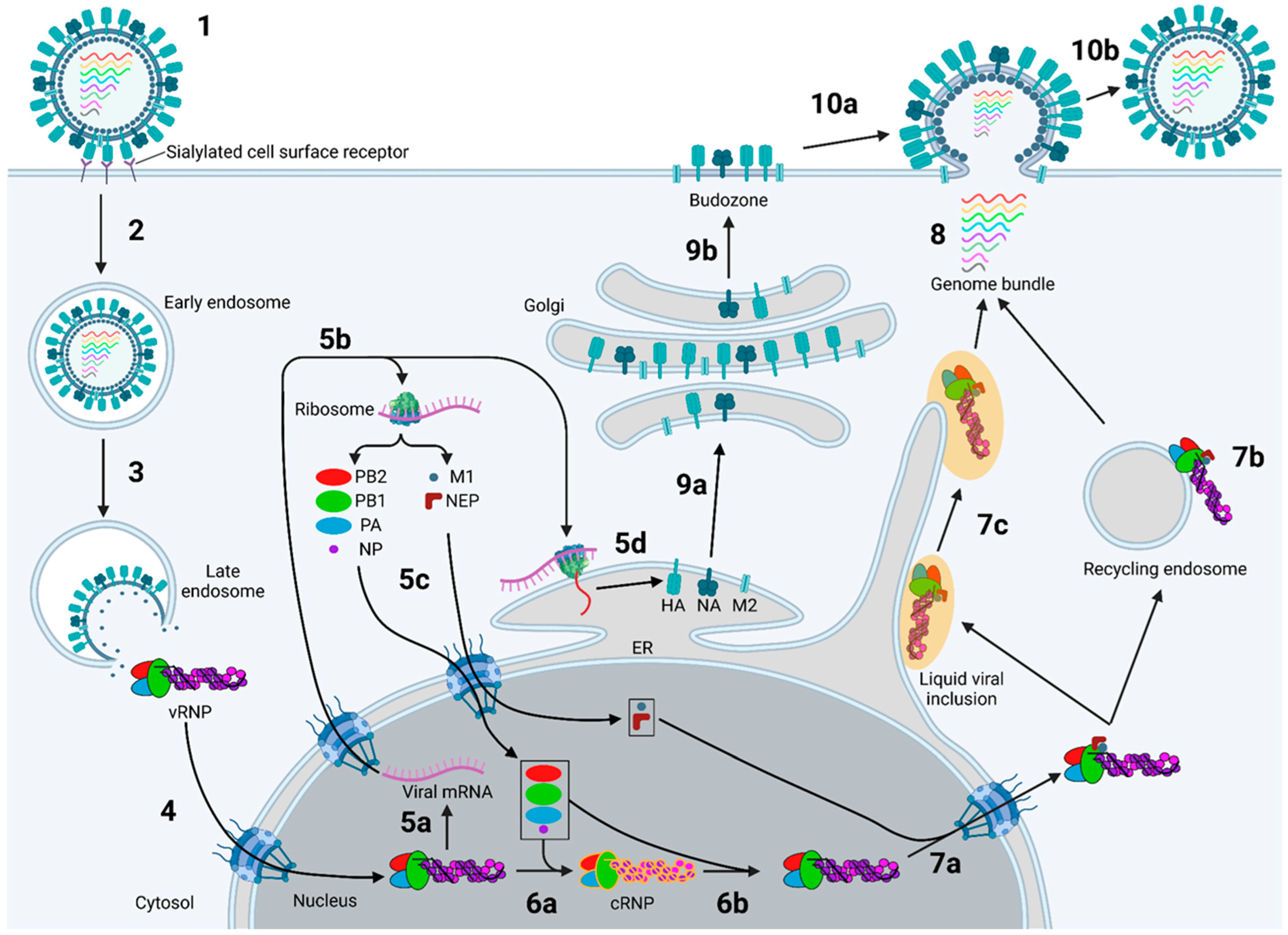
2. Binding and Endocytosis
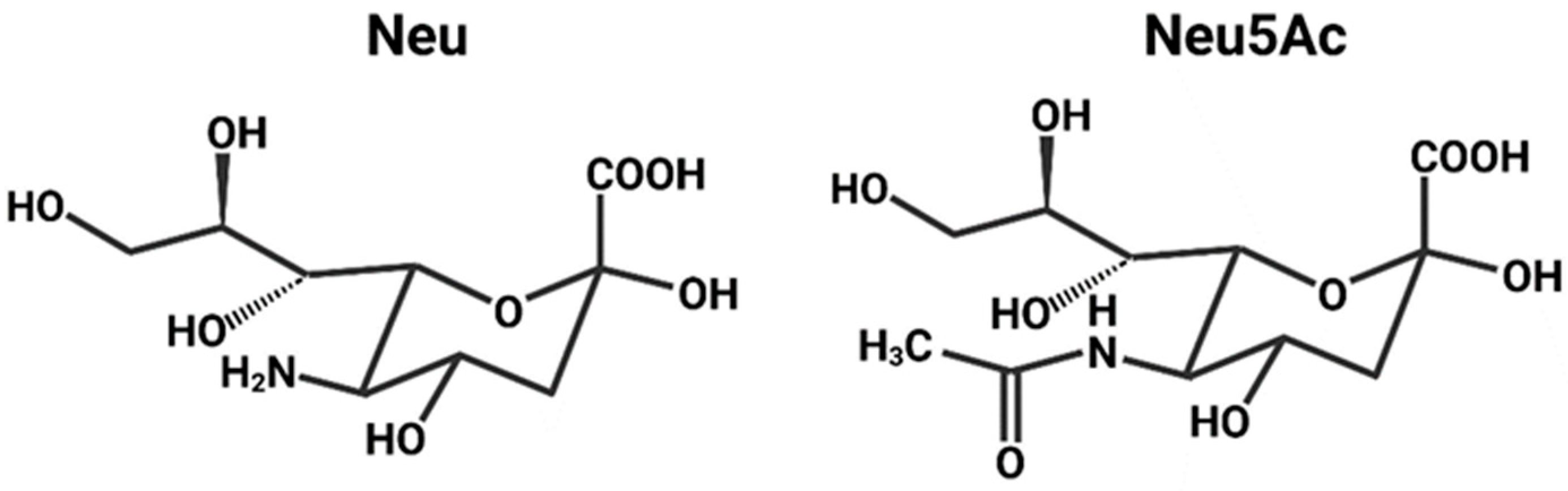
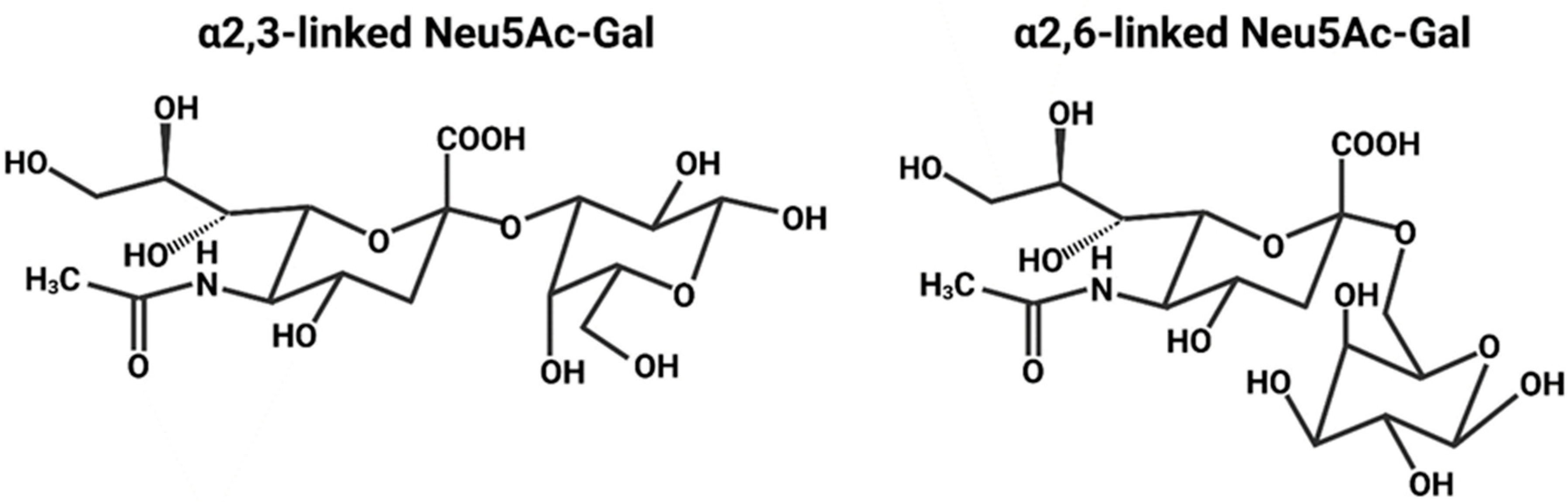
2.1. Sialic Acid
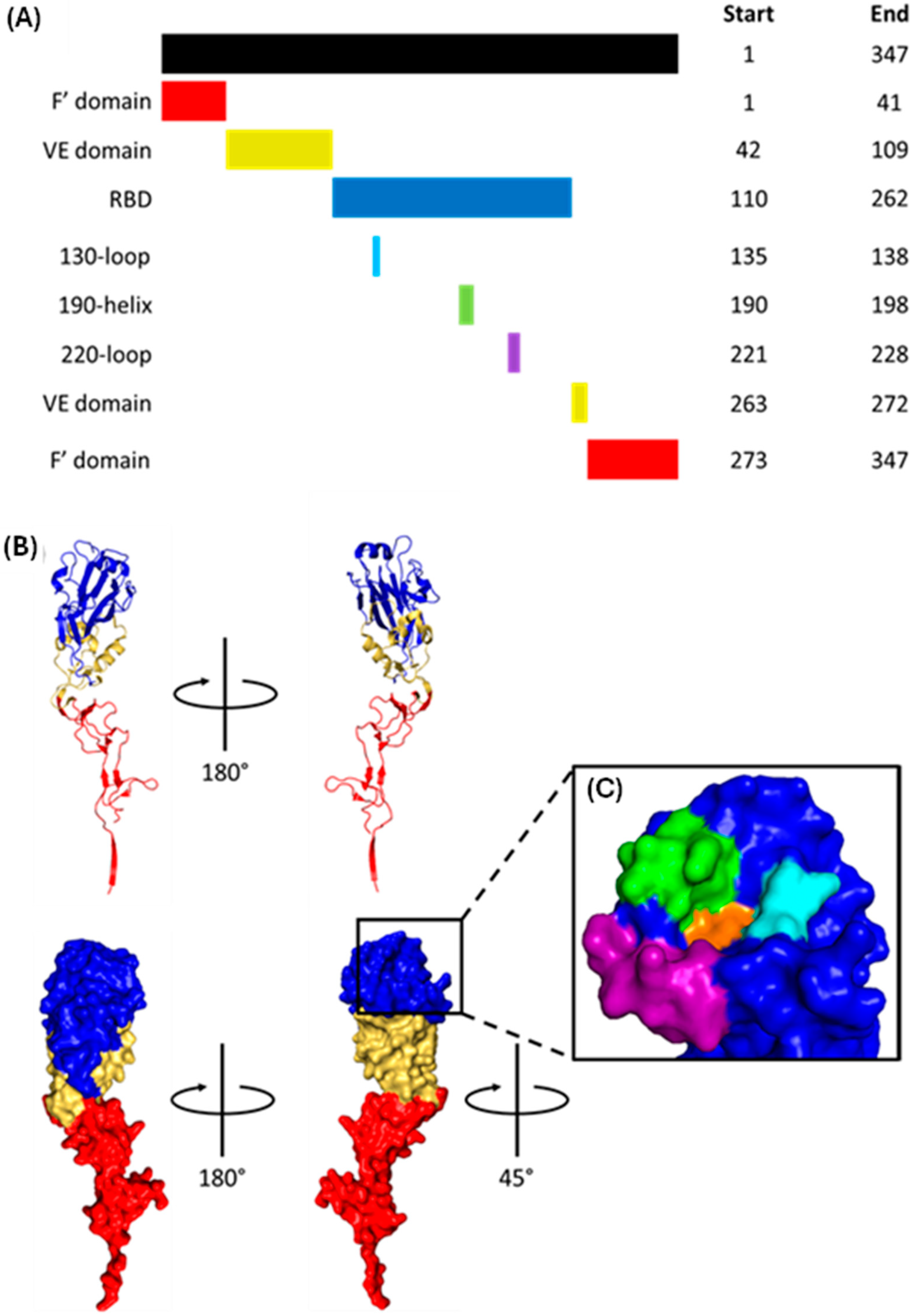
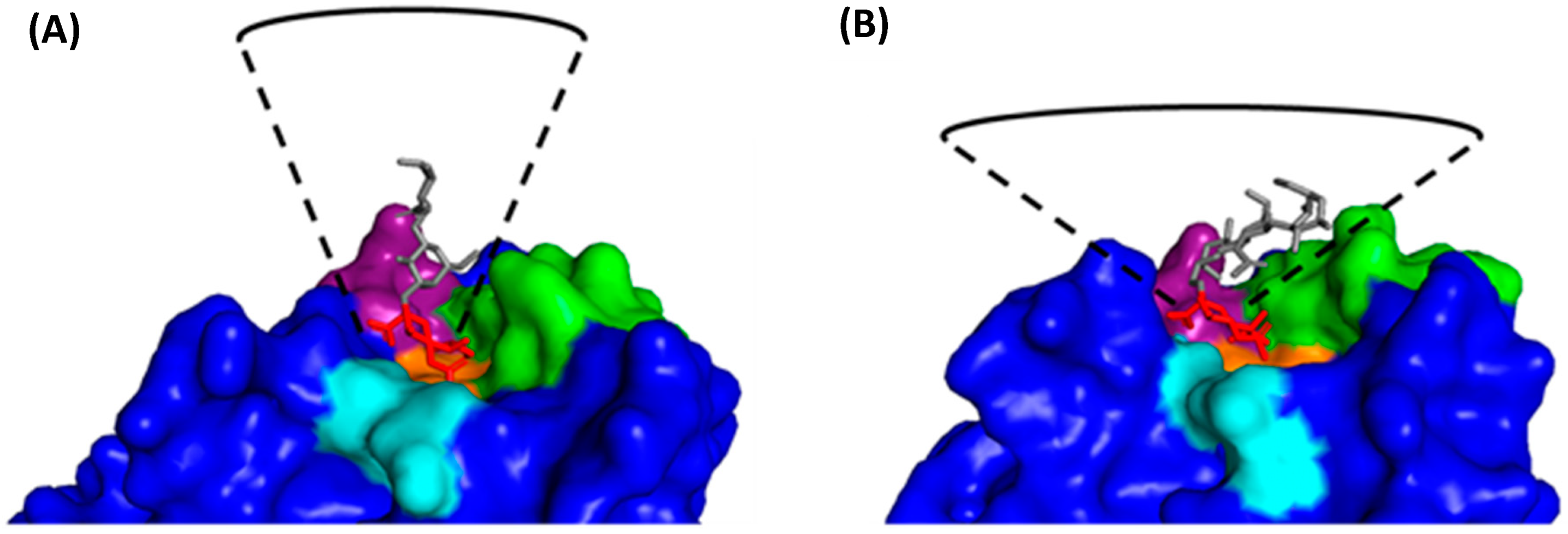
2.2. HA1 Structure and Receptor Binding
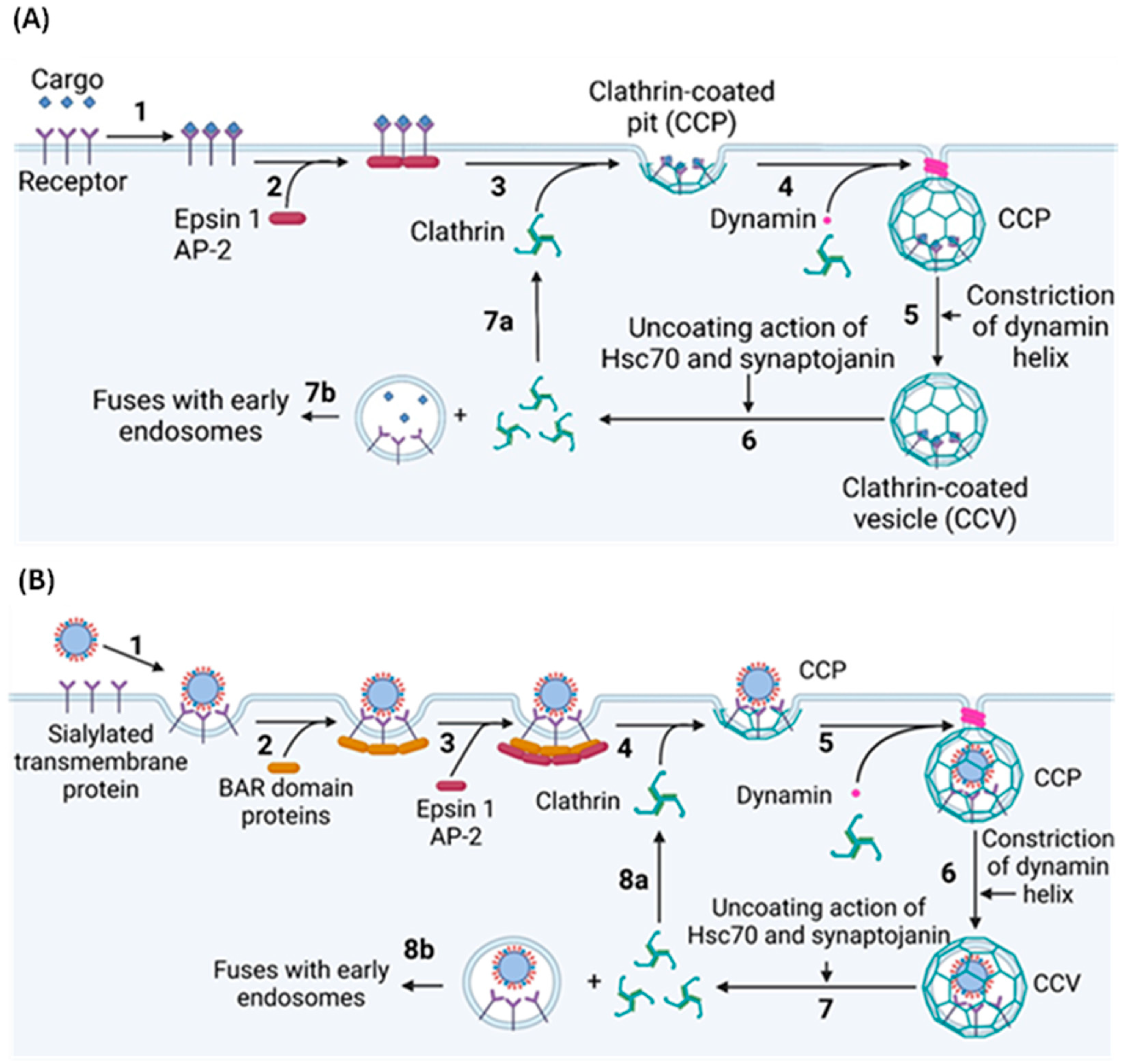
2.3. Clathrin-Mediated Endocytosis

2.4. Clathrin-Independent Endocytosis
3. Fusion of the Viral and Endosomal Membranes
3.1. Virion Acidification
3.2. Virial–Endosomal Membrane Fusion
3.2.1. HA1 Dissociation
3.2.2. Formation of the HA2 Extended Intermediate
3.2.3. Refolding of the HA2 Extended Intermediate
3.3. Matrix Layer Dissociation and vRNP Release
4. Nuclear Import of vRNPs
5. Viral mRNA Transcription and Translation of Viral Cytosolic Proteins
5.1. Cap Snatching by PB2 and PA
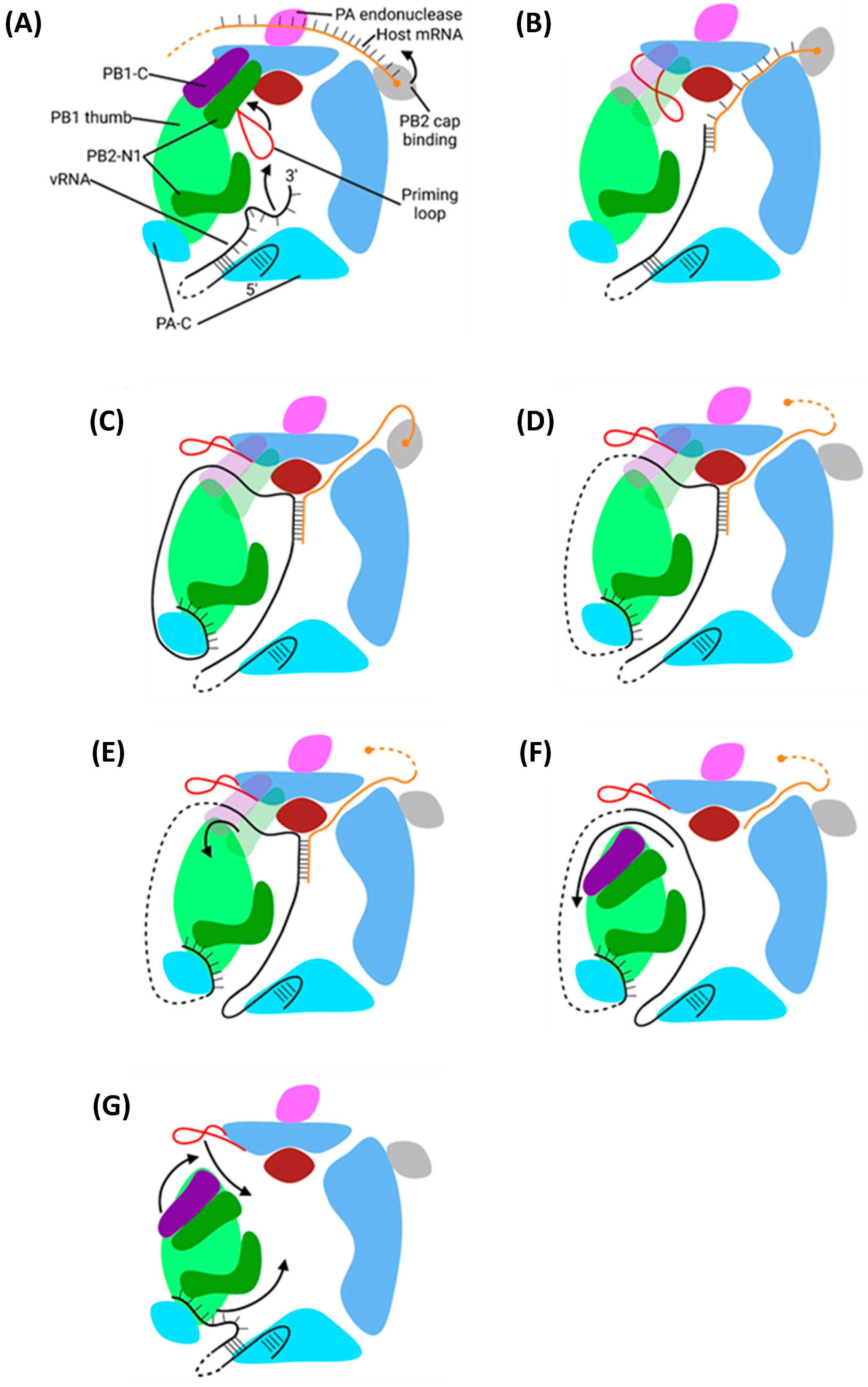
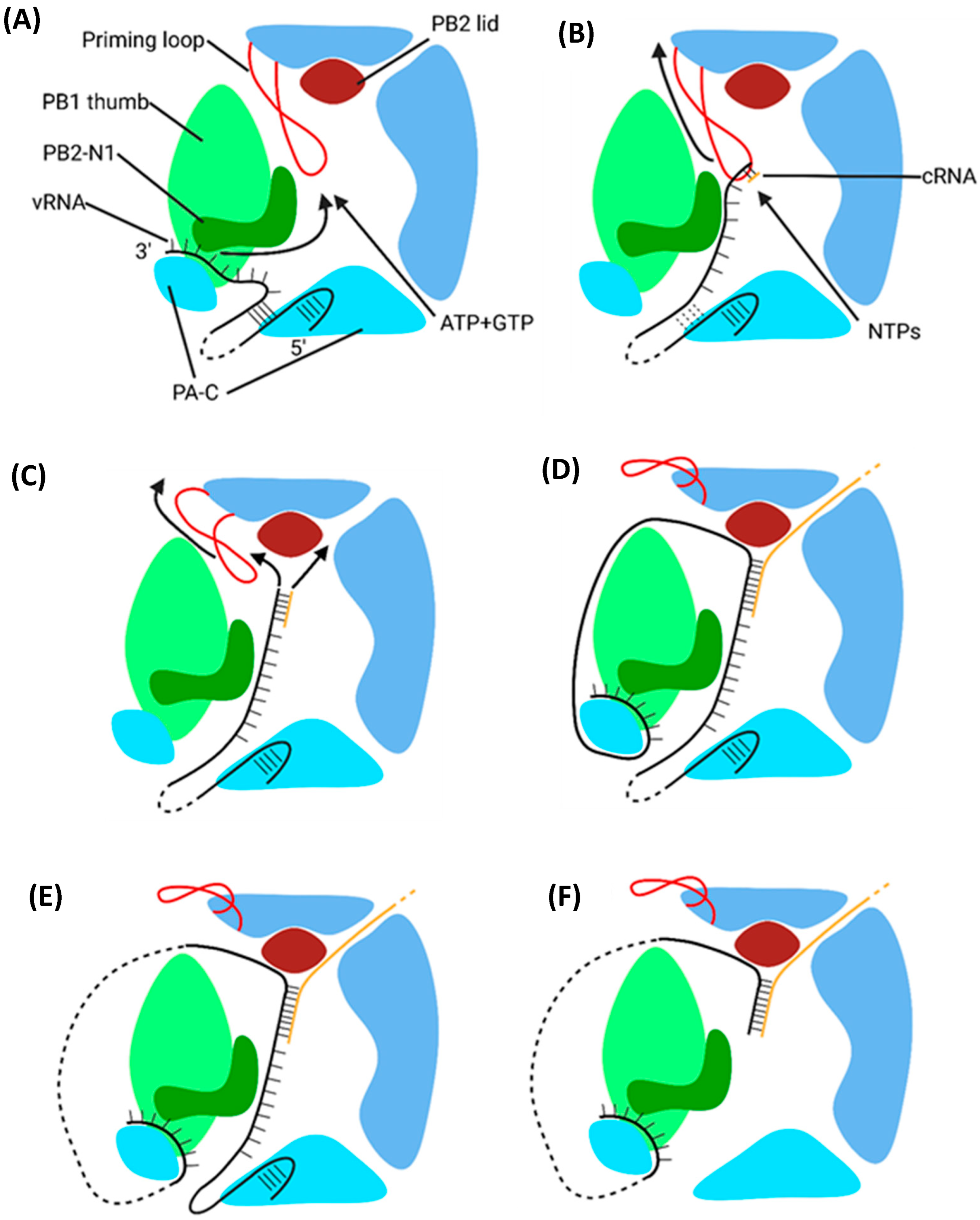
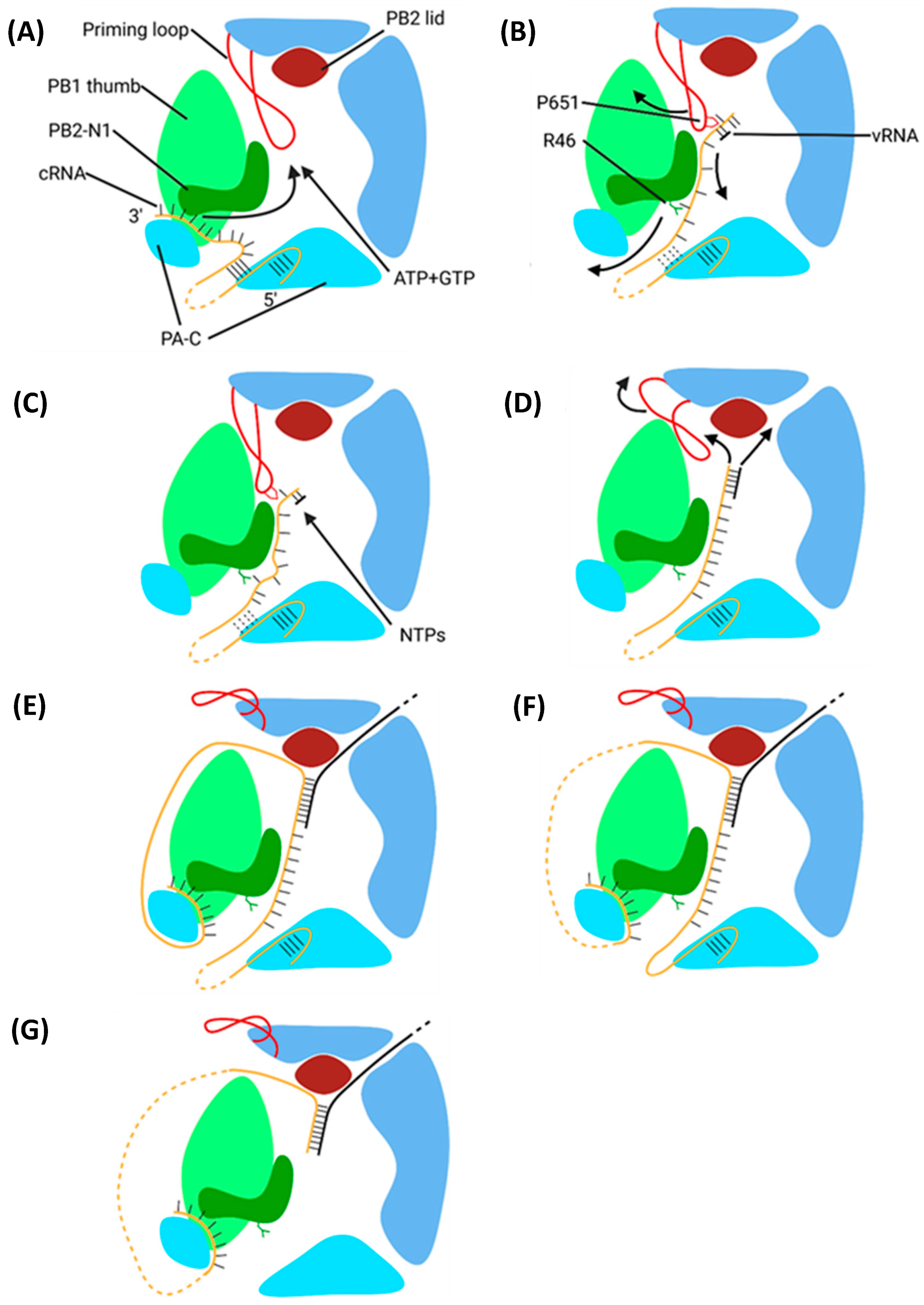
5.2. Mechanism of Transcription by PB1
5.2.1. Initiation
5.2.2. Elongation
5.2.3. Polyadenylation and Termination
5.2.4. Post-Transcriptional Processing and Translation of Soluble Viral Proteins
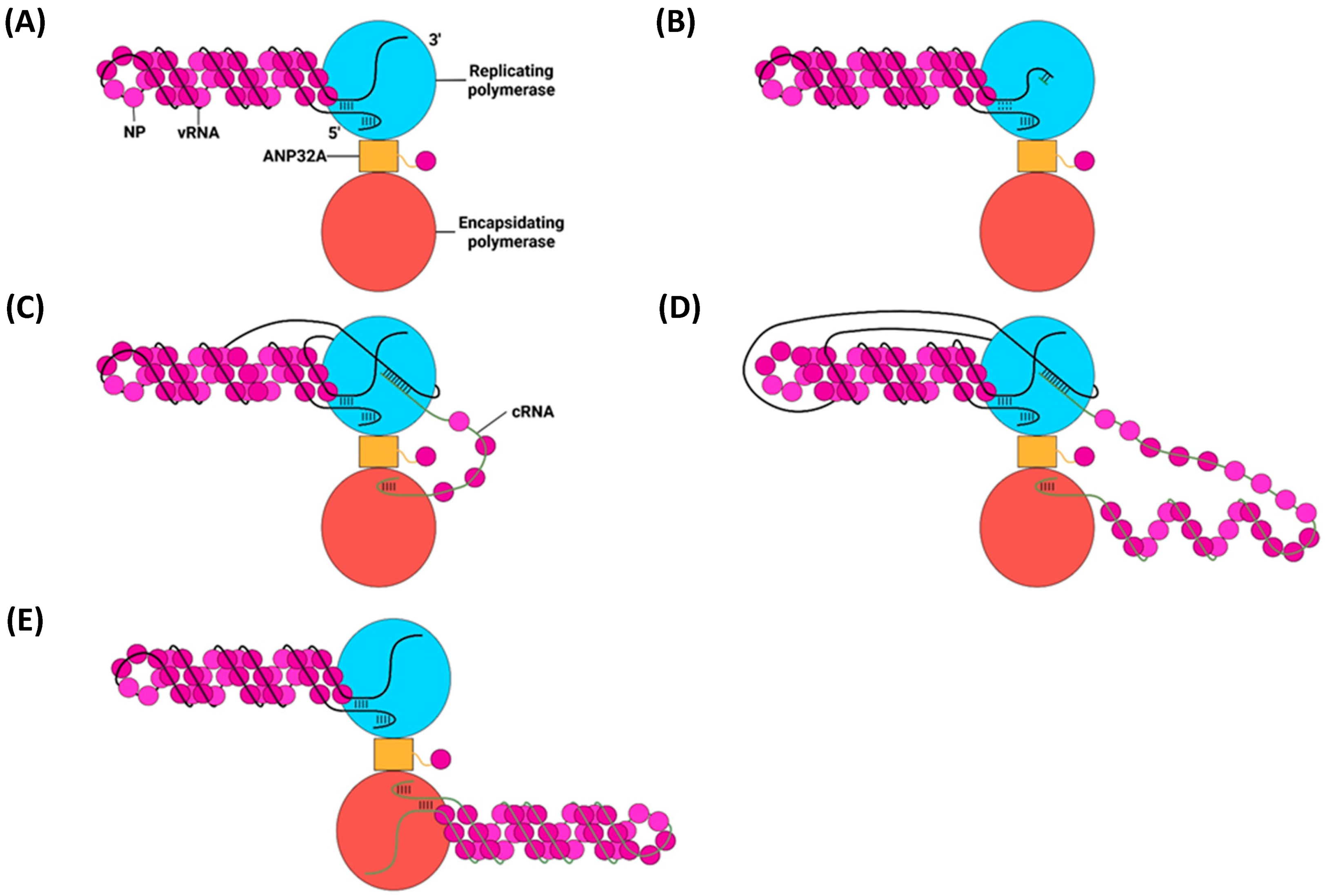
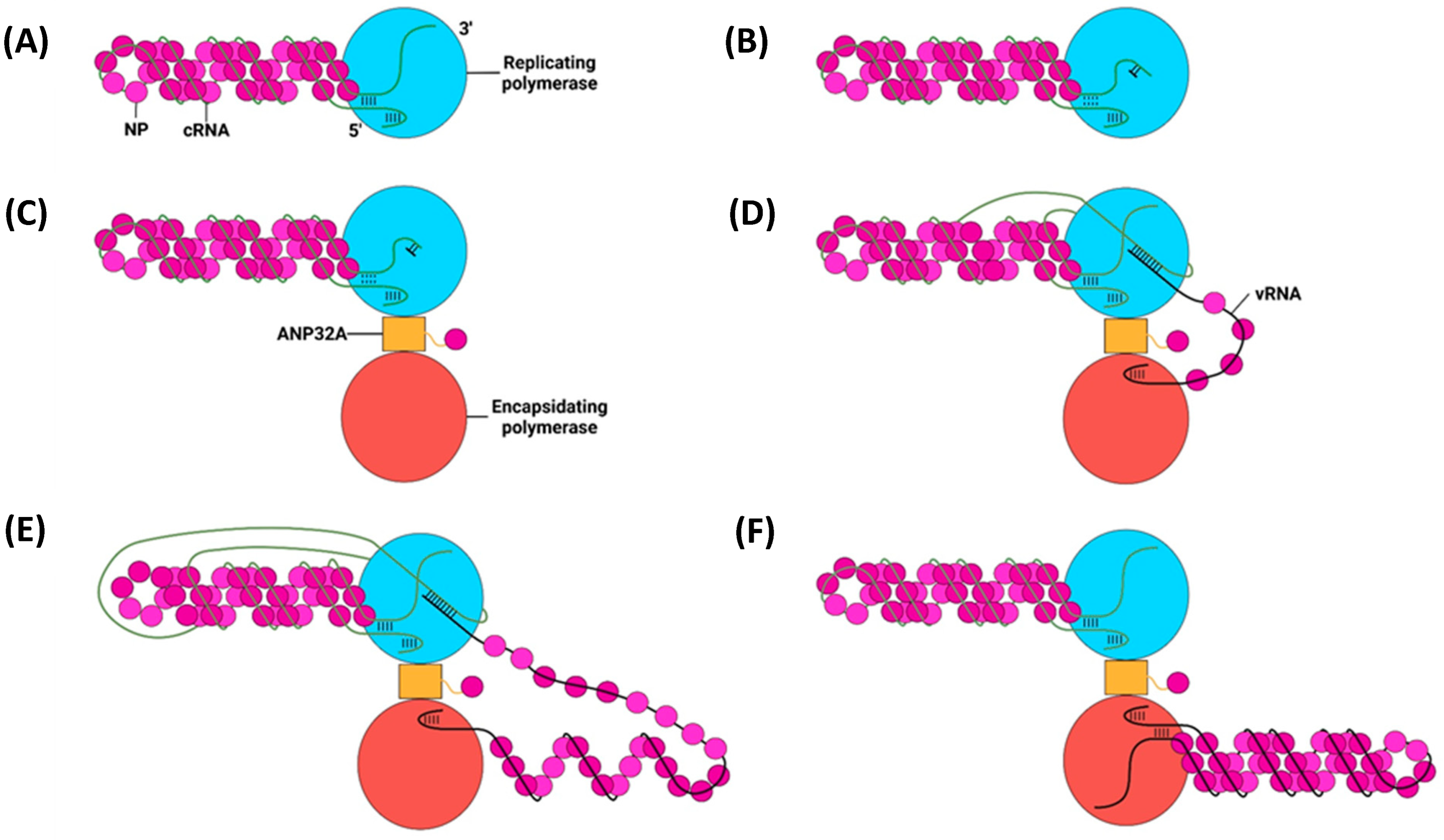
6. Replication of vRNPs
6.1. Primary Replication
cRNP Assembly
6.2. Secondary Replication
6.3. The E627K Mutation
7. Nuclear Export and Intracellular Trafficking of Progeny vRNPs
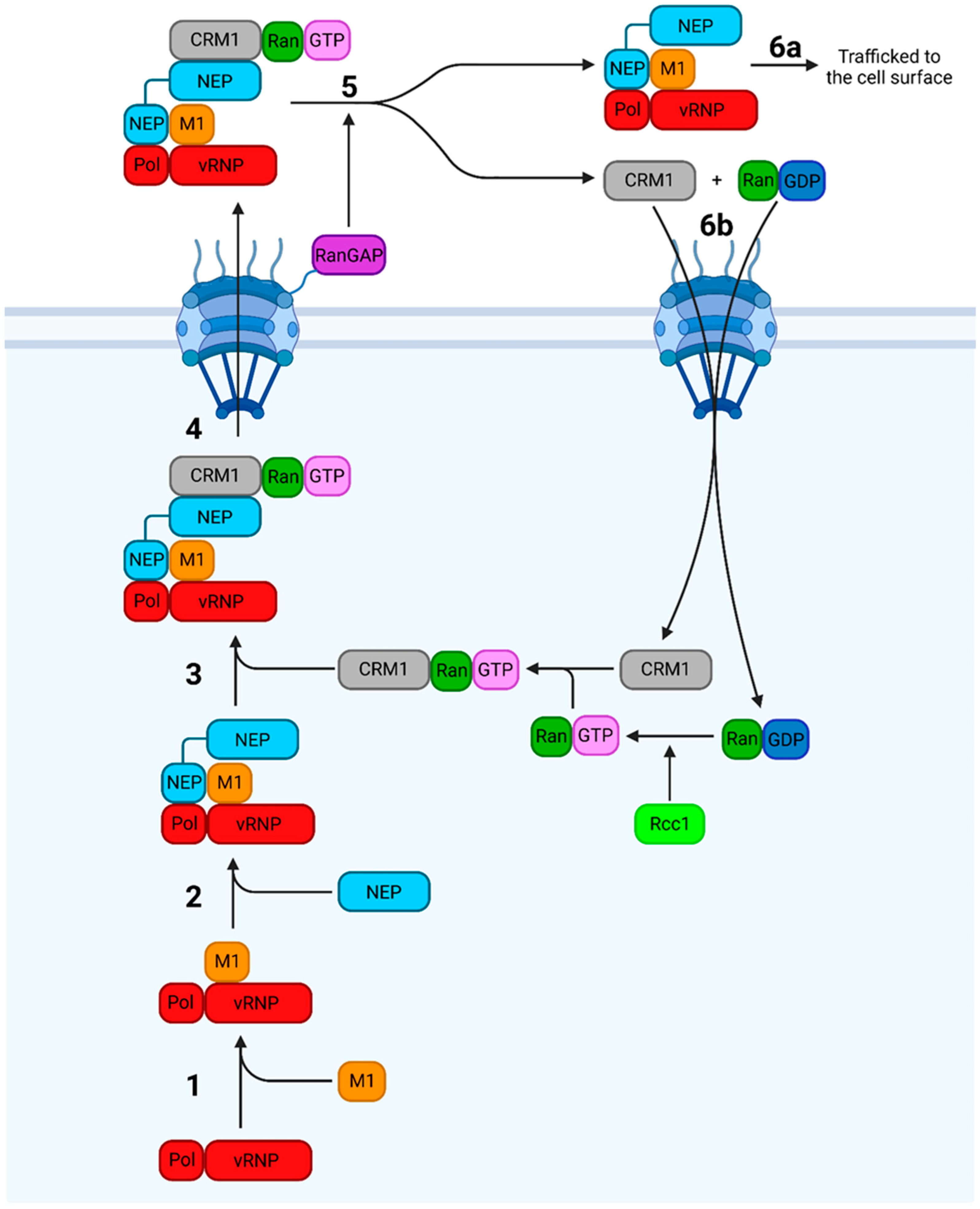
7.1. Nuclear Export
7.2. Intracellular Trafficking
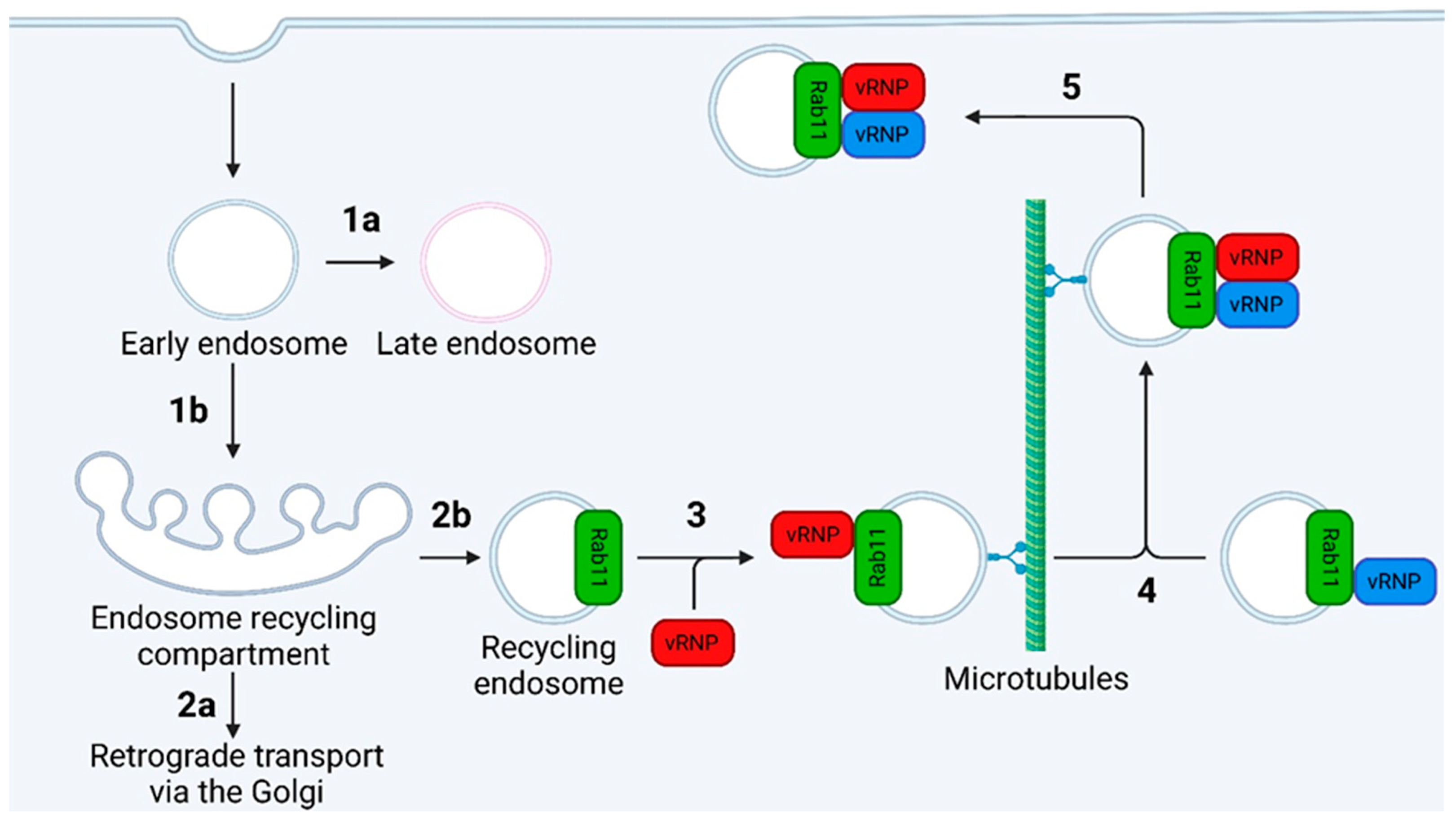
7.2.1. The Recycling Endosome Model
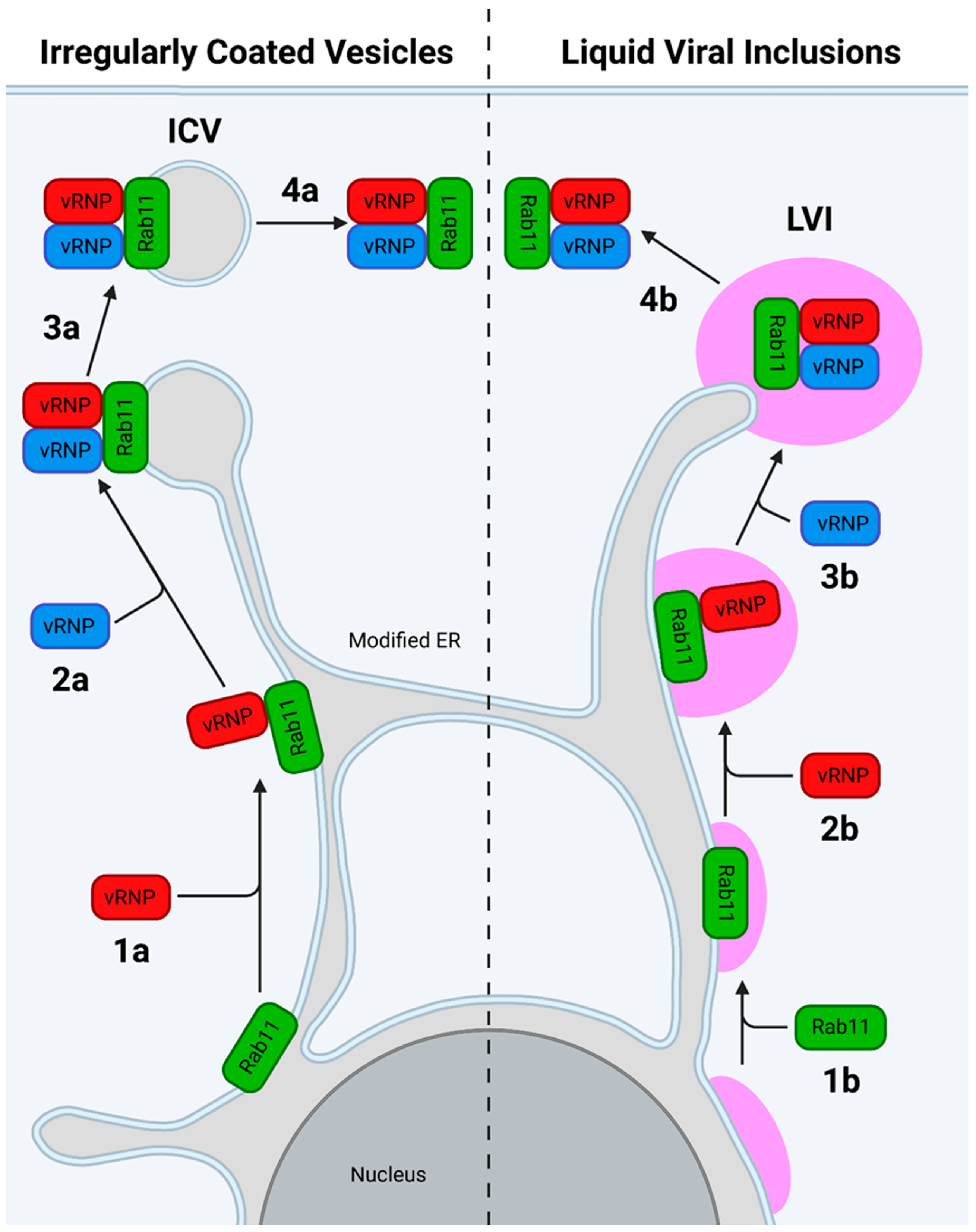
7.2.2. The Modified ER Model
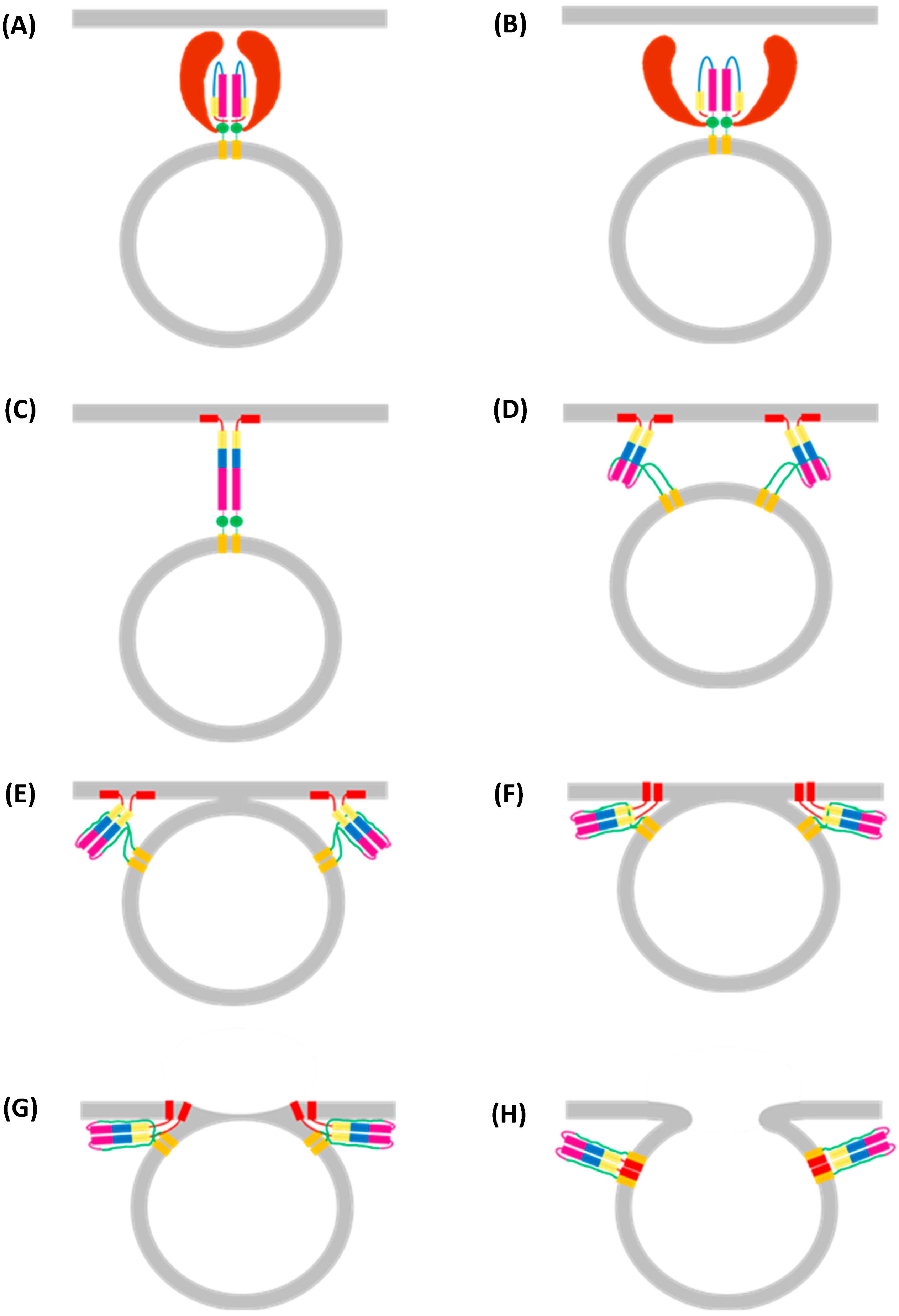
8. Assembly of the IAV Genome
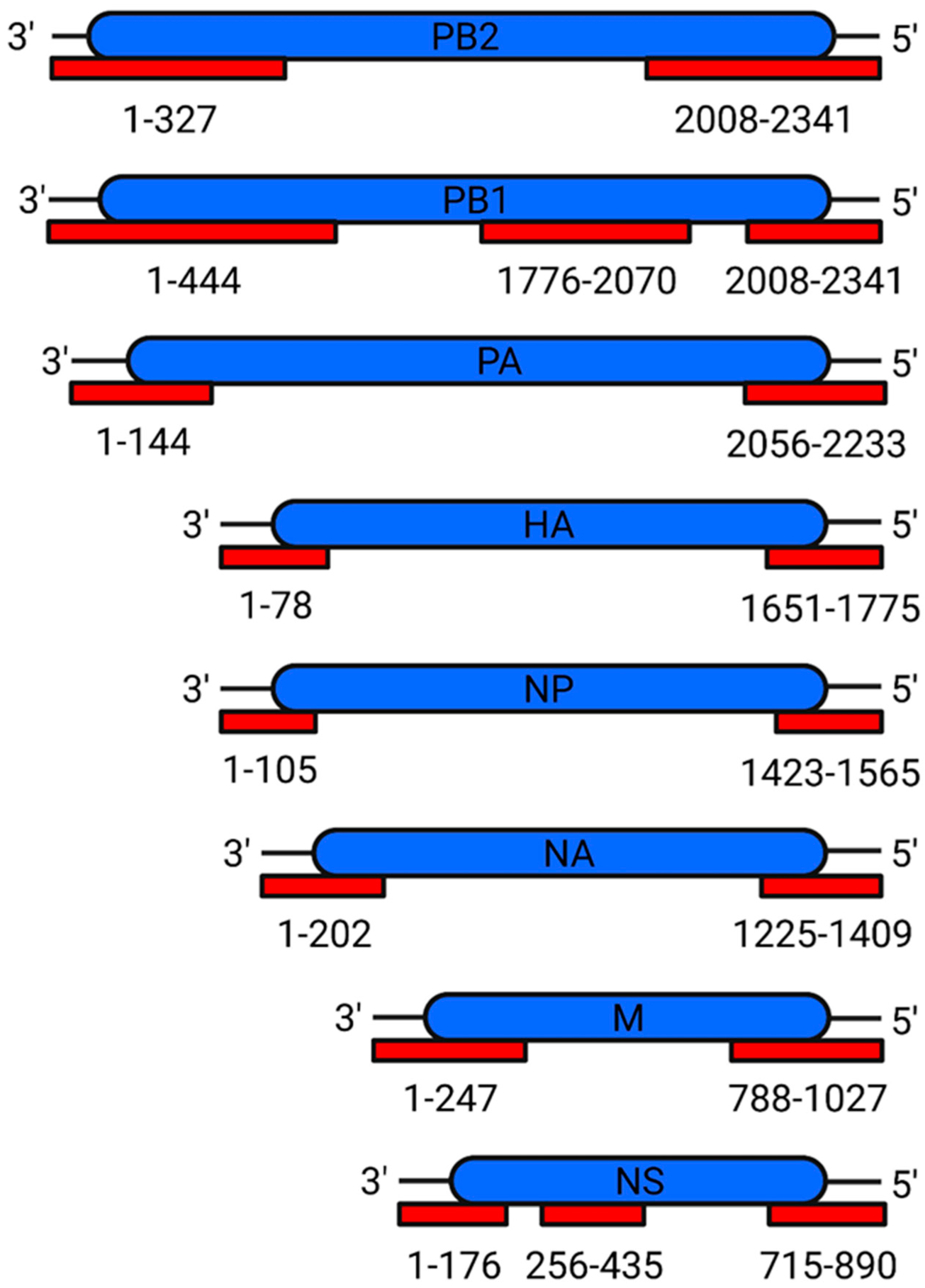
Mechanism of Packaging Signals
9. Translation and Trafficking of IAV Membrane Proteins
9.1. Translation and Folding
9.1.1. HA
9.1.2. NA
9.1.3. M2
9.2. Trafficking to the Plasma Membrane
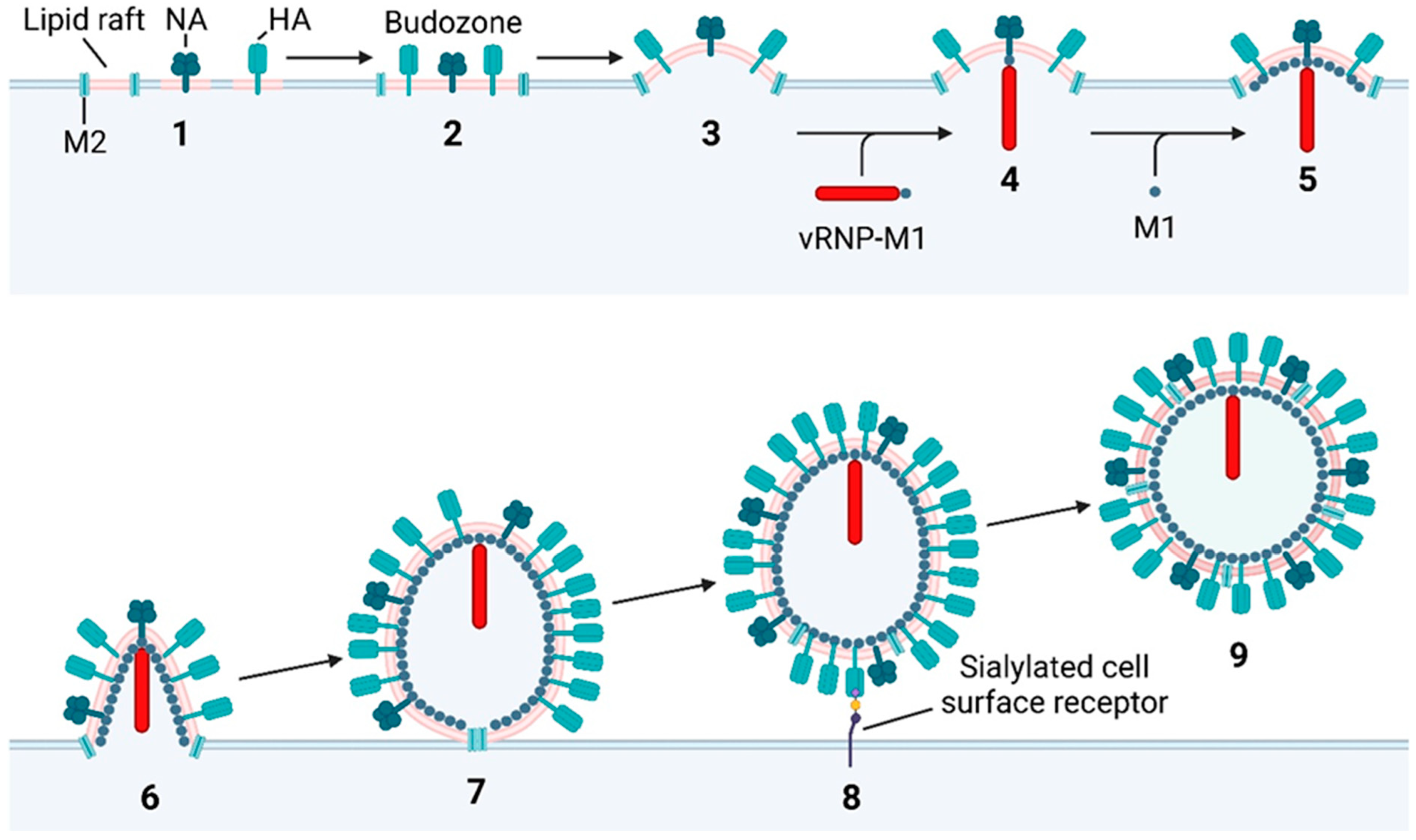
10. Budding of Progeny IAV Virions
10.1. Association of IAV Transmembrane Proteins with Lipid Rafts
10.2. Induction of Budding
10.3. Packaging of vRNPs
10.4. M1 Oligomerisation
10.5. M2-Mediated Scission
10.6. NA-Mediated Release
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kilbourne, E.D. Taxonomy and Comparative Virology of the Influenza Viruses; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Shinya, K.; Ebina, M.; Yamada, S.; Ono, M.; Kasai, N.; Kawaoka, Y. Avian flu: Influenza virus receptors in the human airway. Nature 2006, 440, 435–436. [Google Scholar] [CrossRef] [PubMed]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Couceiro, J.N.S.S.; Kelm, S.; Baum, L.G.; Krauss, S.; Castrucci, M.R.; Donatelli, I.; Kida, H.; Paulson, J.C.; Webster, R.G.; et al. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J. Virol. 1998, 72, 7367–7373. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, Y.; Tefsen, B.; Shi, Y.; Gao, G.F. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 2014, 22, 183–191. [Google Scholar] [CrossRef]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Annu. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef]
- Monto, A.S.; Fukuda, K. Lessons From Influenza Pandemics of the Last 100 Years. Clin. Infect. Dis. 2020, 70, 951–957. [Google Scholar] [CrossRef]
- Spreeuwenberg, P.; Kroneman, M.; Paget, J. Reassessing the Global Mortality Burden of the 1918 Influenza Pandemic. Am. J. Epidemiol. 2018, 187, 2561–2567. [Google Scholar] [CrossRef]
- Chu, C.M.; Dawson, I.M.; Elford, W.J. Filamentous forms associated with newly isolated influenza virus. Lancet 1949, 1, 602. [Google Scholar] [CrossRef]
- Kilbourne, E.D.; Murphy, J.S. Genetic studies of influenza viruses. I. Viral morphology and growth capacity as exchangeable genetic traits. Rapid in ovo adaptation of early passage Asian strain isolates by combination with PR8. J. Exp. Med. 1960, 111, 387–406. [Google Scholar] [CrossRef]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Balannik, V.; Pinto, L.H.; Lamb, R.A. Influenza virus m2 ion channel protein is necessary for filamentous virion formation. J. Virol. 2010, 84, 5078–5088. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef]
- Martin, K.; Helenius, A. Transport of incoming influenza virus nucleocapsids into the nucleus. J. Virol. 1991, 65, 232–244. [Google Scholar] [CrossRef]
- Zebedee, S.L.; Lamb, R.A. Influenza A virus M2 protein: Monoclonal antibody restriction of virus growth and detection of M2 in virions. J. Virol. 1988, 62, 2762–2772. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A virus cell entry, replication, virion assembly and movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Tao, Y.J. Structure and assembly of the influenza A virus ribonucleoprotein complex. FEBS Lett. 2013, 587, 1206–1214. [Google Scholar] [CrossRef]
- Arranz, R.; Coloma, R.; Chichón, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortín, J.; Martín-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef]
- Ortega, J.; Martín-Benito, J.; Zürcher, T.; Valpuesta, J.M.; Carrascosa, J.L.; Ortín, J. Ultrastructural and functional analyses of recombinant influenza virus ribonucleoproteins suggest dimerization of nucleoprotein during virus amplification. J. Virol. 2000, 74, 156–163. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef]
- Pinto, R.M.; Lycett, S.; Gaunt, E.; Digard, P. Accessory Gene Products of Influenza A Virus. Cold Spring Harb. Perspect. Med. 2020, 11, a038380. [Google Scholar] [CrossRef]
- Lee, K.K. Architecture of a nascent viral fusion pore. EMBO J. 2010, 29, 1299–1311. [Google Scholar] [CrossRef]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Chou, Y.Y.; Heaton, N.S.; Gao, Q.; Palese, P.; Singer, R.H.; Lionnet, T. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Plotch, S.J.; Bouloy, M.; Ulmanen, I.; Krug, R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [Google Scholar] [CrossRef]
- Hay, A.; Lomniczi, B.; Bellamy, A.; Skehel, J. Transcription of the influenza virus genome. Virology 1977, 83, 337–355. [Google Scholar] [CrossRef]
- Alenquer, M.; Vale-Costa, S.; Etibor, T.A.; Ferreira, F.; Sousa, A.L.; Amorim, M.J. Influenza A virus ribonucleoproteins form liquid organelles at endoplasmic reticulum exit sites. Nat. Commun. 2019, 10, 1629. [Google Scholar] [CrossRef]
- Amorim, M.J.; Bruce, E.A.; Read, E.K.C.; Foeglein, A.; Mahen, R.; Stuart, A.D.; Digard, P. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, J.; Chen, Z. A second CRM1-dependent nuclear export signal in the influenza A virus NS2 protein contributes to the nuclear export of viral ribonucleoproteins. J. Virol. 2013, 87, 767–778. [Google Scholar] [CrossRef]
- Lakdawala, S.S.; Wu, Y.; Wawrzusin, P.; Kabat, J.; Broadbent, A.J.; Lamirande, E.W.; Fodor, E.; Altan-Bonnet, N.; Shroff, H.; Subbarao, K. Influenza a virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 2014, 10, e1003971. [Google Scholar] [CrossRef]
- Vale-Costa, S.; Etibor, T.A.; Brás, D.; Sousa, A.L.; Ferreira, M.; Martins, G.G.; Mello, V.H.; Amorim, M.J. ATG9A regulates the dissociation of recycling endosomes from microtubules to form liquid influenza A virus inclusions. PLoS Biol. 2023, 21, e3002290. [Google Scholar] [CrossRef]
- Petrich, A.; Dunsing, V.; Bobone, S.; Chiantia, S. Influenza A M2 recruits M1 to the plasma membrane: A fluorescence fluctuation microscopy study. Biophys. J. 2021, 120, 5478–5490. [Google Scholar] [CrossRef]
- Sato, R.; Okura, T.; Kawahara, M.; Takizawa, N.; Momose, F.; Morikawa, Y. Apical Trafficking Pathways of Influenza A Virus HA and NA via Rab17- and Rab23-Positive Compartments. Front. Microbiol. 2019, 10, 1857. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef]
- Gottschalk, A. Neuraminidase: The specific enzyme of influenza virus and Vibrio cholerae. Biochim. Biophys. Acta 1957, 23, 645–646. [Google Scholar] [CrossRef]
- Skehel, J.J.; Bayley, P.M.; Brown, E.B.; Martin, S.R.; Waterfield, M.D.; White, J.M.; Wilson, I.A.; Wiley, D.C. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA 1982, 79, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Datta, K.; Wolkerstorfer, A.; Szolar, O.H.J.; Cusack, S.; Klumpp, K. Characterization of PA-N terminal domain of Influenza A polymerase reveals sequence specific RNA cleavage. Nucleic Acids Res. 2013, 41, 8289–8299. [Google Scholar] [CrossRef] [PubMed]
- Wandzik, J.M.; Kouba, T.; Karuppasamy, M.; Pflug, A.; Drncova, P.; Provaznik, J.; Azevedo, N.; Cusack, S. A Structure-Based Model for the Complete Transcription Cycle of Influenza Polymerase. Cell 2020, 181, 877–893.e21. [Google Scholar] [CrossRef]
- Walker, A.P.; Fodor, E. Interplay between Influenza Virus and the Host RNA Polymerase II Transcriptional Machinery. Trends Microbiol. 2019, 27, 398–407. [Google Scholar] [CrossRef]
- Dou, D.; da Silva, D.V.; Nordholm, J.; Wang, H.; Daniels, R. Type II transmembrane domain hydrophobicity dictates the cotranslational dependence for inversion. Mol. Biol. Cell 2014, 25, 3363–3374. [Google Scholar] [CrossRef]
- Zhu, Z.; Fodor, E.; Keown, J.R. A structural understanding of influenza virus genome replication. Trends Microbiol. 2023, 31, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.P.; Lamb, R.A. Influenza virus assembly and budding at the viral budozone. Adv. Virus Res. 2005, 64, 383–416. [Google Scholar] [PubMed]
- Chlanda, P.; Schraidt, O.; Kummer, S.; Riches, J.; Oberwinkler, H.; Prinz, S.; Kräusslich, H.G.; Briggs, J.A. Structural Analysis of the Roles of Influenza A Virus Membrane-Associated Proteins in Assembly and Morphology. J. Virol. 2015, 89, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
- Calder, L.J.; Wasilewski, S.; Berriman, J.A.; Rosenthal, P.B. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 10685–10690. [Google Scholar] [CrossRef] [PubMed]
- Wandzik, J.M.; Kouba, T.; Cusack, S. Structure and Function of Influenza Polymerase. Cold Spring Harb. Perspect. Med. 2021, 11, a038372. [Google Scholar] [CrossRef]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef]
- Kouba, T.; Drncová, P.; Cusack, S. Structural snapshots of actively transcribing influenza polymerase. Nat. Struct. Mol. Biol. 2019, 26, 460–470. [Google Scholar] [CrossRef]
- Thierry, E.; Guilligay, D.; Kosinski, J.; Bock, T.; Gaudon, S.; Round, A.; Pflug, A.; Hengrung, N.; El Omari, K.; Baudin, F.; et al. Influenza Polymerase Can Adopt an Alternative Configuration Involving a Radical Repacking of PB2 Domains. Mol. Cell 2016, 61, 125–137. [Google Scholar] [CrossRef]
- McCauley, J.; Bye, J.; Elder, K.; Gething, M.; Skehel, J.; Smith, A. Influenza virus haemagglutinin signal sequence. FEBS Lett. 1979, 108, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Nans, A.; Calder, L.J.; Turner, J.; Neu, U.; Lin, Y.P.; Ketelaars, E.; Kallewaard, N.L.; Corti, D.; Lanzavecchia, A.; et al. Influenza hemagglutinin membrane anchor. Proc. Natl. Acad. Sci. USA 2018, 115, 10112–10117. [Google Scholar] [CrossRef] [PubMed]
- Boulay, F.; Doms, R.W.; Webster, R.G.; Helenius, A. Posttranslational oligomerization and cooperative acid activation of mixed influenza hemagglutinin trimers. J. Cell Biol. 1988, 106, 629–639. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J.; Waterfield, M. Evidence from studies with a cross-linking reagent that the haemagglutinin of influenza virus is a trimer. Virology 1977, 79, 446–448. [Google Scholar] [CrossRef]
- Burke, D.F.; Smith, D.J. A recommended numbering scheme for influenza A HA subtypes. PLoS ONE 2014, 9, e112302. [Google Scholar] [CrossRef]
- Coudert, E.; Gehant, S.; de Castro, E.; Pozzato, M.; Baratin, D.; Neto, T.; Sigrist, C.J.A.; Redaschi, N.; Bridge, A.; Aimo, L.; et al. Annotation of biologically relevant ligands in UniProtKB using ChEBI. Bioinformatics 2023, 39, btac793. [Google Scholar] [CrossRef]
- Ng, A.K.; Zhang, H.; Tan, K.; Li, Z.; Liu, J.; Chan, P.K.; Li, S.; Chan, W.; Au, S.W.; Joachimiak, A.; et al. Structure of the influenza virus A H5N1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008, 22, 3638–3647. [Google Scholar] [CrossRef] [PubMed]
- Cros, J.F.; García-Sastre, A.; Palese, P. An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 2005, 6, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.-H.; Ng, A.K.-L.; Robb, N.C.; Lam, M.K.-H.; Chan, P.K.-S.; Au, S.W.-N.; Wang, J.-H.; Fodor, E.; Shaw, P.-C. Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. J. Virol. 2010, 84, 7337–7345. [Google Scholar] [CrossRef]
- Vreede, F.T.; Jung, T.E.; Brownlee, G.G. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J. Virol. 2004, 78, 9568–9572. [Google Scholar] [CrossRef]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Palese, P.; Compans, R.W. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): Mechanism of action. J. Gen. Virol. 1976, 33, 159–163. [Google Scholar] [CrossRef]
- Selzer, L.; Su, Z.; Pintilie, G.D.; Chiu, W.; Kirkegaard, K. Full-length three-dimensional structure of the influenza A virus M1 protein and its organization into a matrix layer. PLoS Biol. 2020, 18, e3000827. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [PubMed]
- Baudin, F.; Petit, I.; Weissenhorn, W.; Ruigrok, R.W. In vitro dissection of the membrane and RNP binding activities of influenza virus M1 protein. Virology 2001, 281, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Lai, C.J.; Choppin, P.W. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: Colinear and interrupted mRNAs code for overlapping proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 4170–4174. [Google Scholar] [CrossRef] [PubMed]
- Sugrue, R.J.; Hay, A.J. Structural characteristics of the M2 protein of influenza A viruses: Evidence that it forms a tetrameric channel. Virology 1991, 180, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Manzoor, R.; Igarashi, M.; Takada, A. Influenza A Virus M2 Protein: Roles from Ingress to Egress. Int. J. Mol. Sci. 2017, 18, 2649. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.W.; Mishra, A.; Wang, J.; DeGrado, W.F.; Wong, G.C.L. Influenza virus A M2 protein generates negative Gaussian membrane curvature necessary for budding and scission. J. Am. Chem. Soc. 2013, 135, 13710–13719. [Google Scholar] [CrossRef]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Müller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef]
- Lamb, R.A.; Choppin, P.W.; Chanock, R.M.; Lai, C.J. Mapping of the two overlapping genes for polypeptides NS1 and NS2 on RNA segment 8 of influenza virus genome. Proc. Natl. Acad. Sci. USA 1980, 77, 1857–1861. [Google Scholar] [CrossRef]
- Porter, A.G.; Smith, J.C.; Emtage, J.S. Nucleotide sequence of influenza virus RNA segment 8 indicates that coding regions for NS1 and NS2 proteins overlap. Proc. Natl. Acad. Sci. USA 1980, 77, 5074–5078. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Yi, M.; Dong, H.; Qin, H.; Peterson, E.; Busath, D.D.; Zhou, H.-X.; Cross, T.A. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 2010, 330, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki-Horimoto, K.; Horimoto, T.; Fujii, Y.; Kawaoka, Y. Generation of influenza A virus NS2 (NEP) mutants with an altered nuclear export signal sequence. J. Virol. 2004, 78, 10149–10155. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Brand-Miller, J. The role and potential of sialic acid in human nutrition. Eur. J. Clin. Nutr. 2003, 57, 1351–1369. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ito, T.; Suzuki, T.; Holland, R.E.; Chambers, T.M.; Kiso, M.; Ishida, H.; Kawaoka, Y. Sialic acid species as a determinant of the host range of influenza A viruses. J. Virol. 2000, 74, 11825–11831. [Google Scholar] [CrossRef]
- Byrd-Leotis, L.; Cummings, R.D.; Steinhauer, D.A. The Interplay between the Host Receptor and Influenza Virus Hemagglutinin and Neuraminidase. Int. J. Mol. Sci. 2017, 18, 1541. [Google Scholar] [CrossRef]
- Connor, R.J.; Kawaoka, Y.; Webster, R.G.; Paulson, J.C. Receptor specificity in human, avian, and equine H2 and H3 influenza virus isolates. Virology 1994, 205, 17–23. [Google Scholar] [CrossRef]
- Traving, C.; Schauer, R. Structure, function and metabolism of sialic acids. Cell. Mol. Life Sci. 1998, 54, 1330–1349. [Google Scholar] [CrossRef]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef]
- Kathan, R.H.; Winzler, R.J.; Johnson, C.A. Preparation of an inhibitor of viral hemagglutination from human erythrocytes. J. Exp. Med. 1961, 113, 37–45. [Google Scholar] [CrossRef]
- Scholtissek, C.; Bürger, H.; Kistner, O.; Shortridge, K. The nucleoprotein as a possible major factor in determining host specificity of influenza H3N2 viruses. Virology 1985, 147, 287–294. [Google Scholar] [CrossRef]
- Claas, E.C.; Kawaoka, Y.; de Jong, J.C.; Masurel, N.; Webster, R.G. Infection of children with avian-human reassortant influenza virus from pigs in europe. Virology 1994, 204, 453–457. [Google Scholar] [CrossRef]
- Kida, H.; Ito, T.; Yasuda, J.; Shimizu, Y.; Itakura, C.; Shortridge, K.F.; Kawaoka, Y.; Webster, R.G. Potential for transmission of avian influenza viruses to pigs. J. Gen. Virol. 1994, 75, 2183–2188. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Srinivasan, A.; Raman, R.; Viswanathan, K.; Raguram, S.; Tumpey, T.M.; Sasisekharan, V.; Sasisekharan, R. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat. Biotechnol. 2008, 26, 107–113. [Google Scholar] [CrossRef]
- Zheng, Z.; Paul, S.S.; Mo, X.; Yuan, Y.-R.A.; Tan, Y.-J. The Vestigial Esterase Domain of Haemagglutinin of H5N1 Avian Influenza A Virus: Antigenicity and Contribution to Viral Pathogenesis. Vaccines 2018, 6, 53. [Google Scholar] [CrossRef]
- Russell, R.J.; Stevens, D.J.; Haire, L.F.; Gamblin, S.J.; Skehel, J.J. Avian and human receptor binding by hemagglutinins of influenza A viruses. Glycoconj. J. 2006, 23, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc. Natl. Acad. Sci. USA 2001, 98, 11181–11186. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.N.; D’Souza, B.L. Receptor binding properties of human and animal H1 influenza virus isolates. Virology 1989, 173, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Sabesanc, S.; Skehel, J.J.; Wiley, D.C. Binding of the influenza A virus to cell-surface receptors: Structures of five hemagglutinin–sialyloligosaccharide complexes determined by X-ray crystallography. Virology 1997, 232, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. X-ray structure of the hemagglutinin of a potential H3 avian progenitor of the 1968 Hong Kong pandemic influenza virus. Virology 2003, 309, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B 2012, 88, 226–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Stevens, D.J.; Haire, L.F.; Walker, P.A.; Coombs, P.J.; Russell, R.J.; Gamblin, S.J.; Skehel, J.J. Structures of receptor complexes formed by hemagglutinins from the Asian Influenza pandemic of 1957. Proc. Natl. Acad. Sci. USA 2009, 106, 17175–17180. [Google Scholar] [CrossRef]
- Bitsikas, V.; Corrêa, I.R., Jr.; Nichols, B.J. Clathrin-independent pathways do not contribute significantly to endocytic flux. eLife 2014, 3, e03970. [Google Scholar] [CrossRef]
- Ehrlich, M.; Boll, W.; van Oijen, A.; Hariharan, R.; Chandran, K.; Nibert, M.L.; Kirchhausen, T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 2004, 118, 591–605. [Google Scholar] [CrossRef]
- Drake, M.T.; Downs, M.A.; Traub, L.M. Epsin binds to clathrin by associating directly with the clathrin-terminal domain: Evidence for cooperative binding through two discrete sites. J. Biol. Chem. 2000, 275, 6479–6489. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.P.; Kelly, B.T.; McCoy, A.J.; Gaffry, T.; James, L.C.; Collins, B.M.; Höning, S.; Evans, P.R.; Owen, D.J. A Large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 2010, 141, 1220–1229. [Google Scholar] [CrossRef]
- Kelly, B.T.; McCoy, A.J.; Späte, K.; Miller, S.E.; Evans, P.R.; Höning, S.; Owen, D.J. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 2008, 456, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhuang, X. Epsin 1 is a cargo-specific adaptor for the clathrin-mediated endocytosis of the influenza virus. Proc. Natl. Acad. Sci. USA 2008, 105, 11790–11795. [Google Scholar] [CrossRef]
- Kelly, B.T.; Graham, S.C.; Liska, N.; Dannhauser, P.N.; Höning, S.; Ungewickell, E.J.; Owen, D.J. AP2 controls clathrin polymerization with a membrane-activated switch. Science 2014, 345, 459–463. [Google Scholar] [CrossRef]
- Bulley, S.J.; Clarke, J.H.; Droubi, A.; Giudici, M.-L.; Irvine, R.F. Exploring phosphatidylinositol 5-phosphate 4-kinase function. Adv. Biol. Regul. 2014, 57, 193–202. [Google Scholar] [CrossRef]
- Ford, M.G.J.; Mills, I.G.; Peter, B.J.; Vallis, Y.; Praefcke, G.J.K.; Evans, P.R.; McMahon, H.T. Curvature of clathrin-coated pits driven by epsin. Nature 2002, 419, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Pearse, B.M. Coated vesicles from pig brain: Purification and biochemical characterization. J. Mol. Biol. 1975, 97, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Bashkirov, P.V.; Akimov, S.A.; Evseev, A.I.; Schmid, S.L.; Zimmerberg, J.; Frolov, V.A. GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell 2008, 135, 1276–1286. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.-M.M.; Parker, J.S.; Parrish, C.R.; Whittaker, G.R. Early stages of influenza virus entry into Mv-1 lung cells: Involvement of dynamin. Virology 2000, 267, 17–28. [Google Scholar] [CrossRef]
- Braell, W.A.; Schlossman, D.M.; Schmid, S.L.; Rothman, J.E. Dissociation of clathrin coats coupled to the hydrolysis of ATP: Role of an uncoating ATPase. J. Cell Biol. 1984, 99, 734–741. [Google Scholar] [CrossRef]
- Barouch, W.; Prasad, K.; Greene, L.E.; Eisenberg, E. ATPase activity associated with the uncoating of clathrin baskets by Hsp70. J. Biol. Chem. 1994, 269, 28563–28568. [Google Scholar] [CrossRef]
- Lee, D.-W.; Wu, X.; Eisenberg, E.; Greene, L.E. Recruitment dynamics of GAK and auxilin to clathrin-coated pits during endocytosis. J. Cell Sci. 2006, 119, 3502–3512. [Google Scholar] [CrossRef]
- Newmyer, S.L.; Christensen, A.; Sever, S. Auxilin-dynamin interactions link the uncoating ATPase chaperone machinery with vesicle formation. Dev. Cell 2003, 4, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza a viruses (IAV) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Leser, G.P.; Lamb, R.A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 2012, 86, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Lanzetti, L.; Palamidessi, A.; Areces, L.; Scita, G.; Di Fiore, P.P. Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 2004, 429, 309–314. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Holsinger, L.J.; Nichani, D.; Pinto, L.H.; Lamb, R.A. Influenza A virus M2 ion channel protein: A structure-function analysis. J. Virol. 1994, 68, 1551–1563. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Schnell, J.R.; Chou, J.J. Structure and mechanism of the M2 proton channel of influenza A virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef]
- Tang, Y.; Zaitseva, F.; Lamb, R.A.; Pinto, L.H. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 2002, 277, 39880–39886. [Google Scholar] [CrossRef]
- Williams, J.K.; Zhang, Y.; Schmidt-Rohr, K.; Hong, M. pH-dependent conformation, dynamics, and aromatic interaction of the gating tryptophan residue of the influenza M2 proton channel from solid-state NMR. Biophys. J. 2013, 104, 1698–1708. [Google Scholar] [CrossRef]
- Liang, R.; Swanson, J.M.J.; Madsen, J.J.; Hong, M.; DeGrado, W.F.; Voth, G.A. Acid activation mechanism of the influenza A M2 proton channel. Proc. Natl. Acad. Sci. USA 2016, 113, E6955–E6964. [Google Scholar] [CrossRef]
- Thomaston, J.L.; Alfonso-Prieto, M.; Woldeyes, R.A.; Fraser, J.S.; Klein, M.L.; Fiorin, G.; DeGrado, W.F. High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 14260–14265. [Google Scholar] [CrossRef]
- Acharya, R.; Carnevale, V.; Fiorin, G.; Levine, B.G.; Polishchuk, A.L.; Balannik, V.; Samish, I.; Lamb, R.A.; Pinto, L.H.; DeGrado, W.F.; et al. Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 15075–15080. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Dieckmann, G.R.; Gandhi, C.S.; Papworth, C.G.; Braman, J.; Shaughnessy, M.A.; Lear, J.D.; Lamb, R.A.; DeGrado, W.F. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. USA 1997, 94, 11301–11306. [Google Scholar] [CrossRef] [PubMed]
- Kass, I.; Arkin, I.T. How pH opens a H+ channel: The gating mechanism of influenza A M2. Structure 2005, 13, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.-S.; Dyer, R.B. Proton Transport Mechanism of M2 Proton Channel Studied by Laser-Induced pH Jump. J. Am. Chem. Soc. 2017, 139, 6621–6628. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Lorieau, J.L.; Louis, J.M.; Bax, A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl. Acad. Sci. USA 2010, 107, 11341–11346. [Google Scholar] [CrossRef] [PubMed]
- Luo, M. Influenza virus entry. Adv. Exp. Med. Biol. 2012, 726, 201–221. [Google Scholar] [PubMed]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Cross, K.J.; Langley, W.A.; Russell, R.J.; Skehel, J.J.; Steinhauer, D.A. Composition and functions of the influenza fusion peptide. Protein Pept. Lett. 2009, 16, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.J.; Gamblin, S.J.; Rosenthal, P.B.; Skehel, J.J. Structural transitions in influenza haemagglutinin at membrane fusion pH. Nature 2020, 583, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Blijleven, J.S.; Boonstra, S.; Onck, P.R.; van der Giessen, E.; van Oijen, A.M. Mechanisms of influenza viral membrane fusion. Semin. Cell Dev. Biol. 2016, 60, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Antanasijevic, A.; Durst, M.A.; Lavie, A.; Caffrey, M. Identification of a pH sensor in Influenza hemagglutinin using X-ray crystallography. J. Struct. Biol. 2019, 209, 107412. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Opitz, R.; Knapp, E.-W.; Herrmann, A. Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys. J. 2002, 82, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Lousa, D.; Soares, C.M. Molecular mechanisms of the influenza fusion peptide: Insights from experimental and simulation studies. FEBS Open Bio 2021, 11, 3253–3261. [Google Scholar] [CrossRef]
- Park, H.E.; Gruenke, J.A.; White, J.M. Leash in the groove mechanism of membrane fusion. Nat. Struct. Mol. Biol. 2003, 10, 1048–1053. [Google Scholar] [CrossRef]
- Chlanda, P.; Mekhedov, E.; Waters, H.; Schwartz, C.L.; Fischer, E.R.; Ryham, R.J.; Cohen, F.S.; Blank, P.S.; Zimmerberg, J. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 2016, 1, 16050. [Google Scholar] [CrossRef]
- Pabis, A.; Rawle, R.J.; Kasson, P.M. Influenza hemagglutinin drives viral entry via two sequential intramembrane mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 7200–7207. [Google Scholar] [CrossRef] [PubMed]
- Harter, C.; James, P.; Bächi, T.; Semenza, G.; Brunner, J. Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the “fusion peptide”. J. Biol. Chem. 1989, 264, 6459–6464. [Google Scholar] [CrossRef]
- Skehel, J.J.; Waterfield, M.D. Studies on the primary structure of the influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA 1975, 72, 93–97. [Google Scholar] [CrossRef]
- Doms, R.W.; Helenius, A.; White, J. Membrane fusion activity of the influenza virus hemagglutinin. The low pH-induced conformational change. J. Biol. Chem. 1985, 260, 2973–2981. [Google Scholar] [CrossRef]
- Godley, L.; Pfeifer, J.; Steinhauer, D.; Ely, B.; Shaw, G.; Kaufmann, R.; Suchanek, E.; Pabo, C.; Skehel, J.; Wiley, D.; et al. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 1992, 68, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Kemble, G.W.; Bodian, D.L.; Rosé, J.; Wilson, I.A.; White, J.M. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 1992, 66, 4940–4950. [Google Scholar] [CrossRef]
- White, J.; Helenius, A.; Gething, M.-J. Haemagglutinin of influenza virus expressed from a cloned gene promotes membrane fusion. Nature 1982, 300, 658–659. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, M.; Lavie, A. pH-Dependent Mechanisms of Influenza Infection Mediated by Hemagglutinin. Front. Mol. Biosci. 2021, 8, 777095. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.; Downie, J.; Hay, A.; Knossow, M.; Skehel, J.; Wang, M.; Wiley, D. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 1985, 40, 431–439. [Google Scholar] [CrossRef]
- Stegmann, T.; Delfino, J.; Richards, F.; Helenius, A. The HA2 subunit of influenza hemagglutinin inserts into the target membrane prior to fusion. J. Biol. Chem. 1991, 266, 18404–18410. [Google Scholar] [CrossRef]
- Ivanovic, T.; Choi, J.L.; Whelan, S.P.; van Oijen, A.M.; Harrison, S.C. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2013, 2, e00333. [Google Scholar] [CrossRef]
- Calder, L.J.; Rosenthal, P.B. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat. Struct. Mol. Biol. 2016, 23, 853–858. [Google Scholar] [CrossRef]
- Gui, L.; Ebner, J.L.; Mileant, A.; Williams, J.A.; Lee, K.K. Visualization and Sequencing of Membrane Remodeling Leading to Influenza Virus Fusion. J. Virol. 2016, 90, 6948–6962. [Google Scholar] [CrossRef]
- Peukes, J.; Xiong, X.; Erlendsson, S.; Qu, K.; Wan, W.; Calder, L.J.; Schraidt, O.; Kummer, S.; Freund, S.M.V.; Kräusslich, H.-G.; et al. The native structure of the assembled matrix protein 1 of influenza A virus. Nature 2020, 587, 495–498. [Google Scholar] [CrossRef]
- Bui, M.; Whittaker, G.; Helenius, A. Effect of M1 protein and low pH on nuclear transport of influenza virus ribonucleoproteins. J. Virol. 1996, 70, 8391–8401. [Google Scholar] [CrossRef]
- Lott, K.; Cingolani, G. The importin β binding domain as a master regulator of nucleocytoplasmic transport. Biochim. Biophys. Acta 2011, 1813, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Mattaj, I.W.; Englmeier, L. Nucleocytoplasmic transport: The soluble phase. Annu. Rev. Biochem. 1998, 67, 265–306. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.Y.H.; Huang, N.-P.; Köser, J.; Deng, J.; Lau, K.H.A.; Schwarz-Herion, K.; Fahrenkrog, B.; Aebi, U. Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 2006, 103, 9512–9517. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Kuramochi, M.; Miyafusa, T. New Family Members of FG Repeat Proteins and Their Unexplored Roles during Phase Separation. Front. Cell Dev. Biol. 2021, 9, 708702. [Google Scholar] [CrossRef] [PubMed]
- Kemler, I.; Whittaker, G.; Helenius, A. Nuclear import of microinjected influenza virus ribonucleoproteins. Virology 1994, 202, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Jaskunas, R.; Blobel, G.; Palese, P.; Moroianu, J. Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J. Biol. Chem. 1995, 270, 22701–22704. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Yoneda, Y. Importin α: Functions as a nuclear transport factor and beyond. Proc. Jpn. Acad. Ser. B 2018, 94, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Weis, K. Regulating access to the genome: Nucleocytoplasmic transport throughout the cell cycle. Cell 2003, 112, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Gaudon, S.; Resa-Infante, P.; Lethier, M.; Reich, S.; Schulze, W.M.; Cusack, S. Capped RNA primer binding to influenza polymerase and implications for the mechanism of cap-binding inhibitors. Nucleic Acids Res. 2017, 46, 956–971. [Google Scholar] [CrossRef]
- Coloma, R.; Arranz, R.; de la Rosa-Trevín, J.M.; Sorzano, C.O.S.; Munier, S.; Carlero, D.; Naffakh, N.; Ortín, J.; Martín-Benito, J. Structural insights into influenza A virus ribonucleoproteins reveal a processive helical track as transcription mechanism. Nat. Microbiol. 2020, 5, 727–734. [Google Scholar] [CrossRef]
- Mahy, B.W.J.; Hastie, N.D.; Armstrong, S.J. Inhibition of influenza virus replication by α-amanitin: Mode of action. Proc. Natl. Acad. Sci. USA 1972, 69, 1421–1424. [Google Scholar] [CrossRef]
- Lukarska, M.; Fournier, G.; Pflug, A.; Resa-Infante, P.; Reich, S.; Naffakh, N.; Cusack, S. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature 2016, 541, 117–121. [Google Scholar] [CrossRef]
- Palancade, B.; Bensaude, O. Investigating RNA polymerase II carboxyl-terminal domain (CTD) phosphorylation. J. Biol. Inorg. Chem. 2003, 270, 3859–3870. [Google Scholar] [CrossRef]
- Engelhardt, O.G.; Smith, M.; Fodor, E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 2005, 79, 5812–5818. [Google Scholar] [CrossRef]
- Martínez-Alonso, M.; Hengrung, N.; Fodor, E. RNA-Free and Ribonucleoprotein-Associated Influenza Virus Polymerases Directly Bind the Serine-5-Phosphorylated Carboxyl-Terminal Domain of Host RNA Polymerase II. J. Virol. 2016, 90, 6014–6021. [Google Scholar] [CrossRef] [PubMed]
- Blass, D.; Patzelt, E.; Kuechler, E. Identification of the cap binding protein of influenza virus. Nucleic Acids Res. 1982, 10, 4803–4812. [Google Scholar] [CrossRef]
- Ulmanen, I.; Broni, B.A.; Krug, R.M. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7 GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. USA 1981, 78, 7355–7359. [Google Scholar] [CrossRef]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.H.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef]
- Li, M.; Ramirez, B.C.; Krug, R.M. RNA-dependent activation of primer RNA production by influenza virus polymerase: Different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J. 1998, 17, 5844–5852. [Google Scholar] [CrossRef]
- Li, M.; Rao, P.; Krug, R.M. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 2001, 20, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Crow, M.; Mingay, L.J.; Deng, T.; Sharps, J.; Fechter, P.; Brownlee, G.G. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 2002, 76, 8989–9001. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lou, Z.; Guo, Y.; Ma, M.; Chen, Y.; Liang, S.; Zhang, L.; Chen, S.; Li, X.; Liu, Y.; et al. Nucleoside monophosphate complex structures of the endonuclease domain from the influenza virus polymerase PA subunit reveal the substrate binding site inside the catalytic center. J. Virol. 2009, 83, 9024–9030. [Google Scholar] [CrossRef][Green Version]
- Reich, S.; Guilligay, D.; Cusack, S. An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res. 2017, 45, 3353–3368. [Google Scholar]
- Jácome, R.; Becerra, A.; de León, S.P.; Lazcano, A. Structural Analysis of Monomeric RNA-Dependent Polymerases: Evolutionary and Therapeutic Implications. PLoS ONE 2015, 10, e0139001. [Google Scholar] [CrossRef]
- Shu, B.; Gong, P. Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc. Natl. Acad. Sci. USA 2016, 113, E4005–E4014. [Google Scholar] [CrossRef]
- Ye, Q.; Guu, T.S.Y.; Mata, D.A.; Kuo, R.-L.; Smith, B.; Krug, R.M.; Tao, Y.J. Biochemical and structural evidence in support of a coherent model for the formation of the double-helical influenza a virus ribonucleoprotein. mBio 2013, 4, e00467-12. [Google Scholar] [CrossRef] [PubMed]
- Poon, L.L.M.; Pritlove, D.C.; Fodor, E.; Brownlee, G.G. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J. Virol. 1999, 73, 3473–3476. [Google Scholar] [CrossRef] [PubMed]
- Pritlove, D.C.; Poon, L.L.M.; Devenish, L.J.; Leahy, M.B.; Brownlee, G.G. A hairpin loop at the 5′ end of influenza A virus virion RNA is required for synthesis of poly(A)+ mRNA in vitro. J. Virol. 1999, 73, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Pritlove, D.C.; Poon, L.L.M.; Fodor, E.; Sharps, J.; Brownlee, G.G. Polyadenylation of influenza virus mRNA transcribed in vitro from model virion RNA templates: Requirement for 5′ conserved sequences. J. Virol. 1998, 72, 1280–1286. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zheng, M.; Wang, P.; Mok, B.W.-Y.; Liu, S.; Lau, S.-Y.; Chen, P.; Liu, Y.-C.; Liu, H.; Chen, Y.; et al. An NS-segment exonic splicing enhancer regulates influenza A virus replication in mammalian cells. Nat. Commun. 2017, 8, 14751. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Choppin, P.W. Identification of a second protein (M2) encoded by RNA segment 7 of influenza virus. Virology 1981, 112, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Lai, C.-J. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 1980, 21, 475–485. [Google Scholar] [CrossRef]
- De Magistris, P. The Great Escape: mRNA Export through the Nuclear Pore Complex. Int. J. Mol. Sci. 2021, 22, 11767. [Google Scholar] [CrossRef] [PubMed]
- de Rozières, C.M.; Pequeno, A.; Shahabi, S.; Lucas, T.M.; Godula, K.; Ghosh, G.; Joseph, S. PABP1 Drives the Selective Translation of Influenza A Virus mRNA. J. Mol. Biol. 2022, 434, 167460. [Google Scholar] [CrossRef] [PubMed]
- Mukaigawa, J.; Nayak, D.P. Two signals mediate nuclear localization of influenza virus (A/WSN/33) polymerase basic protein 2. J. Virol. 1991, 65, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Sharps, J.; Fodor, E.; Brownlee, G.G. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 2005, 79, 8669–8674. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Engelhardt, O.G.; Thomas, B.; Akoulitchev, A.V.; Brownlee, G.G.; Fodor, E. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 2006, 80, 11911–11919. [Google Scholar] [CrossRef]
- Jäkel, S.; Görlich, D. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J. 1998, 17, 4491–4502. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Orr, O.E.; Liu, S.M.; Engelhardt, O.G.; Fodor, E. Characterization of the interaction between the influenza A virus polymerase subunit PB1 and the host nuclear import factor Ran-binding protein 5. J. Gen. Virol. 2011, 92, 1859–1869. [Google Scholar] [CrossRef]
- Nieto, A.; de la Luna, S.; Bárcena, J.; Portela, A.; Ortín, J. Complex structure of the nuclear translocation signal of influenza virus polymerase PA subunit. J. Gen. Virol. 1994, 75, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tarendeau, F.; Boudet, J.; Guilligay, D.; Mas, P.J.; Bougault, C.M.; Boulo, S.; Baudin, F.; Ruigrok, R.W.H.; Daigle, N.; Ellenberg, J.; et al. Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat. Struct. Mol. Biol. 2007, 14, 229–233. [Google Scholar] [CrossRef]
- Davey, J.; Dimmock, N.; Colman, A. Identification of the sequence responsible for the nuclear accumulation of the influenza virus nucleoprotein in Xenopus oocytes. Cell 1985, 40, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Palese, P.; O’Neill, R.E. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza a virus nucleoprotein NP is a nonconventional nuclear localization signal. J. Virol. 1997, 71, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Kochs, G.; Gruber, S.; Haller, O. A classical bipartite nuclear localization signal on Thogoto and influenza A virus nucleoproteins. Virology 1998, 250, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, F.; Tan, L.; Bai, C.; Chen, B.; Liu, J.; Liang, J.; Liu, C.; Zhang, S.; Lu, G.; et al. Host Protein Moloney Leukemia Virus 10 (MOV10) Acts as a Restriction Factor of Influenza A Virus by Inhibiting the Nuclear Import of the Viral Nucleoprotein. J. Virol. 2016, 90, 3966–3980. [Google Scholar] [CrossRef]
- Ye, Z.; Robinson, D.; Wagner, R.R. Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J. Virol. 1995, 69, 1964–1970. [Google Scholar] [CrossRef]
- Boulo, S.; Akarsu, H.; Ruigrok, R.W.; Baudin, F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007, 124, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.; Skehel, J.; McCauley, J. Characterization of influenza virus RNA complete transcripts. Virology 1982, 116, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, Y.; Li, M.; Guo, L.; Gao, Y.; Wang, Q.; Zhang, J.; Lai, Z.; Zhang, X.; Zhu, L.; et al. An intermediate state allows influenza polymerase to switch smoothly between transcription and replication cycles. Nat. Struct. Mol. Biol. 2023, 30, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Sheppard, C.M.; Mistry, B.; Staller, E.; Barclay, W.S.; Grimes, J.M.; Fodor, E.; Fan, H. The C-terminal LCAR of host ANP32 proteins interacts with the influenza A virus nucleoprotein to promote the replication of the viral RNA genome. Nucleic Acids Res. 2022, 50, 5713–5725. [Google Scholar] [CrossRef]
- Robb, N.C.; Velthuis, A.J.W.T.; Wieneke, R.; Tampé, R.; Cordes, T.; Fodor, E.; Kapanidis, A.N. Single-molecule FRET reveals the pre-initiation and initiation conformations of influenza virus promoter RNA. Nucleic Acids Res. 2016, 44, 10304–10315. [Google Scholar] [CrossRef]
- Vreede, F.T.; Gifford, H.; Brownlee, G.G. Role of initiating nucleoside triphosphate concentrations in the regulation of influenza virus replication and transcription. J. Virol. 2008, 82, 6902–6910. [Google Scholar] [CrossRef]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- Carrique, L.; Fan, H.; Walker, A.P.; Keown, J.R.; Sharps, J.; Staller, E.; Barclay, W.S.; Fodor, E.; Grimes, J.M. Host ANP32A mediates the assembly of the influenza virus replicase. Nature 2020, 587, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Mizumoto, K.; Ishihama, A. Identification of the 5′ terminal structure of influenza virus genome RNA by a newly developed enzymatic method. Virus Res. 1998, 55, 199–206. [Google Scholar] [CrossRef] [PubMed]
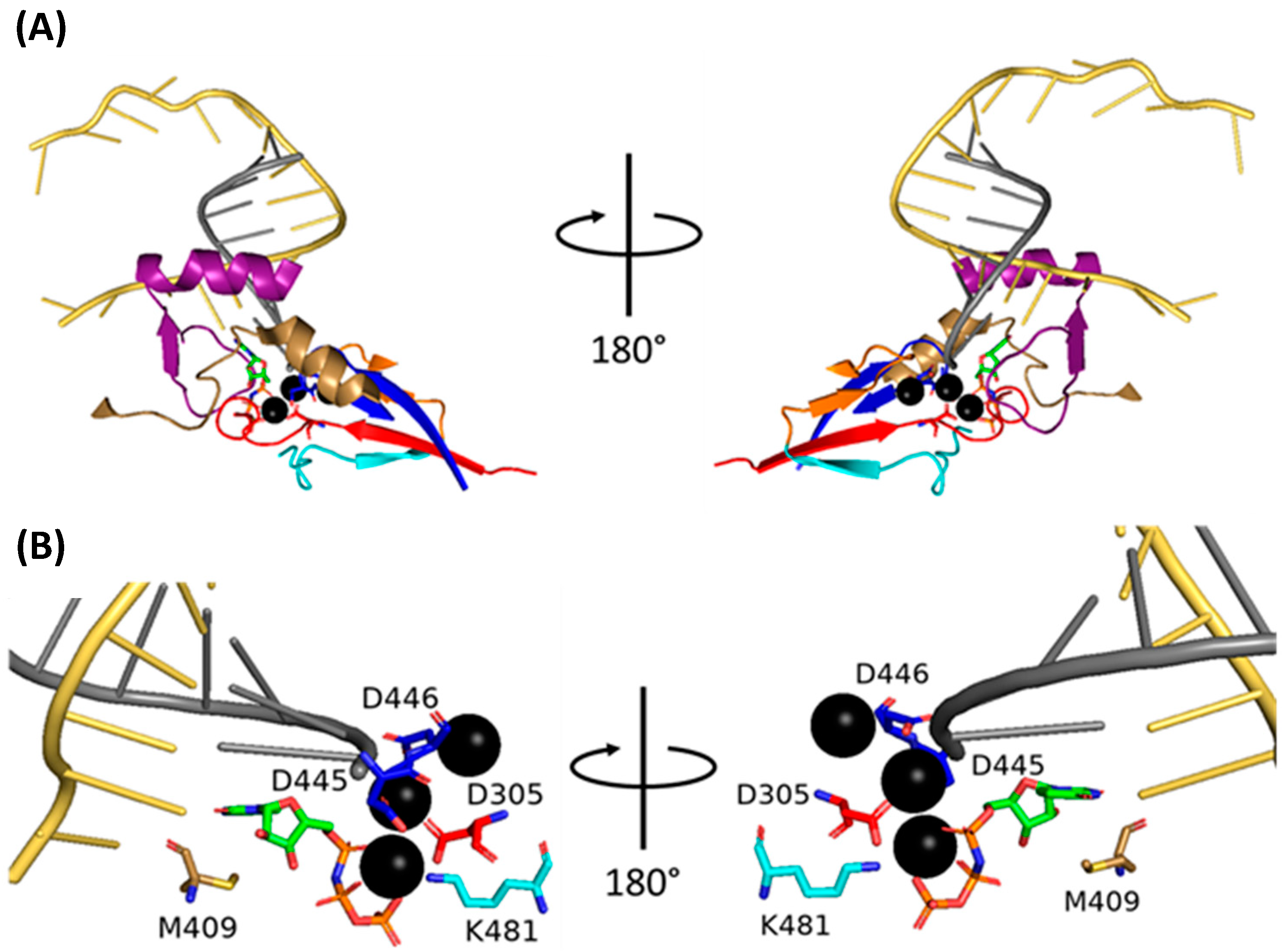
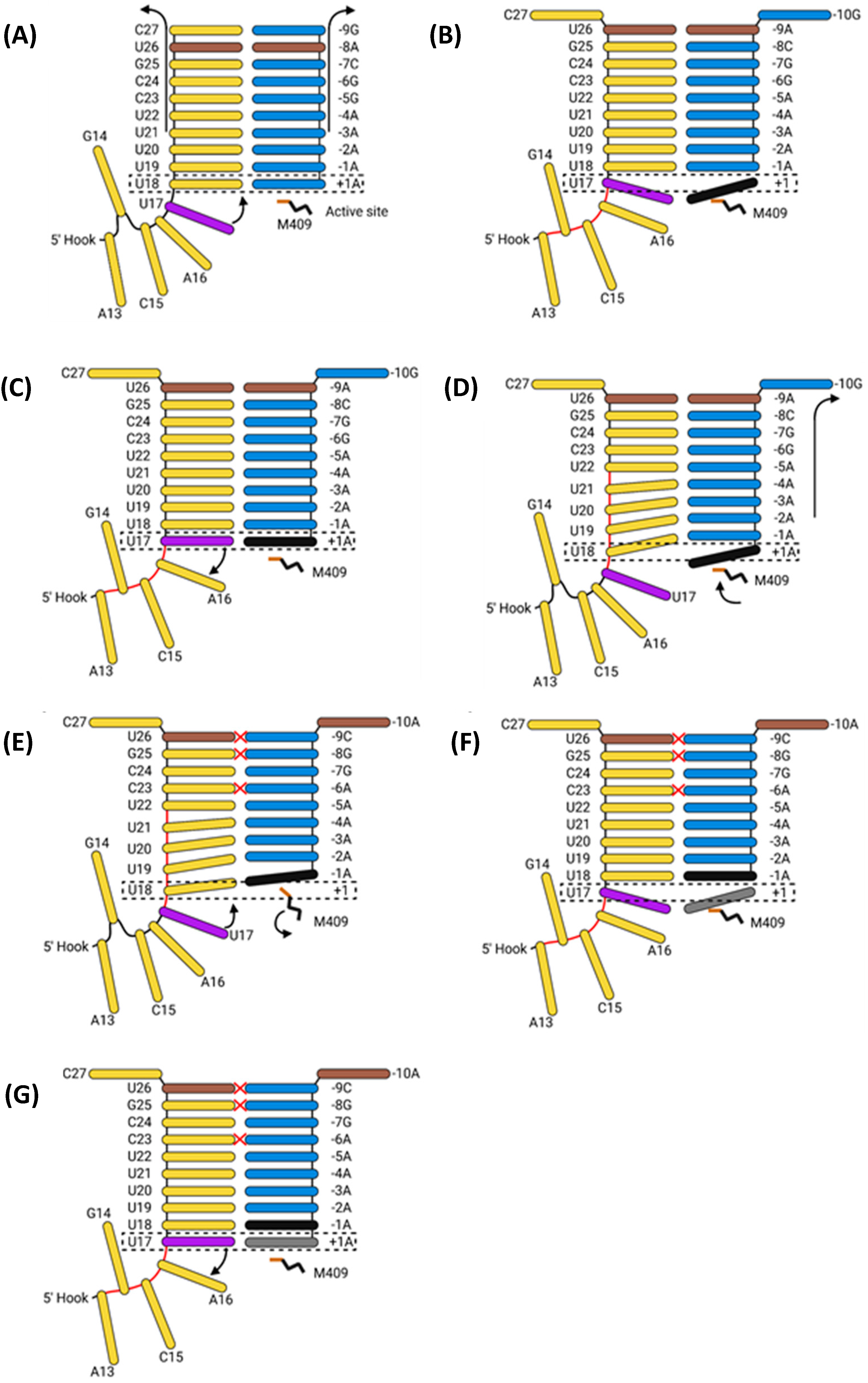
- Velthuis, A.J.W.T.; Robb, N.C.; Kapanidis, A.N.; Fodor, E. The role of the priming loop in influenza A virus RNA synthesis. Nat. Microbiol. 2016, 1, 16029. [Google Scholar] [CrossRef] [PubMed]
- Oymans, J.; Velthuis, A.J.W.T. A Mechanism for Priming and Realignment during Influenza A Virus Replication. J. Virol. 2018, 92, e01773-17. [Google Scholar] [CrossRef] [PubMed]
- Prokudina-Kantorovich, E.; Semenova, N. Intracellular oligomerization of influenza virus nucleoprotein. Virology 1996, 223, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Krug, R.M. Influenza virus RNA replication in vitro: Synthesis of viral template RNAs and virion RNAs in the absence of an added primer. J. Virol. 1988, 62, 2285–2290. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.; Medcalf, L.; Bishop, K.; Harrison, D.; Digard, P. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J. Virol. 1999, 73, 7357–7367. [Google Scholar] [CrossRef]
- Tang, Y.-S.; Xu, S.; Chen, Y.-W.; Wang, J.-H.; Shaw, P.-C. Crystal structures of influenza nucleoprotein complexed with nucleic acid provide insights into the mechanism of RNA interaction. Nucleic Acids Res. 2021, 49, 4144–4154. [Google Scholar] [CrossRef]
- Chenavier, F.; Estrozi, L.F.; Teulon, J.-M.; Zarkadas, E.; Freslon, L.-L.; Pellequer, J.-L.; Ruigrok, R.W.H.; Schoehn, G.; Ballandras-Colas, A.; Crépin, T. Cryo-EM structure of influenza helical nucleocapsid reveals NP-NP and NP-RNA interactions as a model for the genome encapsidation. Sci. Adv. 2023, 9, eadj9974. [Google Scholar] [CrossRef]
- Chenavas, S.; Estrozi, L.F.; Slama-Schwok, A.; Delmas, B.; Di Primo, C.; Baudin, F.; Li, X.; Crépin, T.; Ruigrok, R.W.H. Monomeric nucleoprotein of influenza A virus. PLoS Pathog. 2013, 9, e1003275. [Google Scholar] [CrossRef]
- Mondal, A.; Potts, G.K.; Dawson, A.R.; Coon, J.J.; Mehle, A. Phosphorylation at the homotypic interface regulates nucleoprotein oligomerization and assembly of the influenza virus replication machinery. PLoS Pathog. 2015, 11, e1004826. [Google Scholar] [CrossRef]
- Turrell, L.; Hutchinson, E.C.; Vreede, F.T.; Fodor, E. Regulation of influenza A virus nucleoprotein oligomerization by phosphorylation. J. Virol. 2015, 89, 1452–1455. [Google Scholar] [CrossRef]
- York, A.; Hengrung, N.; Vreede, F.T.; Huiskonen, J.T.; Fodor, E. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc. Natl. Acad. Sci. USA 2013, 110, E4238–E4245. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Vreede, F.T.; Brownlee, G.G. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J. Virol. 2006, 80, 2337–2348. [Google Scholar] [CrossRef]
- Robb, N.C.; Velthuis, A.J.W.T.; Fodor, E.; Kapanidis, A.N. Real-time analysis of single influenza virus replication complexes reveals large promoter-dependent differences in initiation dynamics. Nucleic Acids Res. 2019, 47, 6466–6477. [Google Scholar] [CrossRef]
- Fan, H.; Walker, A.P.; Carrique, L.; Keown, J.R.; Martin, I.S.; Karia, D.; Sharps, J.; Hengrung, N.; Pardon, E.; Steyaert, J.; et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 2019, 573, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Resa-Infante, P.; Jorba, N.; Coloma, R.; Ortin, J. The influenza virus RNA synthesis machine: Advances in its structure and function. RNA Biol. 2011, 8, 207–215. [Google Scholar] [CrossRef]
- Labadie, K.; Afonso, E.D.S.; Rameix-Welti, M.-A.; van der Werf, S.; Naffakh, N. Host-range determinants on the PB2 protein of influenza A viruses control the interaction between the viral polymerase and nucleoprotein in human cells. Virology 2007, 362, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, E.K.; London, W.; Murphy, B.R. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 1993, 67, 1761–1764. [Google Scholar] [CrossRef]
- Staller, E.; Sheppard, C.M.; Neasham, P.J.; Mistry, B.; Peacock, T.P.; Goldhill, D.H.; Long, J.S.; Barclay, W.S. ANP32 Proteins Are Essential for Influenza Virus Replication in Human Cells. J. Virol. 2019, 93, e00217-19. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Zarco, A.R.; Kalayil, S.; Maurin, D.; Salvi, N.; Delaforge, E.; Milles, S.; Jensen, M.R.; Hart, D.J.; Cusack, S.; Blackledge, M. Molecular basis of host-adaptation interactions between influenza virus polymerase PB2 subunit and ANP32A. Nat. Commun. 2020, 11, 3656. [Google Scholar] [CrossRef] [PubMed]
- Brunotte, L.; Flies, J.; Bolte, H.; Reuther, P.; Vreede, F.; Schwemmle, M. The nuclear export protein of H5N1 influenza A viruses recruits Matrix 1 (M1) protein to the viral ribonucleoprotein to mediate nuclear export. J. Biol. Chem. 2014, 289, 20067–20077. [Google Scholar] [CrossRef] [PubMed]
- Chase, G.P.; Rameix-Welti, M.-A.; Zvirbliene, A.; Zvirblis, G.; Götz, V.; Wolff, T.; Naffakh, N.; Schwemmle, M. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog. 2011, 7, e1002187. [Google Scholar] [CrossRef]
- Shimizu, T.; Takizawa, N.; Watanabe, K.; Nagata, K.; Kobayashi, N. Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP. FEBS Lett. 2010, 585, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Bui, M.; Wills, E.G.; Helenius, A.; Whittaker, G.R. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J. Virol. 2000, 74, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Tchatalbachev, S.; Flick, R.; Hobom, G. The packaging signal of influenza viral RNA molecules. RNA 2001, 7, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, N.; Watanabe, K.; Nouno, K.; Kobayashi, N.; Nagata, K. Association of functional influenza viral proteins and RNAs with nuclear chromatin and sub-chromatin structure. Microbes Infect. 2006, 8, 823–833. [Google Scholar] [CrossRef]
- Chua, M.A.; Schmid, S.; Perez, J.T.; Langlois, R.A.; Tenoever, B.R. Influenza A virus utilizes suboptimal splicing to coordinate the timing of infection. Cell Rep. 2013, 3, 23–29. [Google Scholar] [CrossRef]
- Rodriguez Boulan, E.; Sabatini, D.D. Asymmetric budding of viruses in epithelial monlayers: A model system for study of epithelial polarity. Proc. Natl. Acad. Sci. USA 1978, 75, 5071–5075. [Google Scholar] [CrossRef]
- De Castro Martin, I.F.; Fournier, G.; Sachse, M.; Pizarro-Cerda, J.; Risco, C.; Naffakh, N. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat. Commun. 2017, 8, 1396. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Kawakami, E.; Watanabe, T.; Neumann, G.; Kawaoka, Y. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J. Virol. 2011, 85, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Horgan, C.P.; Oleksy, A.; Zhdanov, A.V.; Lall, P.Y.; White, I.J.; Khan, A.R.; Futter, C.E.; McCaffrey, J.G.; McCaffrey, M.W. Rab11-FIP3 is critical for the structural integrity of the endosomal recycling compartment. Traffic 2007, 8, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; DiFiglia, M. The recycling endosome and its role in neurological disorders. Prog. Neurobiol. 2012, 97, 127–141. [Google Scholar] [CrossRef]
- Ullrich, O.; Reinsch, S.; Urbé, S.; Zerial, M.; Parton, R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996, 135, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Veler, H.; Fan, H.; Keown, J.R.; Sharps, J.; Fournier, M.; Grimes, J.M.; Fodor, E. The C-Terminal Domains of the PB2 Subunit of the Influenza A Virus RNA Polymerase Directly Interact with Cellular GTPase Rab11a. J. Virol. 2022, 96, e0197921. [Google Scholar] [CrossRef]
- Kawaguchi, A.; Hirohama, M.; Harada, Y.; Osari, S.; Nagata, K. Influenza Virus Induces Cholesterol-Enriched Endocytic Recycling Compartments for Budozone Formation via Cell Cycle-Independent Centrosome Maturation. PLoS Pathog. 2015, 11, e1005284. [Google Scholar] [CrossRef]
- Bhagwat, A.R.; Le Sage, V.; Nturibi, E.; Kulej, K.; Jones, J.; Guo, M.; Tae Kim, E.; Garcia, B.A.; Weitzman, M.D.; Shroff, H.; et al. Quantitative live cell imaging reveals influenza virus manipulation of Rab11A transport through reduced dynein association. Nat. Commun. 2020, 11, 23. [Google Scholar] [CrossRef]
- Amorim, M.J. A Comprehensive Review on the Interaction Between the Host GTPase Rab11 and Influenza A Virus. Front. Cell Dev. Biol. 2019, 6, 176. [Google Scholar] [CrossRef]
- Chou, Y.-Y.; Vafabakhsh, R.; Doğanay, S.; Gao, Q.; Ha, T.; Palese, P. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc. Natl. Acad. Sci. USA 2012, 109, 9101–9106. [Google Scholar] [CrossRef]
- McGeoch, D.; Fellner, P.; Newton, C. Influenza virus genome consists of eight distinct RNA species. Proc. Natl. Acad. Sci. USA 1976, 73, 3045–3049. [Google Scholar] [CrossRef]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef]
- Noda, T.; Sugita, Y.; Aoyama, K.; Hirase, A.; Kawakami, E.; Miyazawa, A.; Sagara, H.; Kawaoka, Y. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat. Commun. 2012, 3, 639. [Google Scholar] [CrossRef]
- Noda, T.; Murakami, S.; Nakatsu, S.; Imai, H.; Muramoto, Y.; Shindo, K.; Sagara, H.; Kawaoka, Y. Importance of the 1+7 configuration of ribonucleoprotein complexes for influenza A virus genome packaging. Nat. Commun. 2018, 9, 54. [Google Scholar] [CrossRef]
- Bergmann, M.; Muster, T. The relative amount of an influenza A virus segment present in the viral particle is not affected by a reduction in replication of that segment. J. Gen. Virol. 1995, 76, 3211–3215. [Google Scholar] [CrossRef]
- Duhaut, S.; Mccauley, J. Defective RNAs inhibit the assembly of influenza virus genome segments in a segment-specific manner. Virology 1996, 216, 326–337. [Google Scholar] [CrossRef]
- Afonso, E.D.S.; Escriou, N.; Leclercq, I.; van der Werf, S.; Naffakh, N. The generation of recombinant influenza A viruses expressing a PB2 fusion protein requires the conservation of a packaging signal overlapping the coding and noncoding regions at the 5′ end of the PB2 segment. Virology 2005, 341, 34–46. [Google Scholar] [CrossRef]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Fujii, Y.; Noda, T.; Muramoto, Y.; Watanabe, T.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005, 79, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ozawa, M.; Iwatsuki-Horimoto, K.; Horimoto, T.; Kawaoka, Y. Incorporation of influenza A virus genome segments does not absolutely require wild-type sequences. J. Gen. Virol. 2009, 90, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, C.; Yver, M.; Isel, C.; Smyth, R.P.; Rosa-Calatrava, M.; Lina, B.; Moulès, V.; Marquet, R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc. Natl. Acad. Sci. USA 2013, 110, 16604–16609. [Google Scholar] [CrossRef]
- Gog, J.R.; Afonso, E.D.S.; Dalton, R.M.; Leclercq, I.; Tiley, L.; Elton, D.; von Kirchbach, J.C.; Naffakh, N.; Escriou, N.; Digard, P. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res. 2007, 35, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Muramoto, Y.; Noda, T.; Kawaoka, Y. The genome-packaging signal of the influenza A virus genome comprises a genome incorporation signal and a genome-bundling signal. J. Virol. 2013, 87, 11316–11322. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hong, Y.; Parslow, T.G. cis-Acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Huang, T.; Ly, H.; Parslow, T.G.; Liang, Y. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J. Virol. 2008, 82, 229–236. [Google Scholar] [CrossRef]
- Marsh, G.A.; Hatami, R.; Palese, P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 2007, 81, 9727–9736. [Google Scholar] [CrossRef]
- Muramoto, Y.; Takada, A.; Fujii, K.; Noda, T.; Iwatsuki-Horimoto, K.; Watanabe, S.; Horimoto, T.; Kida, H.; Kawaoka, Y. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J. Virol. 2006, 80, 2318–2325. [Google Scholar] [CrossRef]
- Ozawa, M.; Fujii, K.; Muramoto, Y.; Yamada, S.; Yamayoshi, S.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007, 81, 30–41. [Google Scholar] [CrossRef]
- Ozawa, M.; Maeda, J.; Iwatsuki-Horimoto, K.; Watanabe, S.; Goto, H.; Horimoto, T.; Kawaoka, Y. Nucleotide sequence requirements at the 5′ end of the influenza A virus M RNA segment for efficient virus replication. J. Virol. 2009, 83, 3384–3388. [Google Scholar] [CrossRef][Green Version]
- Watanabe, T.; Watanabe, S.; Noda, T.; Fujii, Y.; Kawaoka, Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 2003, 77, 10575–10583. [Google Scholar] [CrossRef]
- Wise, H.M.; Barbezange, C.; Jagger, B.W.; Dalton, R.M.; Gog, J.R.; Curran, M.D.; Taubenberger, J.K.; Anderson, E.C.; Digard, P. Overlapping signals for translational regulation and packaging of influenza A virus segment 2. Nucleic Acids Res. 2011, 39, 7775–7790. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gu, M.; Zheng, Q.; Gao, R.; Liu, X. Packaging signal of influenza A virus. Virol. J. 2021, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.-C.; Sirbat, J.-D.; Cavalier, A.; Rolland, J.-P.; Thomas, D.; Lina, B.; Isel, C.; et al. Interaction network linking the human H3N2 influenza A virus genomic RNA segments. Vaccine 2012, 30, 7359–7367. [Google Scholar] [CrossRef]
- Jakob, C.; Paul-Stansilaus, R.; Schwemmle, M.; Marquet, R.; Bolte, H. The influenza A virus genome packaging network—Complex, flexible and yet unsolved. Nucleic Acids Res. 2022, 50, 9023–9038. [Google Scholar] [CrossRef]
- Moreira, A.; Weber, A.; Bolte, H.; Kolesnikova, L.; Giese, S.; Lakdawala, S.; Beer, M.; Zimmer, G.; García-Sastre, A.; Schwemmle, M.; et al. A conserved influenza A virus nucleoprotein code controls specific viral genome packaging. Nat. Commun. 2016, 7, 12861. [Google Scholar] [CrossRef] [PubMed]
- Bolte, H.; Rosu, M.E.; Hagelauer, E.; García-Sastre, A.; Schwemmle, M. Packaging of the Influenza Virus Genome Is Governed by a Plastic Network of RNA- and Nucleoprotein-Mediated Interactions. J. Virol. 2019, 93, e01861-18. [Google Scholar] [CrossRef]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Gavazzi, C.; Isel, C.; Fournier, E.; Moules, V.; Cavalier, A.; Thomas, D.; Lina, B.; Marquet, R. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: Comparison with a human H3N2 virus. Nucleic Acids Res. 2012, 41, 1241–1254. [Google Scholar] [CrossRef]
- Gilmore, R.; Walter, P.; Blobel, G. Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J. Cell Biol. 1982, 95, 470–477. [Google Scholar] [CrossRef]
- Walter, P.; Blobel, G. Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J. Cell Biol. 1981, 91, 557–561. [Google Scholar] [CrossRef]
- Berg, B.v.D.; Clemons, W.M.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein-conducting channel. Nature 2003, 427, 36–44. [Google Scholar] [CrossRef]
- Oliver, J.D.; Roderick, H.L.; Llewellyn, D.H.; High, S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol. Biol. Cell 1999, 10, 2573–2582. [Google Scholar] [CrossRef]
- Sousa, M.; Parodi, A.J. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 1995, 14, 4196–4203. [Google Scholar] [CrossRef]
- Tatu, U.; Hammond, C.; Helenius, A. Folding and oligomerization of influenza hemagglutinin in the ER and the intermediate compartment. EMBO J. 1995, 14, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Hoover-Litty, H.; Wagner, K.R.; Helenius, A. Folding of influenza hemagglutinin in the endoplasmic reticulum. J. Cell Biol. 1991, 114, 401–411. [Google Scholar] [CrossRef]
- Braakman, I.; Helenius, J.; Helenius, A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J. 1992, 11, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.J.; Henneberry, J.M.; Sambrook, J.F.; Gething, M.J. Glycosylation requirements for intracellular transport and function of the hemagglutinin of influenza virus. J. Virol. 1992, 66, 7136–7145. [Google Scholar] [CrossRef] [PubMed]
- Gething, M.-J.; McCammon, K.; Sambrook, J. Expression of wild-type and mutant forms of influenza hemagglutinin: The role of folding in intracellular transport. Cell 1986, 46, 939–950. [Google Scholar] [CrossRef]
- Bos, T.J.; Davis, A.R.; Nayak, D.P. NH2-terminal hydrophobic region of influenza virus neuraminidase provides the signal function in translocation. Proc. Natl. Acad. Sci. USA 1984, 81, 2327–2331. [Google Scholar] [CrossRef]
- Wang, N.; Glidden, E.J.; Murphy, S.R.; Pearse, B.R.; Hebert, D.N. The cotranslational maturation program for the type II membrane glycoprotein influenza neuraminidase. J. Biol. Chem. 2008, 283, 33826–33837. [Google Scholar] [CrossRef]
- Hull, J.D.; Gilmore, R.; Lamb, R.A. Integration of a small integral membrane protein, M2, of influenza virus into the endoplasmic reticulum: Analysis of the internal signal-anchor domain of a protein with an ectoplasmic NH2 terminus. J. Cell Biol. 1988, 106, 1489–1498. [Google Scholar] [CrossRef]
- Martinez-Gil, L.; Mingarro, I. Viroporins, Examples of the Two-Stage Membrane Protein Folding Model. Viruses 2015, 7, 3462–3482. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- OuYang, B.; Chou, J.J. The minimalist architectures of viroporins and their therapeutic implications. Biochim. Biophys. Acta 2014, 1838, 1058–1067. [Google Scholar] [CrossRef]
- Georgieva, E.R.; Borbat, P.P.; Norman, H.D.; Freed, J.H. Mechanism of influenza A M2 transmembrane domain assembly in lipid membranes. Sci. Rep. 2015, 5, 11757. [Google Scholar] [CrossRef] [PubMed]
- Barlowe, C. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Hauri, H.-P.; Schweizer, A. The endoplasmic reticulum—Golgi intermediate compartment. Curr. Opin. Cell Biol. 1992, 4, 600–608. [Google Scholar] [CrossRef]
- McCaughey, J.; Stephens, D.J. COPII-dependent ER export in animal cells: Adaptation and control for diverse cargo. Histochem. Cell Biol. 2018, 150, 119–131. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hauri, H.-P. The ER-Golgi intermediate compartment (ERGIC): In search of its identity and function. J. Cell Sci. 2006, 119, 2173–2183. [Google Scholar] [CrossRef]
- Ben-Tekaya, H.; Miura, K.; Pepperkok, R.; Hauri, H.-P. Live imaging of bidirectional traffic from the ERGIC. J. Cell Sci. 2005, 118, 357–367. [Google Scholar] [CrossRef]
- Klenk, H.-D.; Rott, R.; Orlich, M.; Blödorn, J. Activation of influenza A viruses by trypsin treatment. Virology 1975, 68, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.T.; Rott, R.; Klenk, H.-D. Influenza viruses cause hemolysis and fusion of cells. Virology 1981, 110, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Vey, M.; Orlich, M.; Adler, S.; Klenk, H.-D.; Rott, R.; Garten, W. Hemagglutinin activation of pathogenic avian influenza viruses of serotype H7 requires the protease recognition motif R-X-K/R-R. Virology 1992, 188, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Nao, N.; Yamagishi, J.; Miyamoto, H.; Igarashi, M.; Manzoor, R.; Ohnuma, A.; Tsuda, Y.; Furuyama, W.; Shigeno, A.; Kajihara, M.; et al. Genetic Predisposition to Acquire a Polybasic Cleavage Site for Highly Pathogenic Avian Influenza Virus Hemagglutinin. mBio 2017, 8, e02298-16. [Google Scholar] [CrossRef]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef]
- Böttcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.-D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef]
- Schalken, J.A.; Roebroek, A.J.; Oomen, P.P.; Wagenaar, S.S.; Debruyne, F.M.; Bloemers, H.P.; Van de Ven, W.J. fur gene expression as a discriminating marker for small cell and nonsmall cell lung carcinomas. J. Clin. Investig. 1987, 80, 1545–1549. [Google Scholar] [CrossRef]
- Stieneke-Gröber, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.; Garten, W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar] [CrossRef]
- Hughey, P.G.; Compans, R.W.; Zebedee, S.L.; Lamb, R.A. Expression of the influenza A virus M2 protein is restricted to apical surfaces of polarized epithelial cells. J. Virol. 1992, 66, 5542–5552. [Google Scholar] [CrossRef]
- Jones, L.V.; Compans, R.W.; Davis, A.R.; Bos, T.J.; Nayak, D.P. Surface expression of influenza virus neuraminidase, an amino-terminally anchored viral membrane glycoprotein, in polarized epithelial cells. Mol. Cell. Biol. 1985, 5, 2181–2189. [Google Scholar]
- Roth, M.G.; Compans, R.W.; Giusti, L.; Davis, A.R.; Nayak, D.P.; Gething, M.-J.; Sambrook, J. Influenza virus hemagglutinin expression is polarized in cells infected with recombinant SV40 viruses carrying cloned hemagglutinin DNA. Cell 1983, 33, 435–443. [Google Scholar] [CrossRef]
- Lin, S.; Naim, H.Y.; Rodriguez, A.C.; Roth, M.G. Mutations in the middle of the transmembrane domain reverse the polarity of transport of the influenza virus hemagglutinin in MDCK epithelial cells. J. Cell Biol. 1998, 142, 51–57. [Google Scholar] [CrossRef]
- Barman, S.; Nayak, D.P. Analysis of the transmembrane domain of influenza virus neuraminidase, a type II transmembrane glycoprotein, for apical sorting and raft association. J. Virol. 2000, 74, 6538–6545. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J.; Christensen, I.B.; Mogensen, E.N.; Damkier, H.H.; Praetorius, J.; Gallo, L.I.; Dalghi, M.G.; Clayton, D.R.; Ruiz, W.G.; et al. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Grantham, M.L.; Stewart, S.M.; Lalime, E.N.; Pekosz, A. Tyrosines in the influenza A virus M2 protein cytoplasmic tail are critical for production of infectious virus particles. J. Virol. 2010, 84, 8765–8776. [Google Scholar] [CrossRef] [PubMed]
- Palese, P.; Tobita, K.; Ueda, M.; Compans, R.W. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 1974, 61, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Leser, G.P.; Ma, C.; Lamb, R.A. The amphipathic helix of influenza A virus M2 protein is required for filamentous bud formation and scission of filamentous and spherical particles. J. Virol. 2013, 87, 9973–9982. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza virus hemagglutinin concentrates in lipid raft microdomains for efficient viral fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef]
- Kordyukova, L.V.; Konarev, P.V.; Fedorova, N.V.; Shtykova, E.V.; Ksenofontov, A.L.; Loshkarev, N.A.; Dadinova, L.A.; Timofeeva, T.A.; Abramchuk, S.S.; Moiseenko, A.V.; et al. The Cytoplasmic Tail of Influenza A Virus Hemagglutinin and Membrane Lipid Composition Change the Mode of M1 Protein Association with the Lipid Bilayer. Membranes 2021, 11, 772. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef]
- Rodriguez-Boulan, E.; Kreitzer, G.; Müsch, A. Organization of vesicular trafficking in epithelia. Nat. Rev. Mol. Cell Biol. 2005, 6, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Scheiffele, P.; Rietveld, A.; Wilk, T.; Simons, K. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 1999, 274, 2038–2044. [Google Scholar] [CrossRef] [PubMed]
- Scheiffele, P.; Roth, M.G.; Simons, K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997, 16, 5501–5508. [Google Scholar] [CrossRef]
- Zhang, J.; Pekosz, A.; Lamb, R.A. Influenza virus assembly and lipid raft microdomains: A role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 2000, 74, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Takeda, M.; Lamb, R.A. Influenza virus hemagglutinin (H3 subtype) requires palmitoylation of its cytoplasmic tail for assembly: M1 proteins of two subtypes differ in their ability to support assembly. J. Virol. 2005, 79, 13673–13684. [Google Scholar] [CrossRef]
- Veit, M.; Thaa, B. Association of influenza virus proteins with membrane rafts. Adv. Virol. 2011, 2011, 370606. [Google Scholar] [CrossRef]
- Marjuki, H.; Alam, M.I.; Ehrhardt, C.; Wagner, R.; Planz, O.; Klenk, H.D.; Ludwig, S.; Pleschka, S. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Calpha-mediated activation of ERK signaling. J. Biol. Chem. 2006, 281, 16707–16715. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Barge, A.; Durrer, P.; Brunner, J.; Ma, K.; Whittaker, G.R. Membrane interaction of influenza virus M1 protein. Virology 2000, 267, 289–298. [Google Scholar] [CrossRef]
- Wang, D.; Harmon, A.; Jin, J.; Francis, D.H.; Christopher-Hennings, J.; Nelson, E.; Montelaro, R.C.; Li, F. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J. Virol. 2010, 84, 4673–4681. [Google Scholar] [CrossRef]
- Chen, B.J.; Leser, G.P.; Morita, E.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007, 81, 7111–7123. [Google Scholar] [CrossRef]
- McCown, M.F.; Pekosz, A. Distinct domains of the influenza a virus M2 protein cytoplasmic tail mediate binding to the M1 protein and facilitate infectious virus production. J. Virol. 2006, 80, 8178–8189. [Google Scholar] [CrossRef]
- Chen, B.J.; Leser, G.P.; Jackson, D.; Lamb, R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008, 82, 10059–10070. [Google Scholar] [CrossRef]
- Elkins, M.R.; Williams, J.K.; Gelenter, M.D.; Dai, P.; Kwon, B.; Sergeyev, I.V.; Pentelute, B.L.; Hong, M. Cholesterol-binding site of the influenza M2 protein in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 2017, 114, 12946–12951. [Google Scholar] [CrossRef]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef]
- Noton, S.L.; Medcalf, E.; Fisher, D.; Mullin, A.E.; Elton, D.; Digard, P. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J. Gen. Virol. 2007, 88, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Liu, T.; Offringa, D.P.; McInnis, J.; Levandowski, R.A. Association of influenza virus matrix protein with ribonucleoproteins. J. Virol. 1999, 73, 7467–7473. [Google Scholar] [CrossRef] [PubMed]
- Nermut, M.V. Further investigation on the fine structure of influenza virus. J. Gen. Virol. 1972, 17, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.; Calder, L.J.; Wharton, S.A. Electron microscopy of the influenza virus submembranal structure. Virology 1989, 173, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, N.G. Electron microscopy of influenza virus. Br. Med. Bull. 1979, 35, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Peukes, J.; Xiong, X.; Briggs, J.A.G. New structural insights into the multifunctional influenza A matrix protein 1. FEBS Lett. 2021, 595, 2535–2543. [Google Scholar] [CrossRef]
- Martyna, A.; Bahsoun, B.; Madsen, J.J.; Jackson, F.S.J.S.; Badham, M.D.; Voth, G.A.; Rossman, J.S. Cholesterol Alters the Orientation and Activity of the Influenza Virus M2 Amphipathic Helix in the Membrane. J. Phys. Chem. B 2020, 124, 6738–6747. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Tomana, M.; Compans, R.W. Sialic acid is incorporated into influenza hemagglutinin glycoproteins in the absence of viral neuraminidase. Virus Res. 1985, 2, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Zhang, X.-Q.; Senaati, H.P.; Chen, H.-W.; Varki, N.M.; Schooley, R.T.; Gagneux, P. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol. J. 2013, 10, 321. [Google Scholar] [CrossRef] [PubMed]




























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carter, T.; Iqbal, M. The Influenza A Virus Replication Cycle: A Comprehensive Review. Viruses 2024, 16, 316. https://doi.org/10.3390/v16020316
Carter T, Iqbal M. The Influenza A Virus Replication Cycle: A Comprehensive Review. Viruses. 2024; 16(2):316. https://doi.org/10.3390/v16020316
Chicago/Turabian StyleCarter, Toby, and Munir Iqbal. 2024. "The Influenza A Virus Replication Cycle: A Comprehensive Review" Viruses 16, no. 2: 316. https://doi.org/10.3390/v16020316
APA StyleCarter, T., & Iqbal, M. (2024). The Influenza A Virus Replication Cycle: A Comprehensive Review. Viruses, 16(2), 316. https://doi.org/10.3390/v16020316