

Phylogenetics, Epidemiology and Temporal Patterns of Dengue Virus in Araraquara, São Paulo State
 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Population
2.3. DENV RNA Detection and Sample Selection for Next-Generation Sequencing (NGS)
2.4. DENV-1 Complete Genome Amplification
2.5. Bioinformatics Workflow and Consensus Generation
2.6. Envelope (E) Gene Sequencing
2.7. Multiple Sequence Alignment and ML Phylogenetic Reconstruction
2.8. Coalescent Analysis and Molecular Clock
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Dengue and Severe Dengue. Available online: https://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue (accessed on 17 March 2023).
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Twiddy, S. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 2003, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zanotto, P.M.; Gould, E.A.; Gao, G.F.; Harvey, P.H.; Holmes, E.C. Population dynamics of flaviviruses revealed by molecular phylogenies. Proc. Natl. Acad. Sci. USA 1996, 93, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Villabona-Arenas, C.J.; de Andrade Zanotto, P.M. Worldwide Spread of Dengue Virus Type 1. PLoS ONE 2013, 8, e62649. [Google Scholar] [CrossRef] [PubMed]
- Ministério da Saúde. Óbito por Arboviroses no Brasil, 2008 a 2019. Available online: http://plataforma.saude.gov.br/anomalias-congenitas/boletim-epidemiologico-SVS-33-2020.pdf (accessed on 25 August 2020).
- Souza, C.S.; Romano, C.M. Dengue in the cooling off period of the COVID-19 epidemic in Brazil: From the shadows to the spotlight. Rev. Inst. Med. Trop. São Paulo 2022, 64, e44. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Santiago, G.A.; Maito, R.M.; Meneses, C.A.R.; Nascimento, V.A.; Souza, V.C.; do Nascimento, F.O.; Silva, D.; Mejía, M.; Gonçalves, L.; et al. Reemergence of Dengue Virus Serotype 3, Brazil, 2023. medRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- de Bruycker-Nogueira, F.; Souza, T.M.A.; Chouin-Carneiro, T.; da Costa Faria, N.R.; Santos, J.B.; Torres, M.C.; Ramalho, I.L.C.; de Aguiar, S.F.; Nogueira, R.M.R.; de Filippis, A.M.B.; et al. DENV-1 Genotype V in Brazil: Spatiotemporal dispersion pattern reveals continuous co-circulation of distinct lineages until 2016. Sci. Rep. 2018, 8, 17160. [Google Scholar] [CrossRef] [PubMed]
- Díaz, Y.; Chen-Germán, M.; Quiroz, E.; Carrera, J.-P.; Cisneros, J.; Moreno, B.; Cerezo, L.; Martinez-Torres, A.O.; Moreno, L.; de Mosca, I.B.; et al. Molecular Epidemiology of Dengue in Panama: 25 Years of Circulation. Viruses 2019, 11, 764. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Chiaravalloti Neto, F.; Mondini, A. Dengue in Araraquara, state of São Paulo: Epidemiology, climate and Aedes aegypti infestation. Rev. Saúde Pública 2018, 52, 18. [Google Scholar] [CrossRef]
- Centro de Vigilância Epidemiológica “Prof. Alexandre Vranjac”. Dados Estatísticos. São Paulo: CVE. 2015. Available online: http://portal.saude.sp.gov.br/cve-centro-devigilancia-epidemiologica-prof.-alexandre-vranjac/areas-de-vigilancia/doencas-de-transmissaopor-vetores-e-zoonoses/agravos/dengue/dados-estatisticos (accessed on 15 September 2016).
- Gularte, J.S.; Sacchetto, L.; Demoliner, M.; Girardi, V.; da Silva, M.S.; Filippi, M.; Pereira, V.M.d.A.G.; Hansen, A.W.; da Silva, L.L.; Fleck, J.D.; et al. DENV-1 genotype V linked to the 2022 dengue epidemic in Southern Brazil. J. Clin. Virol. 2023, 168, 105599. [Google Scholar] [CrossRef]
- BRASIL; Ministério da Saúde; Secretaria de Vigilância em Saúde. Sistema de Informação de Agravos de Notificação (SINAN). Available online: http://www.portalsinan.saude.gov.br/sinan-dengue-chikungunya (accessed on 25 January 2023).
- Luna, E.J.; Figueiredo, G.M.; Levi, J.E.; Campos, S.R.; Felix, A.C.; e Souza, N.S.; Figueiredo, W.M.; Costa, A.A.; Cardoso, M.R.; Pannuti, C.S. A cohort study to assess the incidence of dengue, Brazil, 2014–2018. Acta Trop. 2020, 204, 105313. [Google Scholar] [CrossRef] [PubMed]
- Huhtamo, E.; Hasu, E.; Uzcátegui, N.Y.; Erra, E.; Nikkari, S.; Kantele, A.; Vapalahti, O.; Piiparinen, H. Early diagnosis of dengue in travelers: Comparison of a novel real-time RT-PCR, NS1 antigen detection and serology. J. Clin. Virol. 2010, 47, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Figueiredo, G.; Levi, J.; Campos, S.; Felix, A.; Souza, N.; Figueiredo, W.; Costa, A.; Cardoso, M.; Pannuti, C. Data on dengue incidence in South-eastern Brazil, 2014–2018. Data Brief 2020, 29, 105266. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.D.; Wu, S.-J.L.; Dion-Schultz, A.; Mangold, B.E.; Peruski, L.F.; Watts, D.M.; Porter, K.R.; Murphy, G.R.; Suharyono, W.; King, C.-C.; et al. Development and evaluation of serotype- and group-specific fluorogenic reverse transcriptase PCR (TaqMan) assays for dengue virus. J. Clin. Microbiol. 2001, 39, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Claro, I.M. Em tempo Real, Rápida Detecção e Sequenciamento de Arbovírus No Brasil. Ph.D. Thesis, Universidade de São Paulo, São Paulo, Brazil, 2021. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; A Beutler, N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Di, Y.P. Analysis of RNA Sequencing Data Using CLC Genomics Workbench. Methods Mol. Biol. 2020, 2102, 61–113. [Google Scholar] [CrossRef] [PubMed]
- de Bruycker-Nogueira, F.; Nogueira, R.M.R.; Faria, N.R.d.C.; Simões, J.B.S.; Nunes, P.C.G.; de Filippis, A.M.B.; dos Santos, F.B. Insights of the genetic diversity of DENV-1 detected in Brazil in 25years: Analysis of the envelope domain III allows lineages characterization. Infect. Genet. Evol. 2015, 34, 126–136. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. Seaview version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2009, 27, 221–224. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.3.1. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh. 2010. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 20 July 2020).
- Ferreira, M.A.R.; Suchard, M.A. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 2008, 36, 355–368. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Teoh, B.-T.; Sam, S.-S.; Tan, K.-K.; Johari, J.; Shu, M.-H.; Danlami, M.B.; Abd-Jamil, J.; MatRahim, N.; Mahadi, N.M.; AbuBakar, S. Dengue virus type 1 clade replacement in recurring homotypic outbreaks. BMC Evol. Biol. 2013, 13, 213. [Google Scholar] [CrossRef] [PubMed]
- European Center for Disease Control. Dengue Worldwide Overview. Available online: https://www.ecdc.europa.eu/en/dengue-monthly (accessed on 23 August 2023).
- de Jesus, J.G.; Dutra, K.R.; da Silva Sales, F.C.; Claro, I.M.; Terzian, A.C.; da Silva Candido, D.; Hill, S.C.; Thézé, J.; Torres, C.; D’agostini, T.L.; et al. Genomic detection of a virus lineage replacement event of dengue virus serotype 2 in Brazil, 2019. Mem. Inst. Oswaldo Cruz 2020, 115, e190423. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.C.G.; Sampaio, S.A.F.; da Costa, N.R.; de Mendonça, M.C.L.; da Rocha Queiroz Lima, M.; Araujo, S.E.M.; dos Santos, F.B.; Simões, J.B.S.; de Santis Gonçalves, B.; Nogueira, R.M.R.; et al. Dengue severity associated with age and a new lineage of dengue virus-type 2 during an outbreak in Rio De Janeiro, Brazil. J. Med. Virol. 2016, 88, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- de Souza, U.J.B.; Macedo, Y.d.S.M.; dos Santos, R.N.; Cardoso, F.D.P.; Galvão, J.D.; Gabev, E.E.; Franco, A.C.; Roehe, P.M.; Spilki, F.R.; Campos, F.S. Circulation of Dengue Virus Serotype 1 Genotype V and Dengue Virus Serotype 2 Genotype III in Tocantins State, Northern Brazil, 2021–2022. Viruses 2023, 15, 2136. [Google Scholar] [CrossRef] [PubMed]
- Ministério da Saúde. Monitoramento dos Casos de Arboviroses Até a Semana Epidemiológica 51 de 2022. Available online: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/boletins/epidemiologicos/edicoes/2022/boletim-epidemiologico-vol-53-no48/view (accessed on 29 December 2022).
- Thu, H.M.; Lowry, K.; Jiang, L.; Hlaing, T.; Holmes, E.C.; Aaskov, J. Lineage extinction and replacement in dengue type 1 virus populations are due to stochastic events rather than to natural selection. Virology 2005, 336, 163–172. [Google Scholar] [CrossRef]
- Armstrong, P.M.; Rico-Hesse, R. Efficiency of dengue serotype 2 virus strains to infect and disseminate in Aedes aegypti. Am. J. Trop. Med. Hyg. 2003, 68, 539–544. [Google Scholar] [CrossRef]
- Cologna, R.; Rico-Hesse, R. American genotype structures decrease dengue virus output from human monocytes and dendritic cells. J. Virol. 2003, 77, 3929–3938. [Google Scholar] [CrossRef]
- Tan, C.H.; Hapuarachchi, H.C.; Tan, L.K.; Wong, P.S.J.; Li, M.Z.I.; Wong, W.Y.; Ng, L.C. Lineage Replacement Associated with Fitness Gain in Mammalian Cells and Aedes aegypti: A Catalyst for Dengue Virus Type 2 Transmission. Microorganisms 2022, 10, 1100. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Escoto, A.C.; Huang, A.T.; Garcia-Carreras, B.; Chowdhury, N.; Berry, I.M.; Chavez, C.; Buchy, P.; Duong, V.; Dussart, P.; et al. Antigenic evolution of dengue viruses over 20 years. Science 2021, 374, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Goncalvez, A.P.; Escalante, A.A.; Pujol, F.H.; Ludert, J.E.; Tovar, D.; Salas, R.A.; Liprandi, F. Diversity and evolution of the envelope gene of dengue virus type 1. Virology 2002, 303, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 1990, 174, 479–493. [Google Scholar] [CrossRef]
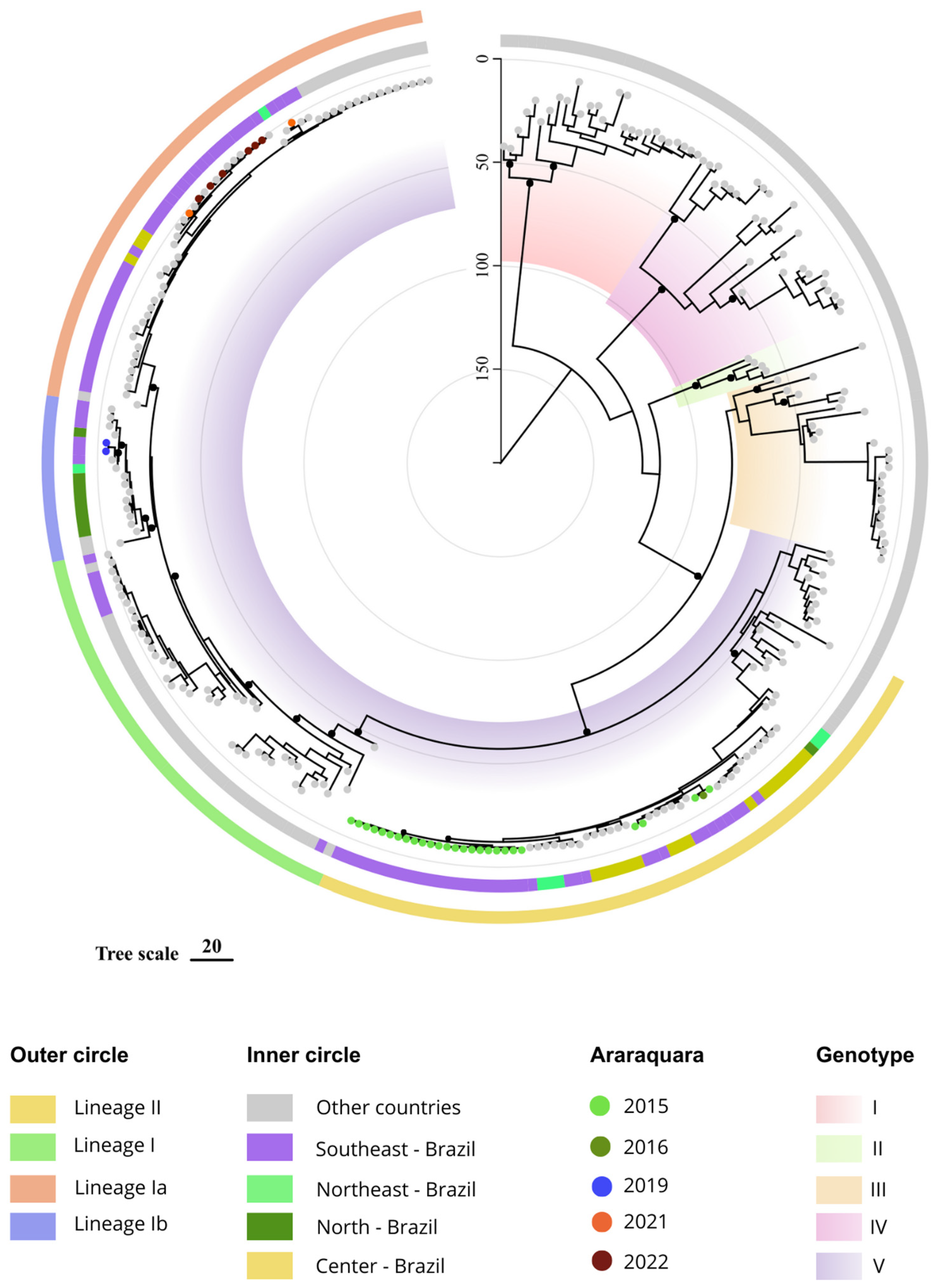

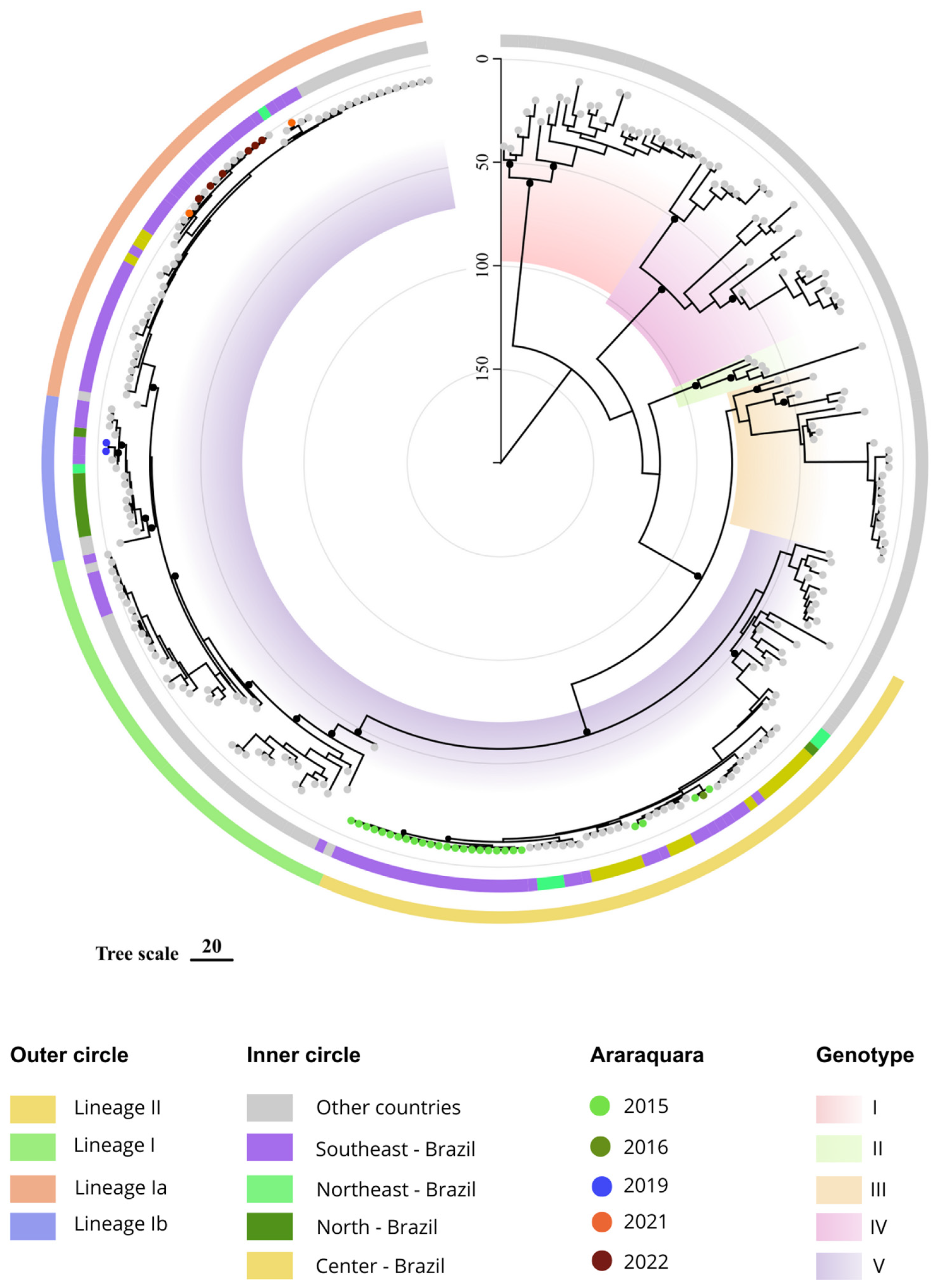{kind=link}
| Group | tMRCA Bayesian Skyline (HPD 95%) | tMRCA Exponential Growth (HPD 95%) |
|---|---|---|
| DENV-1 | 1884 (1833–1920) | 1886 (1842–1916) |
| Genotype I | 1966 (1953–1974) | 1967 (1954–1976) |
| Genotype II | 1931 (1907–1948) | 1930 (1909–1947) |
| Genotype III | 1945 (1927–1958) | 1943 (1926–1955) |
| Genotype IV | 1944 (1921–1959) | 1944 (1923–1959) |
| Genotype V | 1965 (1955–1972) | 1966 (1957–1973) |
| Lineage I | 1977 (1971–1978) | 1976 (1973–1978) |
| Lineage Ia | 2001 (1998–2005) | 2002 (1999–2005) |
| Lineage Ib | 2001 (1998–2005) | 2002 (2000–2005) |
| Lineage II | 1982 (1977–1985) | 1976 (1968–1982) |
| Araraquara 2015 | 2009 (2006–2011) | 2008 (2007–2011) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, C.S.; Caleiro, G.S.; Claro, I.M.; de Jesus, J.G.; Coletti, T.M.; da Silva, C.A.M.; Costa, Â.A.; Inenami, M.; Ribeiro, A.C.; Felix, A.C.; et al. Phylogenetics, Epidemiology and Temporal Patterns of Dengue Virus in Araraquara, São Paulo State. Viruses 2024, 16, 274. https://doi.org/10.3390/v16020274
de Souza CS, Caleiro GS, Claro IM, de Jesus JG, Coletti TM, da Silva CAM, Costa ÂA, Inenami M, Ribeiro AC, Felix AC, et al. Phylogenetics, Epidemiology and Temporal Patterns of Dengue Virus in Araraquara, São Paulo State. Viruses. 2024; 16(2):274. https://doi.org/10.3390/v16020274
Chicago/Turabian Stylede Souza, Caio Santos, Giovana Santos Caleiro, Ingra Morales Claro, Jaqueline Goes de Jesus, Thaís Moura Coletti, Camila Alves Maia da Silva, Ângela Aparecida Costa, Marta Inenami, Andreia C. Ribeiro, Alvina Clara Felix, and et al. 2024. "Phylogenetics, Epidemiology and Temporal Patterns of Dengue Virus in Araraquara, São Paulo State" Viruses 16, no. 2: 274. https://doi.org/10.3390/v16020274
APA Stylede Souza, C. S., Caleiro, G. S., Claro, I. M., de Jesus, J. G., Coletti, T. M., da Silva, C. A. M., Costa, Â. A., Inenami, M., Ribeiro, A. C., Felix, A. C., de Paula, A. V., Figueiredo, W. M., de Albuquerque Luna, E. J., Sabino, E. C., & Romano, C. M. (2024). Phylogenetics, Epidemiology and Temporal Patterns of Dengue Virus in Araraquara, São Paulo State. Viruses, 16(2), 274. https://doi.org/10.3390/v16020274