

Histone H1.2 Inhibited EMCV Replication through Enhancing MDA5-Mediated IFN-β Signaling Pathway

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Mass Spectrometry (MS) Assay
2.3. Plasmids and Antibodies
2.4. RNAi Assay
2.5. EMCV Infection and Infectivity Assays
2.6. RNA Extraction and RT-qPCR
2.7. Co-Immunoprecipitation (Co-IP) Assay
2.8. Nuclear and Cytoplasmic Extraction
2.9. Indirect Immunofluorescent Assay (IFA)
2.10. Statistical Analysis
3. Results
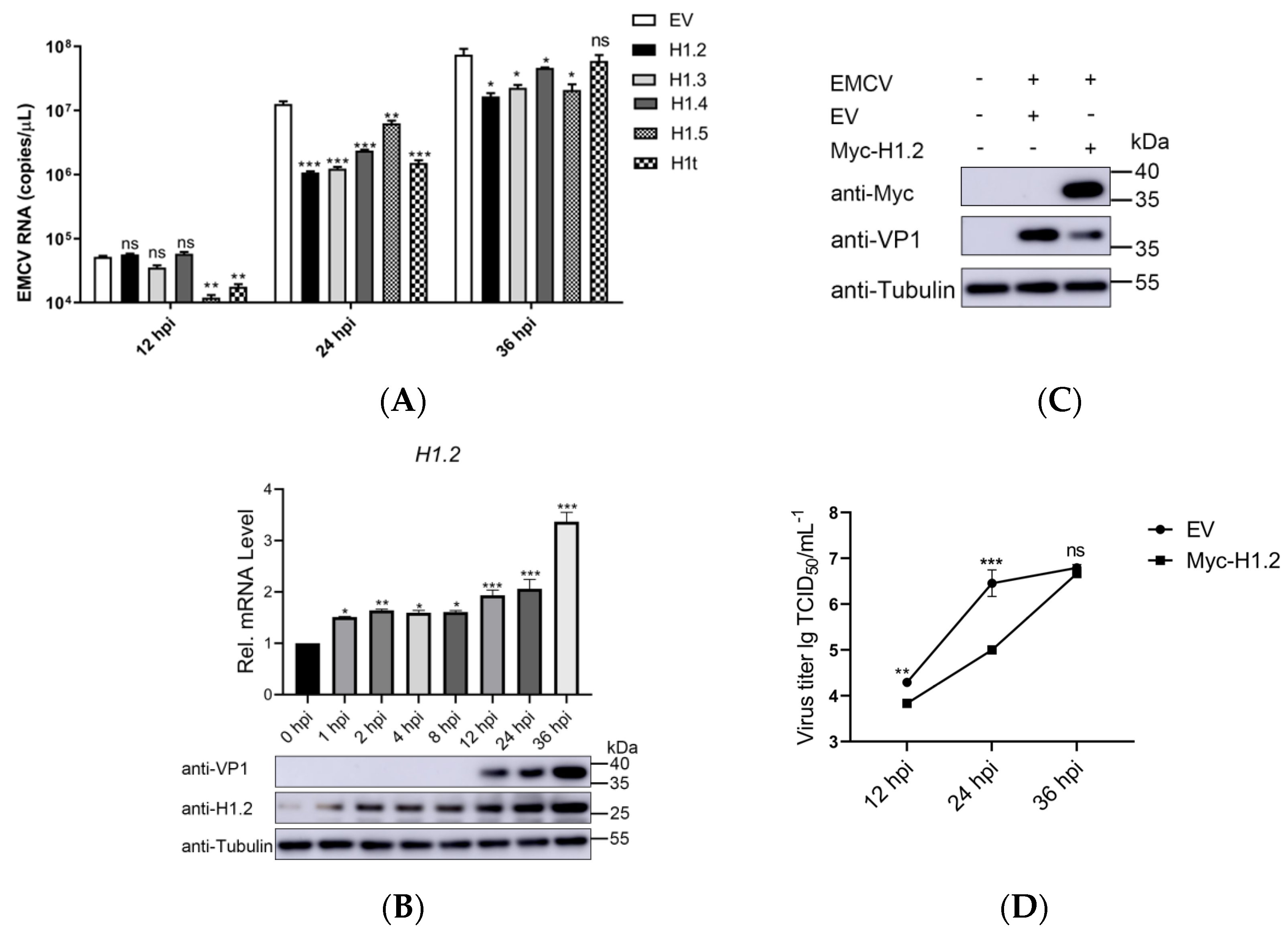
3.1. H1.2 Expression Inhibited EMCV Replication
3.2. Knockdown of H1.2 Promoted EMCV Replication
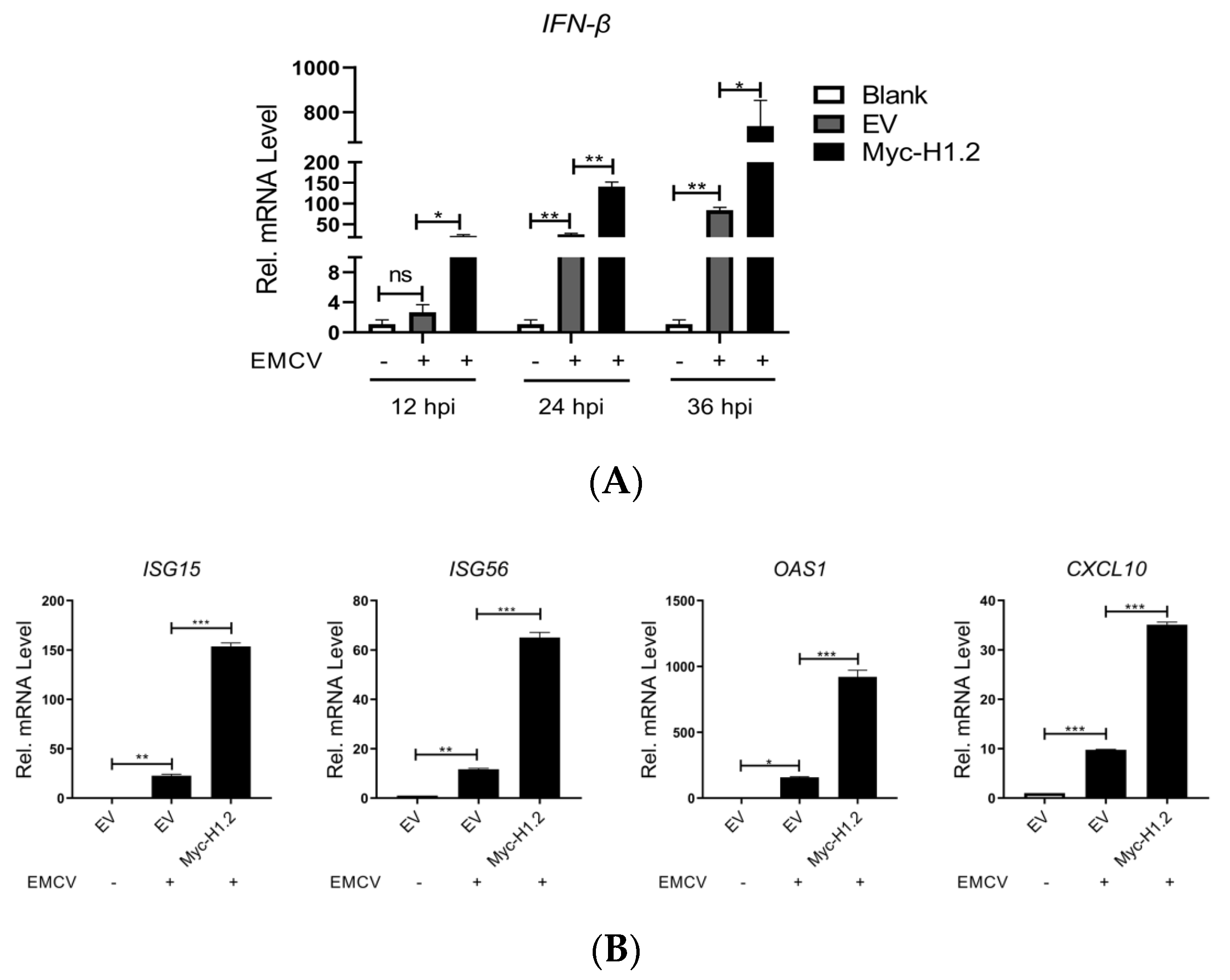
3.3. H1.2 Up-Regulated IFN-β Expression Induced by EMCV
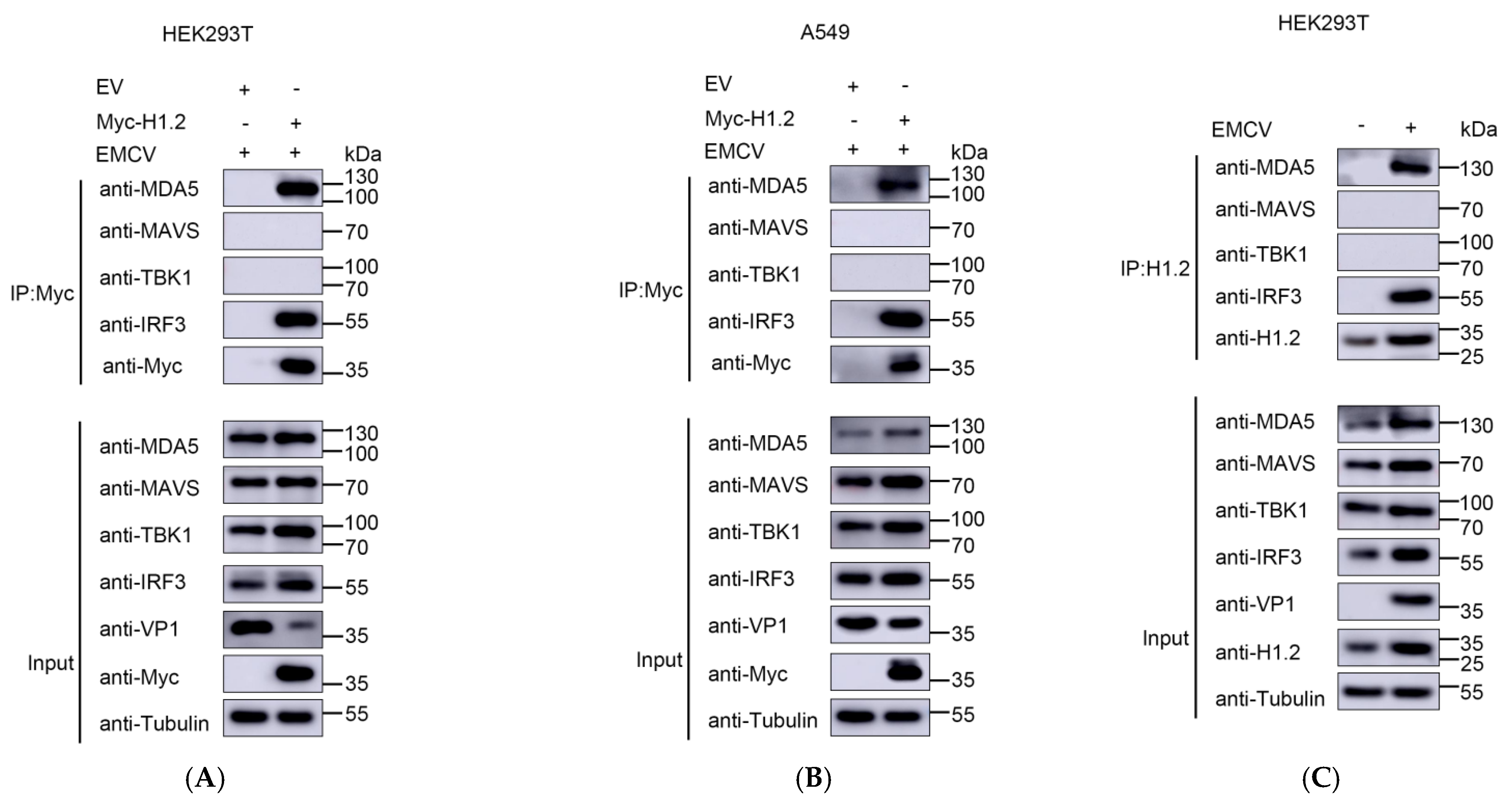
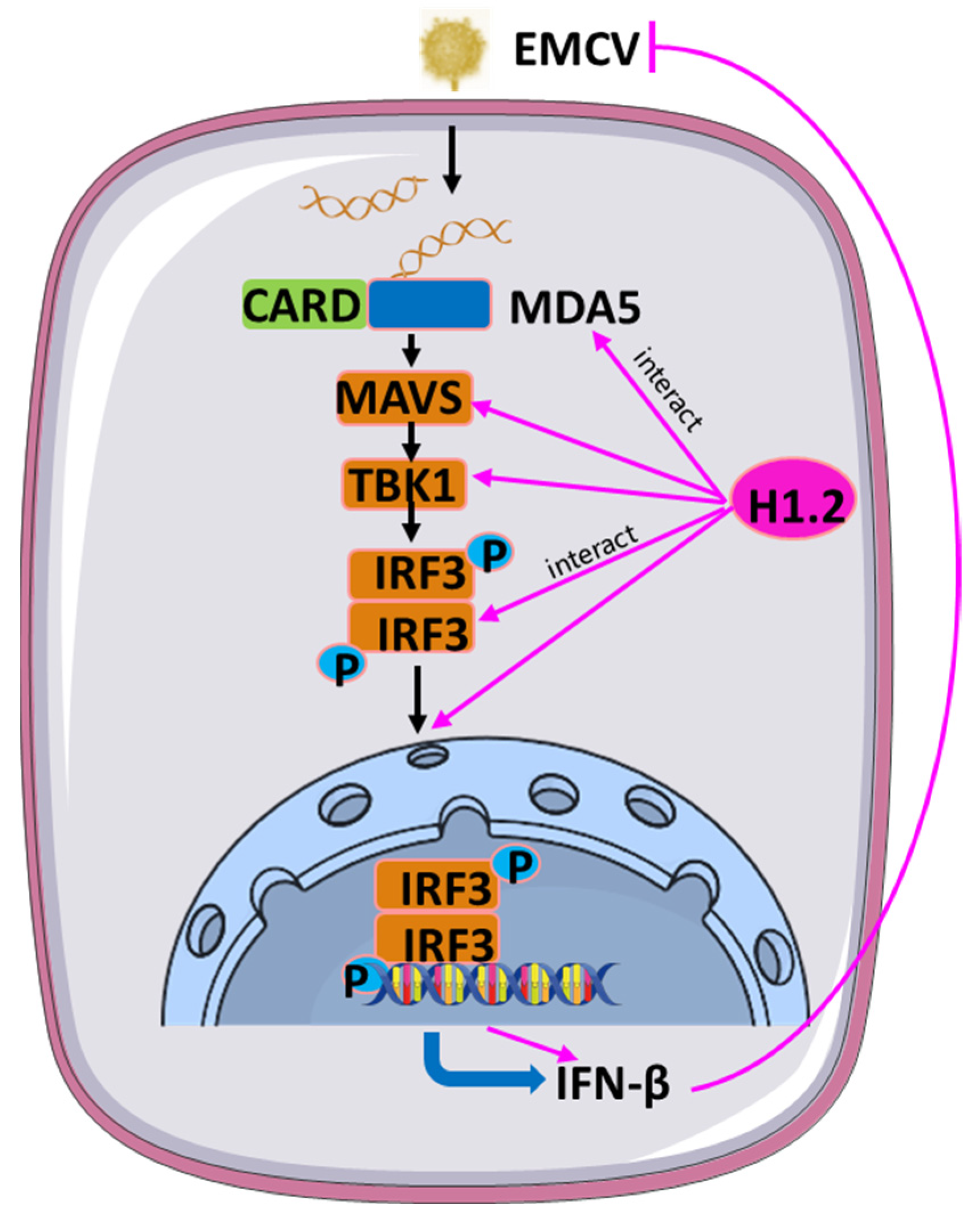
3.4. H1.2 Promoted EMCV-Triggered MDA5 Signaling Pathway
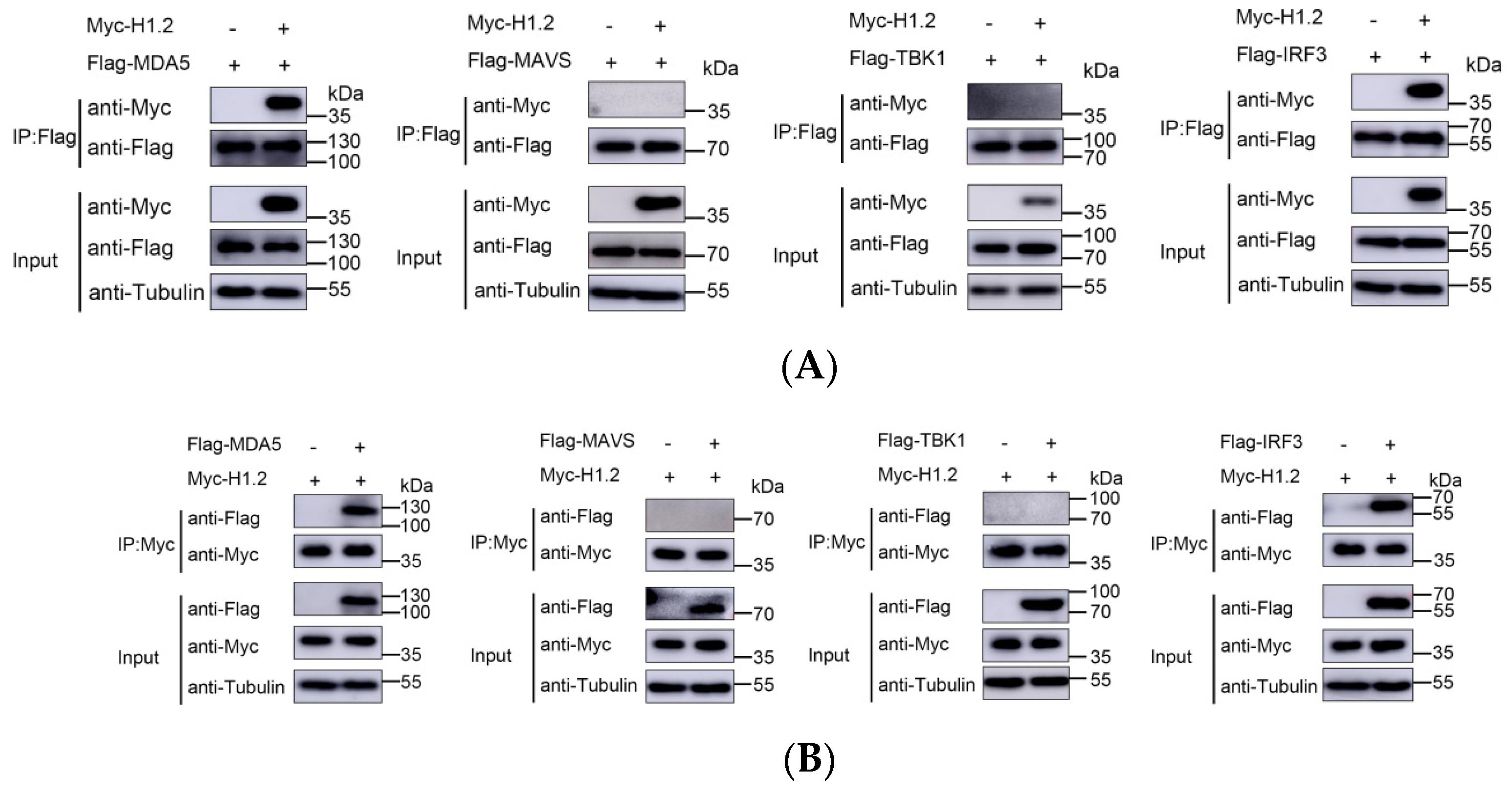
3.5. H1.2 Interacted with MDA5 and IRF3
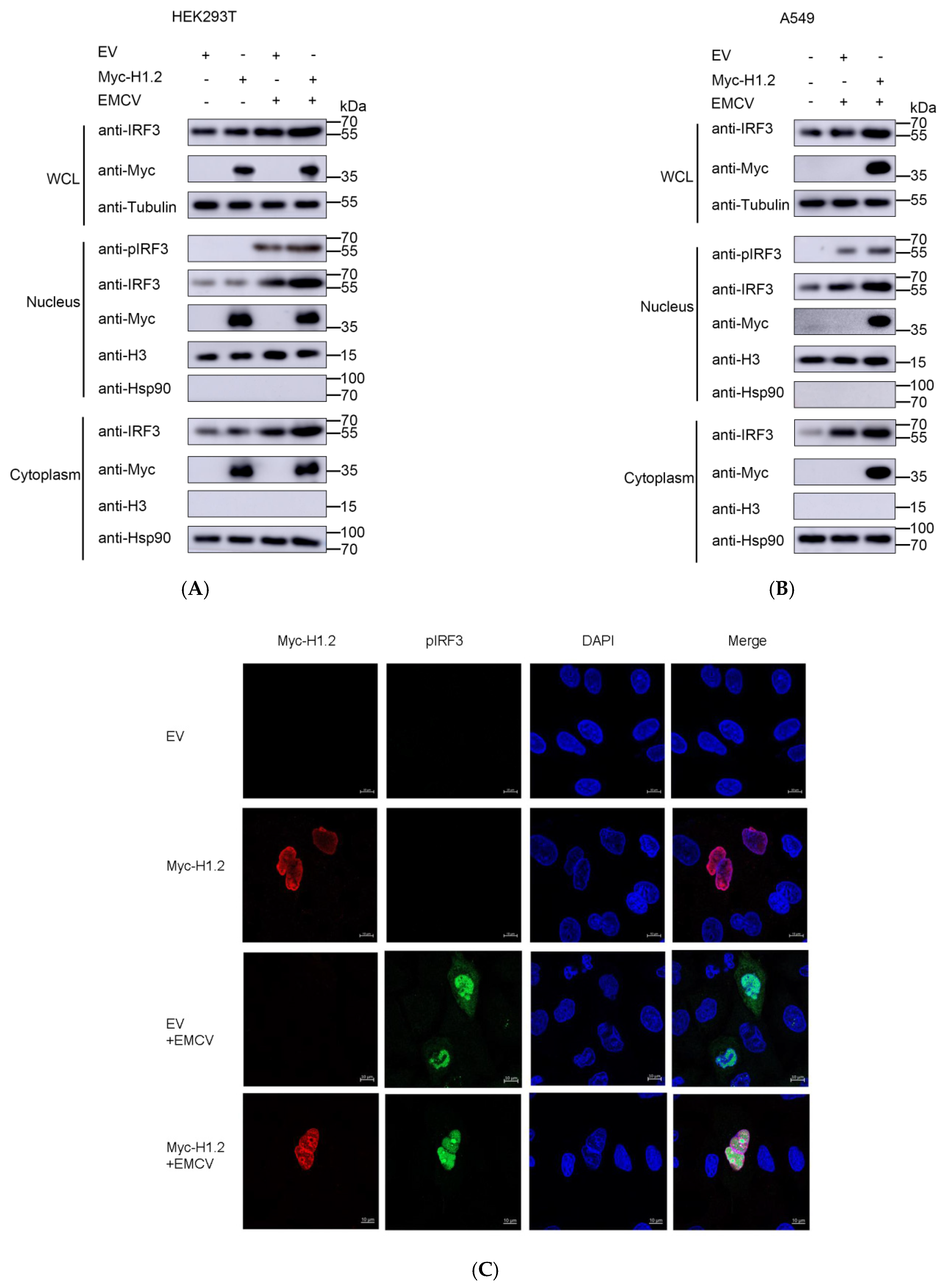
3.6. Overexpression of H1.2 Promoted IRF3 Nuclear Translocation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Czechowicz, J.; Huaman, J.L.; Forshey, B.M.; Morrison, A.C.; Castillo, R.; Huaman, A.; Caceda, R.; Eza, D.; Rocha, C.; Blair, P.J.; et al. Prevalence and Risk Factors for Encephalomyocarditis Virus Infection in Peru. Vector Borne Zoonotic Dis. 2011, 11, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Carocci, M.; Bakkali-Kassimi, L. The Encephalomyocarditis Virus. Virulence 2012, 3, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Cardeti, G.; Mariano, V.; Eleni, C.; Aloisi, M.; Grifoni, G.; Sittinieri, S.; Dante, G.; Antognetti, V.; Foglia, E.A.; Cersini, A.; et al. Encephalomyocarditis virus infection in Macaca sylvanus and Hystrix cristata from an Italian rescue centre for wild and exotic animals. Virol. J. 2016, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Scollo, A.; Mazzoni, C.; Luppi, A. Management of Encephalomyocarditis Virus Infection in Italian Pig Farms: A Case Report. BMC Vet. Res. 2023, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Foglia, E.A.; Pezzoni, G.; Bonilauri, P.; Torri, D.; Grazioli, S.; Brocchi, E. A Recent View about Encephalomyocarditis Virus Circulating in Compartmentalised Animal Population in Northern Italy. Sci. Rep. 2023, 13, 592. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Tan, P.; Li, Y.; Lin, M.; Li, C.; Mao, J.; Cui, J.; Zhao, W.; Wang, H.Y.; Wang, R.-F. DHX29 Functions as an RNA Co-Sensor for MDA5-Mediated EMCV-Specific Antiviral Immunity. PLoS Pathog. 2018, 14, e1006886. [Google Scholar] [CrossRef]
- Visser, L.J.; Aloise, C.; Swatek, K.N.; Medina, G.N.; Olek, K.M.; Rabouw, H.H.; de Groot, R.J.; Langereis, M.A.; de Los Santos, T.; Komander, D.; et al. Dissecting distinct proteolytic activities of FMDV Lpro implicates cleavage and degradation of RLR signaling proteins, not its deISGylase/DUB activity, in type I interferon suppression. PLoS Pathog. 2020, 16, e1008702. [Google Scholar] [CrossRef]
- Li, L.; Fan, H.; Song, Z.; Liu, X.; Bai, J.; Jiang, P. Encephalomyocarditis Virus 2C Protein Antagonizes Interferon-β Signaling Pathway through Interaction with MDA5. Antiviral Res. 2019, 161, 70–84. [Google Scholar] [CrossRef]
- Huang, L.; Xiong, T.; Yu, H.; Zhang, Q.; Zhang, K.; Li, C.; Hu, L.; Zhang, Y.; Zhang, L.; Liu, Q.; et al. Encephalomyocarditis Virus 3C Protease Attenuates Type I Interferon Production through Disrupting the TANK-TBK1-IKKε-IRF3 Complex. Biochem. J. 2017, 474, 2051–2065. [Google Scholar] [CrossRef]
- Han, Y.; Xie, J.; Xu, S.; Bi, Y.; Li, X.; Zhang, H.; Idris, A.; Bai, J.; Feng, R. Encephalomyocarditis Virus Abrogates the Interferon Beta Signaling Pathway via Its Structural Protein VP2. J. Virol. 2021, 95, e1590-20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Leung, C.; Tahan, S.; Wang, D. Entry by multiple picornaviruses is dependent on a pathway that includes TNK2, WASL, and NCK1. Elife 2019, 8, e50276. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Thibaut, H.J.; Hurdiss, D.L.; Wahedi, M.; Marceau, C.D.; van Vliet, A.L.W.; Carette, J.E.; van Kuppeveld, F.J.M. Identification of the Cell-Surface Protease ADAM9 as an Entry Factor for Encephalomyocarditis Virus. Mbio 2019, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, Y.; Xu, S.; Zhao, K.; Ling, Y.; Liu, R.; Ali, A.; Bai, J. Caveolin-1 Is Involved in Encephalomyocarditis Virus Replication in BHK-21 Cells. Virol. J. 2021, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, X.; Wu, B.; Niu, Y.; Ma, R.; Xie, J.; Ali, A.; Feng, R. Host Protein, HSP90β, Antagonizes IFN-β Signaling Pathway and Facilitates the Proliferation of Encephalomyocarditis Virus In Vitro. Virus Res. 2021, 305, 198547. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Feng, L.; Zang, R.; Lei, C.Q.; Yang, Q.; Shu, H.B. ZFYVE1 negatively regulates MDA5- but not RIG-I-mediated innate antiviral response. PLoS Pathog. 2020, 16, e1008457. [Google Scholar] [CrossRef]
- Narayan, K.; Waggoner, L.; Pham, S.T.; Hendricks, G.L.; Waggoner, S.N.; Conlon, J.; Wang, J.P.; Fitzgerald, K.A.; Kang, J. TRIM13 is a negative regulator of MDA5-mediated type I interferon production. J. Virol. 2014, 88, 10748–10757. [Google Scholar] [CrossRef]
- Li, X.; Ma, R.; Wu, B.; Niu, Y.; Li, H.; Li, D.; Xie, J.; Idris, A.; Feng, R. HSP27 Protein Dampens Encephalomyocarditis Virus Replication by Stabilizing Melanoma Differentiation-Associated Gene 5. Front. Microbiol. 2021, 12, 788870. [Google Scholar] [CrossRef]
- Li, M.; Yan, J.; Zhu, H.; Guo, C.; Jiang, X.; Gao, Y.; Liu, X.; Jiang, P.; Bai, J. TRIM7 Inhibits Encephalomyocarditis Virus Replication by Activating Interferon-β Signaling Pathway. Vet. Microbiol. 2023, 281, 109729. [Google Scholar] [CrossRef]
- Lai, S.; Jia, J.; Cao, X.; Zhou, P.-K.; Gao, S. Molecular and Cellular Functions of the Linker Histone H1.2. Front. Cell Dev. Biol. 2021, 9, 773195. [Google Scholar] [CrossRef]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Conn, K.L.; Hendzel, M.J.; Schang, L.M. Linker Histones Are Mobilized during Infection with Herpes Simplex Virus Type 1. J. Virol. 2008, 82, 8629–8646. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, C.; Hu, Y.; Lei, E.; Lin, X.; Zhao, L.; Zou, Z.; Zhang, A.; Zhou, H.; Chen, H.; et al. HIST1H1C Regulates Interferon-β and Inhibits Influenza Virus Replication by Interacting with IRF3. Front. Immunol. 2017, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Wei, J.; Zhang, H.; Fan, J.; Li, X.; Wang, D.; Xie, J.; Qiao, Z.; Li, M.; Bai, J.; et al. National Serosurvey of Encephalomyocarditis Virus in Healthy People and Pigs in China. Arch. Virol. 2015, 160, 2957–2964. [Google Scholar] [CrossRef]
- Miao, M.; Deng, G.; Xiong, X.; Qiu, Y.; Huang, W.; Yuan, M.; Yu, F.; Bai, S.; Zhou, X.; Zhao, X. Enterovirus 71 3C proteolytically processes the histone H3 N-terminal tail during infection. Virol. Sin. 2022, 37, 314–317. [Google Scholar] [CrossRef]
- Sapp, N.; Burge, N.; Cox, K.; Prakash, P.; Balasubramaniam, M.; Thapa, S.; Christensen, D.; Li, M.; Linderberger, J.; Kvaratskhelia, M.; et al. HIV-1 Preintegration Complex Preferentially Integrates the Viral DNA into Nucleosomes Containing Trimethylated Histone 3-Lysine 36 Modification and Flanking Linker DNA. J. Virol. 2022, 96, e0101122. [Google Scholar] [CrossRef]
- Hoeksema, M.; Tripathi, S.; White, M.; Qi, L.; Taubenberger, J.; van Eijk, M.; Haagsman, H.; Hartshorn, K.L. Arginine-rich histones have strong antiviral activity for influenza A viruses. Innate Immun. 2015, 21, 736–745. [Google Scholar] [CrossRef]
- Tamura, M.; Natori, K.; Kobayashi, M.; Miyamura, T.; Takeda, N. Inhibition of attachment of virions of Norwalk virus to mammalian cells by soluble histone molecules. Arch. Virol. 2003, 148, 1659–1670. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Names | Primer Sequences (5′-3′) |
|---|---|
| IFN-β forward | TTGTTGAGAACCTCCTGGCT |
| IFN-β reverse | TGACTATGGTCCAGGCACAG |
| ISG15 forward | GCAGACTGTAGACACGCTTAA |
| ISG15 reverse | TTCCAATGCTATCCCAAA |
| ISG56 forward | TCATCAGGTCAAGGATAGTC |
| ISG56 reverse | CCACACTGTATTTGGTGTCTAGG |
| OAS1 forward | ATGCTTTAAGTACAGGGACG |
| OAS1 reverse | CCAAGACACTCTGGGCTA |
| CXCL10 forward | ATTTGCTGCCTTATCTTTCTGACTCTA |
| CXCL10 reverse | TGGCCTTCGATTCTGGATTCA |
| EMCV-3D forward | GTCATACTATCGTCCAGGGACTCTAT |
| EMCV-3D reverse | CATCTGTACTCCACACTCTCGAATG |
| EMCV probe | CACTTCGATCACTATGCTTGCCGTT |
| GAPDH forward | GTCTCCTCTGACTTCAACAGCG |
| GAPDH reverse | ACCACCCTGTTGCTGTAGCCAA |
| H1.2 forward | CAACTCCGAAGAAGAGCGCTAAG |
| H1.2 reverse | GCGTTCGCCTATTTCTTGG |
| H1.3 forward | CTGGGAAACGCAAAGCATCC |
| H1.3 reverse | GAAGCCGCTTTCTTGTTGAGTTT |
| H1.4 forward | GAGAAGAACAACAGCCGCATCAA |
| H1.4 reverse | GGTCTTCTTGGCGCTCTTCTT |
| H1.5 forward | GTGAAGAAGACTCCGAAGAAGGC |
| H1.5 reverse | GCAGGACTCTTGGTTGCCTTT |
| H1t forward | CCTCTCTGTGTCCAAGTTGATCA |
| H1t reverse | CACTAAGCTCTTGAGGGACAGTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Li, H.; Lian, R.; Dou, X.; Li, S.; Xie, J.; Li, X.; Feng, R.; Li, Z. Histone H1.2 Inhibited EMCV Replication through Enhancing MDA5-Mediated IFN-β Signaling Pathway. Viruses 2024, 16, 174. https://doi.org/10.3390/v16020174
Song Y, Li H, Lian R, Dou X, Li S, Xie J, Li X, Feng R, Li Z. Histone H1.2 Inhibited EMCV Replication through Enhancing MDA5-Mediated IFN-β Signaling Pathway. Viruses. 2024; 16(2):174. https://doi.org/10.3390/v16020174
Chicago/Turabian StyleSong, Yangran, Huixia Li, Ruiya Lian, Xueer Dou, Shasha Li, Jingying Xie, Xiangrong Li, Ruofei Feng, and Zhiqiang Li. 2024. "Histone H1.2 Inhibited EMCV Replication through Enhancing MDA5-Mediated IFN-β Signaling Pathway" Viruses 16, no. 2: 174. https://doi.org/10.3390/v16020174
APA StyleSong, Y., Li, H., Lian, R., Dou, X., Li, S., Xie, J., Li, X., Feng, R., & Li, Z. (2024). Histone H1.2 Inhibited EMCV Replication through Enhancing MDA5-Mediated IFN-β Signaling Pathway. Viruses, 16(2), 174. https://doi.org/10.3390/v16020174