
Optimized Directed Virus Evolution to Accelerate the Generation of Oncolytic Coxsackievirus B3 Adapted to Resistant Colorectal Cancer Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
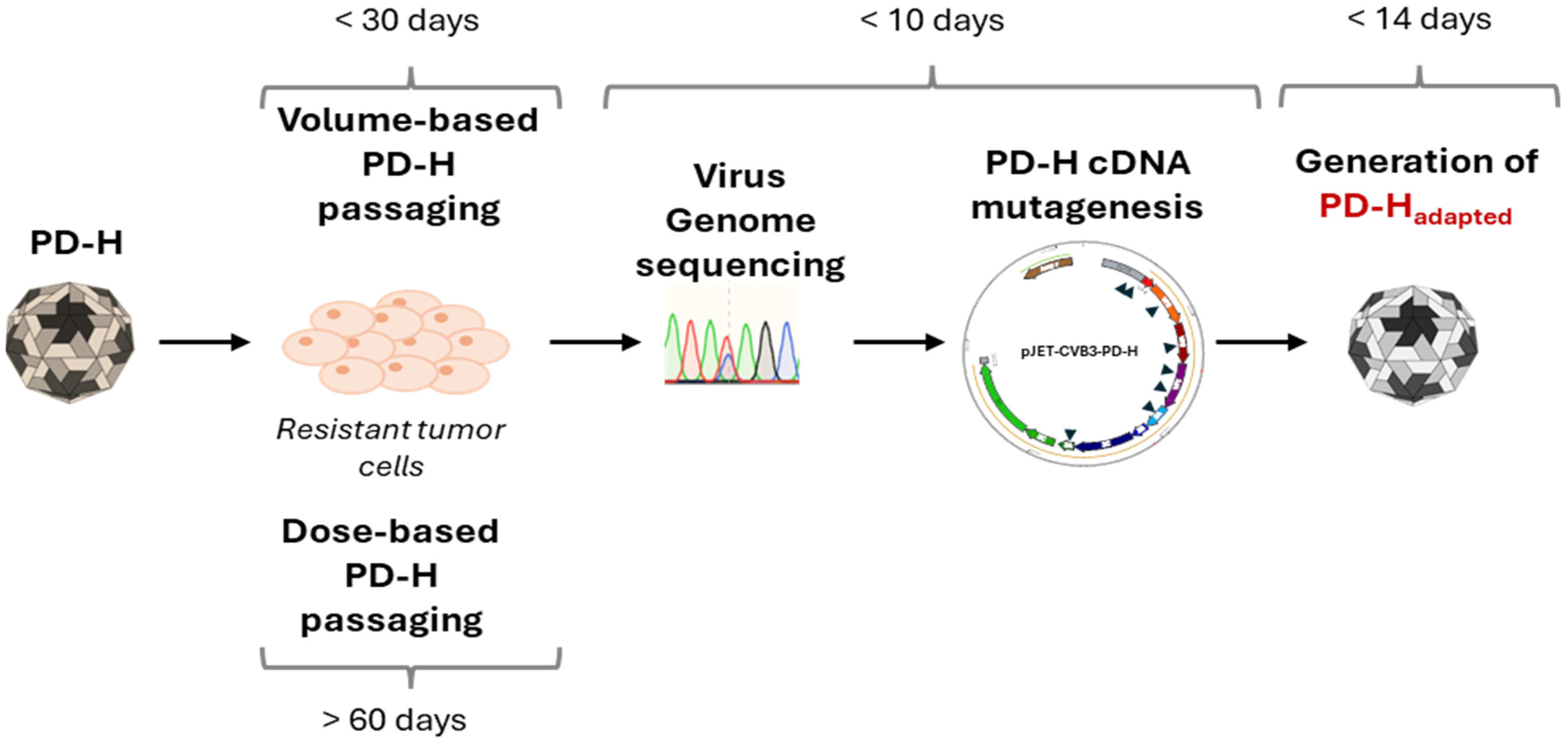
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Serial Passaging of PD-H in MC38 Cells
2.3. Virus Plaque Assay
2.4. Virus Growth Curves
2.5. Cell Viability Assay
2.6. Mutagenesis and Cloning of PD-H cDNA-Containing Plasmids
2.7. Generation of Recombinant PD-H and PD-MC38
2.8. Sequencing of CVB3 Genome
2.9. Comparison of PD-10 with Other CVB3 Strains
2.10. Statistical Analysis
3. Results
3.1. Adaptation of PD-H to MC38 Cells
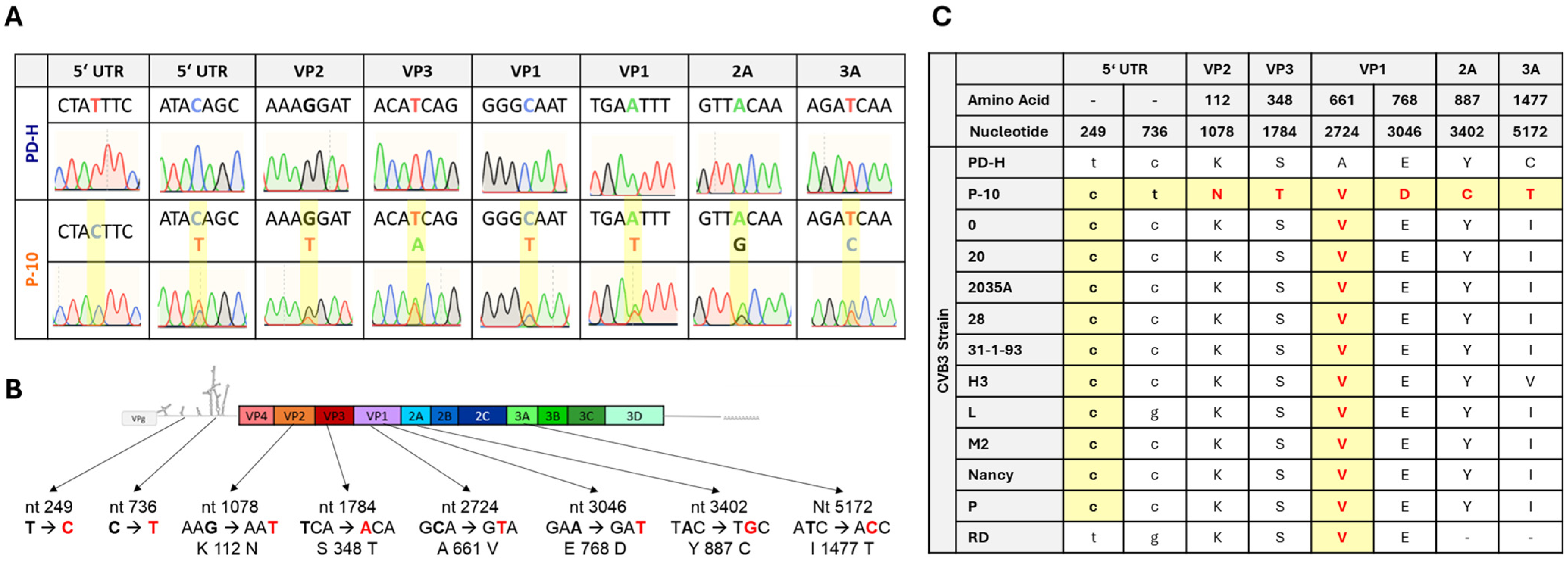
3.2. Sequence Analysis of P-10 and Comparison of P-10 with Other CVB3 Isolates
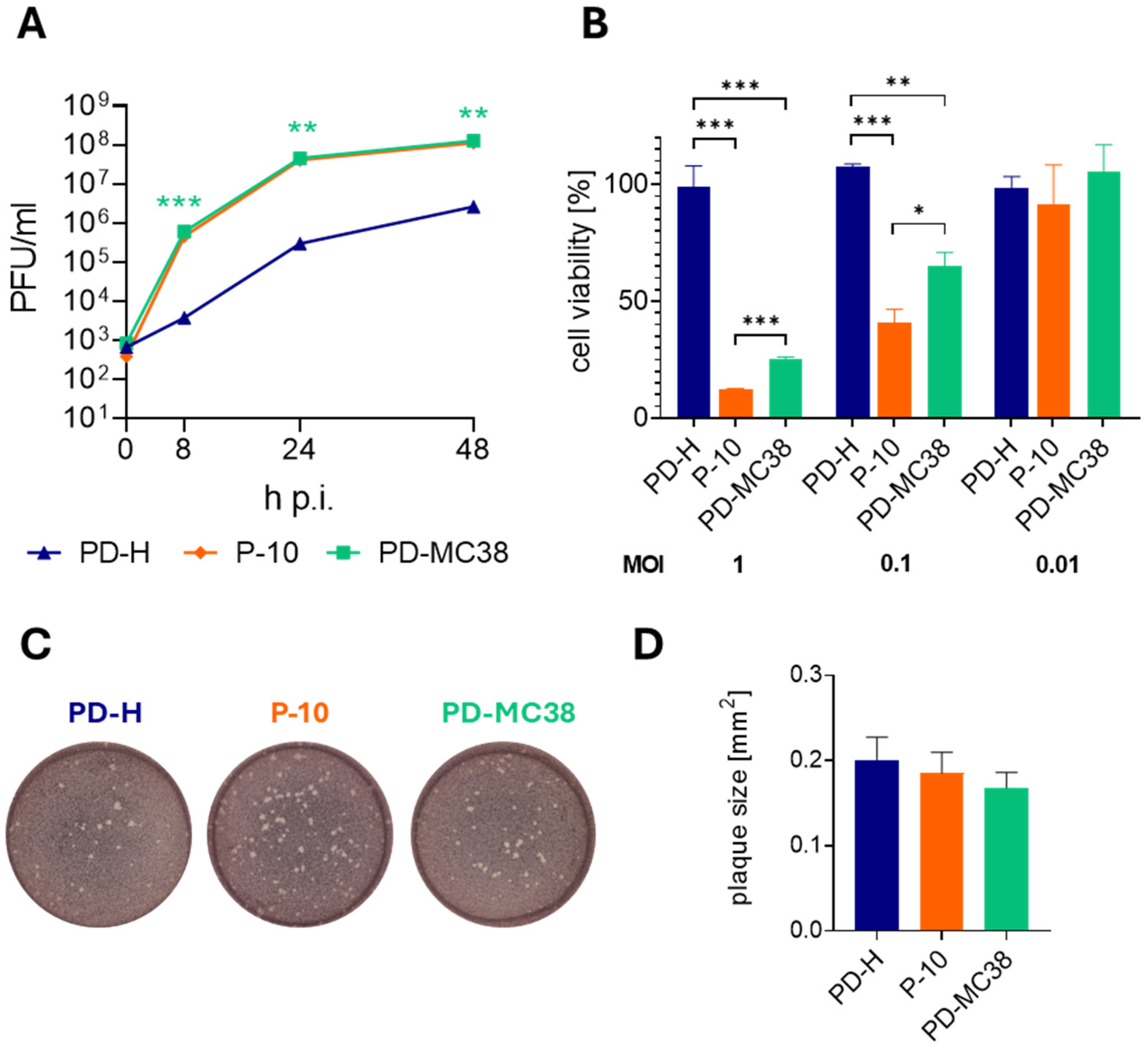
3.3. Replication and Oncolytic Activity of PD-MC38
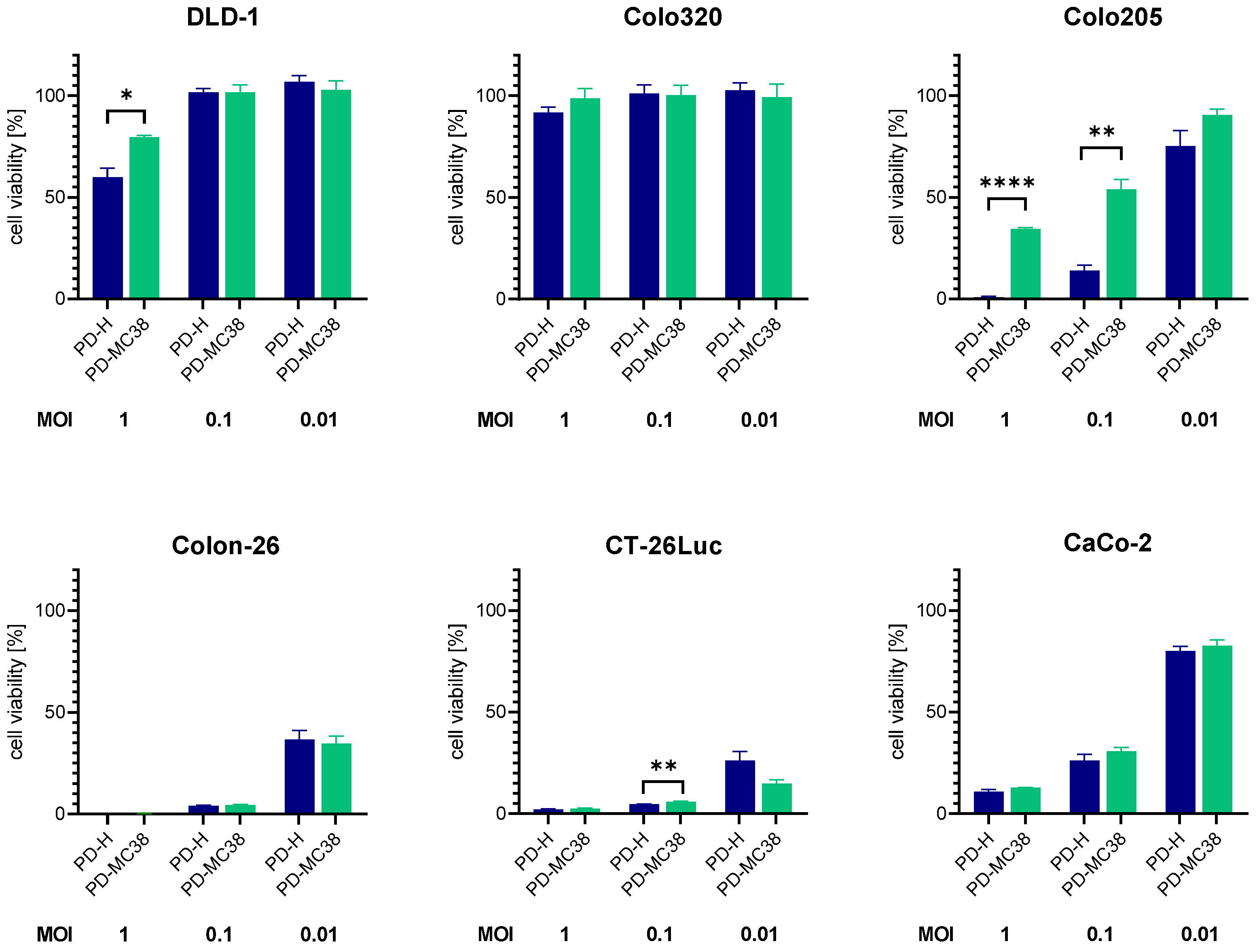
3.4. Oncolytic Efficiency of PD-MC38 in Different Colorectal Cancer Cell Lines
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China Approves World’s First Oncolytic Virus Therapy for Cancer Treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and Its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Sakurai, A.; Noda, S.; Fujiwara, Y.; Okura, N.; Takagi, T.; Asano, J.; Honda, F. Regulatory Issues: PMDA—Review of Sakigake Designation Products: Oncolytic Virus Therapy with Delytact Injection (Teserpaturev) for Malignant Glioma. Oncologist 2023, 28, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical Landscape of Oncolytic Virus Research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Hastie, E.; Grdzelishvili, V.Z. Vesicular Stomatitis Virus as a Flexible Platform for Oncolytic Virotherapy against Cancer. J. Gen. Virol. 2012, 93, 2529–2545. [Google Scholar] [CrossRef]
- Miyamoto, S.; Inoue, H.; Nakamura, T.; Yamada, M.; Sakamoto, C.; Urata, Y.; Okazaki, T.; Marumoto, T.; Takahashi, A.; Takayama, K.; et al. Coxsackievirus B3 Is an Oncolytic Virus with Immunostimulatory Properties That Is Active against Lung Adenocarcinoma. Cancer Res. 2012, 72, 2609–2621. [Google Scholar] [CrossRef]
- Wollmann, G.; Tattersall, P.; van den Pol, A.N. Targeting Human Glioblastoma Cells: Comparison of Nine Viruses with Oncolytic Potential. J. Virol. 2005, 79, 6005–6022. [Google Scholar] [CrossRef] [PubMed]
- Hazini, A.; Pryshliak, M.; Brückner, V.; Klingel, K.; Sauter, M.; Pinkert, S.; Kurreck, J.; Fechner, H. Heparan Sulfate Binding Coxsackievirus B3 Strain PD: A Novel Avirulent Oncolytic Agent Against Human Colorectal Carcinoma. Hum. Gene Ther. 2018, 29, 1301–1314. [Google Scholar] [CrossRef]
- Wollmann, G.; Davis, J.N.; Bosenberg, M.W.; van den Pol, A.N. Vesicular Stomatitis Virus Variants Selectively Infect and Kill Human Melanomas but Not Normal Melanocytes. J. Virol. 2013, 87, 6644–6659. [Google Scholar] [CrossRef] [PubMed]
- Mathis, J.M.; Stoff-Khalili, M.A.; Curiel, D.T. Oncolytic Adenoviruses—Selective Retargeting to Tumor Cells. Oncogene 2005, 24, 7775–7791. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic Viruses for Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, R.; Grdzelishvili, V.Z. Evolution of Oncolytic Viruses. Curr Opin Virol. 2015, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Svyatchenko, V.A.; Ternovoy, V.A.; Kiselev, N.N.; Demina, A.V.; Loktev, V.B.; Netesov, S.V.; Chumakov, P.M. Bioselection of Coxsackievirus B6 Strain Variants with Altered Tropism to Human Cancer Cell Lines. Arch. Virol. 2017, 162, 3355–3362. [Google Scholar] [CrossRef]
- Seegers, S.L.; Frasier, C.; Greene, S.; Nesmelova, I.V.; Grdzelishvili, V.Z. Experimental Evolution Generates Novel Oncolytic Vesicular Stomatitis Viruses with Improved Replication in Virus-Resistant Pancreatic Cancer Cells. J. Virol. 2020, 94, e01643-19. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed Evolution Generates a Novel Oncolytic Virus for the Treatment of Colon Cancer. PLoS ONE 2008, 3, 2409. [Google Scholar] [CrossRef]
- Al-Zaher, A.; Domingo-Calap, P.; Sanjuán, R. Experimental Virus Evolution in Cancer Cell Monolayers, Spheroids, and Tissue Explants. Virus Evol. 2021, 7, veab045. [Google Scholar] [CrossRef]
- Waqqar, S.; Lee, K.; Lawley, B.; Bilton, T.; Quiñones-Mateu, M.E.; Bostina, M.; Burga, L.N. Directed Evolution of Seneca Valley Virus in Tumorsphere and Monolayer Cell Cultures of a Small-Cell Lung Cancer Model. Cancers 2023, 15, 2541. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hu, C.; Liu, Y.; Chen, X.; Song, D.; Shen, R.; Liu, Z.; Jia, X.; Zhang, Q.; Gao, Y.; et al. Directed Natural Evolution Generates a Next-Generation Oncolytic Virus with a High Potency and Safety Profile. Nat. Commun. 2023, 14, 3410. [Google Scholar] [CrossRef]
- Elsner, L.; Heimann, L.; Geisler, A.; Dieringer, B.; Knoch, K.-P.; Hinze, L.; Klingel, K.; Solimena, M.; Kurreck, J.; Fechner, H. Fast Track Adaptation of Oncolytic Coxsackie B3 Virus to Resistant Colorectal Cancer Cells—A Method to Personalize Virotherapy. Biol. Proced. Online 2024, 26, 11. [Google Scholar] [CrossRef]
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 Replication and Pathogenesis. Future Microbiol. 2015, 10, 629653. [Google Scholar] [CrossRef]
- Bahreyni, A.; Mohamud, Y.; Ashraf Nouhegar, S.; Zhang, J.; Luo, H. Synergistic Viro-Chemoimmunotherapy in Breast Cancer Enabled by Bioengineered Immunostimulatory Exosomes and Dual-Targeted Coxsackievirus B3. ACS Nano 2024, 18, 4241–4255. [Google Scholar] [CrossRef] [PubMed]
- Hazini, A.; Dieringer, B.; Klingel, K.; Pryshliak, M.; Geisler, A.; Kobelt, D.; Daberkow, O.; Kurreck, J.; Van Linthout, S.; Fechner, H. Application Route and Immune Status of the Host Determine Safety and Oncolytic Activity of Oncolytic Coxsackievirus B3 Variant PD-H. Viruses 2021, 13, 1918. [Google Scholar] [CrossRef]
- Geisler, A.; Dieringer, B.; Elsner, L.; Klopfleisch, R.; Kurreck, J.; Fechner, H. Oncolytic Coxsackievirus B3 Strain PD-H Is Effective Against a Broad Spectrum of Pancreatic Cancer Cell Lines and Induces a Growth Delay in Pancreatic KPC Cell Tumors In Vivo. Int. J. Mol. Sci. 2024, 25, 11224. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, N.; Yang, C.; Tan, H.; Fang, C.; Yu, K.; Zhao, H.; Xia, N.; Wang, W.; Huang, X.; et al. Oncolytic Activity of a Coxsackievirus B3 Strain in Patient-Derived Cervical Squamous Cell Carcinoma Organoids and Synergistic Effect with Paclitaxel. Virol. J. 2024, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Bahreyni, A.; Liu, H.; Mohamud, Y.; Xue, Y.C.; Zhang, J.; Luo, H. A New miRNA-Modified Coxsackievirus B3 Inhibits Triple Negative Breast Cancer Growth with Improved Safety Profile in Immunocompetent Mice. Cancer Lett. 2022, 548, 215849. [Google Scholar] [CrossRef] [PubMed]
- Sagara, M.; Miyamoto, S.; Itoh, S.; Soda, Y.; Tani, K. Development of New Oncolytic Virotherapy Targeting Breast Cancer Using Coxsackievirus B3. Anticancer Res. 2021, 41, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Miyamoto, S.; Soda, Y.; Takishima, Y.; Sagara, M.; Liao, J.; Hirose, L.; Hijikata, Y.; Miura, Y.; Hara, K.; et al. Extremely Low Organ Toxicity and Strong Antitumor Activity of miR-34-Regulated Oncolytic Coxsackievirus B3. Mol. Ther. Oncolytics 2019, 12, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xue, Y.C.; Deng, H.; Mohamud, Y.; Ng, C.S.; Chu, A.; Lim, C.J.; Lockwood, W.W.; Jia, W.W.G.; Luo, H. MicroRNA Modification of Coxsackievirus B3 Decreases Its Toxicity, While Retaining Oncolytic Potency against Lung Cancer. Mol. Ther. Oncolytics 2020, 16, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Hazini, A.; Dieringer, B.; Pryshliak, M.; Knoch, K.-P.; Heimann, L.; Tolksdorf, B.; Pappritz, K.; El-Shafeey, M.; Solimena, M.; Beling, A.; et al. miR-375- and miR-1-Regulated Coxsackievirus B3 Has No Pancreas and Heart Toxicity but Strong Antitumor Efficiency in Colorectal Carcinomas. Hum. Gene Ther. 2021, 32, 216–230. [Google Scholar] [CrossRef]
- Bahreyni, A.; Liu, H.; Mohamud, Y.; Xue, Y.C.; Fan, Y.M.; Zhang, Y.L.; Luo, H. A Combination of Genetically Engineered Oncolytic Virus and Melittin-CpG for Cancer Viro-Chemo-Immunotherapy. BMC Med. 2023, 21, 193. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Scaltriti, M.; Kapil, A.; Sade, H.; Galbraith, S. Shifting the Paradigm in Personalized Cancer Care through Next-Generation Therapeutics and Computational Pathology. Mol. Oncol. 2024, 18, 2607–2611. [Google Scholar] [CrossRef]
- Mahmood, M.; Taufiq, I.; Mazhar, S.; Hafeez, F.; Malik, K.; Afzal, S. Revolutionizing Personalized Cancer Treatment: The Synergy of next-Generation Sequencing and CRISPR/Cas9. Per. Med. 2024, 21, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Volovat, S.R.; Scripcariu, D.V.; Vasilache, I.A.; Stolniceanu, C.R.; Volovat, C.; Augustin, I.G.; Volovat, C.C.; Ostafe, M.-R.; Andreea-Voichița, S.-G.; Bejusca-Vieriu, T.; et al. Oncolytic Virotherapy: A New Paradigm in Cancer Immunotherapy. Int. J. Mol. Sci. 2024, 25, 1180. [Google Scholar] [CrossRef]
- Bauzon, M.; Hermiston, T.W. Oncolytic Viruses: The Power of Directed Evolution. Adv. Virol. 2012, 2012, 586389. [Google Scholar] [CrossRef]
- Zainutdinov, S.S.; Kochneva, G.V.; Netesov, S.V.; Chumakov, P.M.; Matveeva, O.V. Directed Evolution as a Tool for the Selection of Oncolytic RNA Viruses with Desired Phenotypes. Oncolytic Virother. 2019, 8, 9–26. [Google Scholar] [CrossRef]
- Yan, W.; Kitzes, G.; Dormishian, F.; Hawkins, L.; Sampson-Johannes, A.; Watanabe, J.; Holt, J.; Lee, V.; Dubensky, T.; Fattaey, A.; et al. Developing Novel Oncolytic Adenoviruses through Bioselection. J. Virol. 2003, 77, 2640–2650. [Google Scholar] [CrossRef]
- He, Y.; Chipman, P.R.; Howitt, J.; Bator, C.M.; Whitt, M.A.; Baker, T.S.; Kuhn, R.J.; Anderson, C.W.; Freimuth, P.; Rossmann, M.G. Interaction of Coxsackievirus B3 with the Full Length Coxsackievirus-Adenovirus Receptor. Nat. Struct. Biol. 2001, 8, 874–878. [Google Scholar] [CrossRef]
- Organtini, L.J.; Makhov, A.M.; Conway, J.F.; Hafenstein, S.; Carson, S.D. Kinetic and Structural Analysis of Coxsackievirus B3 Receptor Interactions and Formation of the A-Particle. J. Virol. 2014, 88, 57555765. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, M.; Selinka, H.C.; Heim, A.; Jahn, B.; Tonew, M.; Kandolf, R.; Stelzner, A.; Zell, R. Attachment of Coxsackievirus B3 Variants to Various Cell Lines: Mapping of Phenotypic Differences to Capsid Protein VP1. Virology 2000, 275, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.H.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 Proteases 2A and 3C Induce Apoptotic Cell Death through Mitochondrial Injury and Cleavage of eIF4GI but Not DAP5/P97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell. Physiol. Biochem. 2019, 53, 121140. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsner, L.; Dieringer, B.; Geisler, A.; Girod, M.; Van Linthout, S.; Kurreck, J.; Fechner, H. Optimized Directed Virus Evolution to Accelerate the Generation of Oncolytic Coxsackievirus B3 Adapted to Resistant Colorectal Cancer Cells. Viruses 2024, 16, 1958. https://doi.org/10.3390/v16121958
Elsner L, Dieringer B, Geisler A, Girod M, Van Linthout S, Kurreck J, Fechner H. Optimized Directed Virus Evolution to Accelerate the Generation of Oncolytic Coxsackievirus B3 Adapted to Resistant Colorectal Cancer Cells. Viruses. 2024; 16(12):1958. https://doi.org/10.3390/v16121958
Chicago/Turabian StyleElsner, Leslie, Babette Dieringer, Anja Geisler, Maxim Girod, Sophie Van Linthout, Jens Kurreck, and Henry Fechner. 2024. "Optimized Directed Virus Evolution to Accelerate the Generation of Oncolytic Coxsackievirus B3 Adapted to Resistant Colorectal Cancer Cells" Viruses 16, no. 12: 1958. https://doi.org/10.3390/v16121958
APA StyleElsner, L., Dieringer, B., Geisler, A., Girod, M., Van Linthout, S., Kurreck, J., & Fechner, H. (2024). Optimized Directed Virus Evolution to Accelerate the Generation of Oncolytic Coxsackievirus B3 Adapted to Resistant Colorectal Cancer Cells. Viruses, 16(12), 1958. https://doi.org/10.3390/v16121958