
Genotypic Clustering of H5N1 Avian Influenza Viruses in North America Evaluated by Ordination Analysis

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses Used in This Study
2.1.1. NWHC/USDA-NVSL
2.1.2. GISAID EpiFlu™ Database
2.1.3. GenBank Nucleotide Sequence Database
2.2. Sequence Pre-Processing
2.3. Bioinformatic Data Analyses
2.3.1. Phylogenetic Analysis
2.3.2. Visualization of Evolutionary Distances Using Clustering Analysis
2.3.3. Reference Dataset for H5N1 Virus Ordination and Clustering
3. Results
3.1. Introduction of H5N1 Viruses to North America
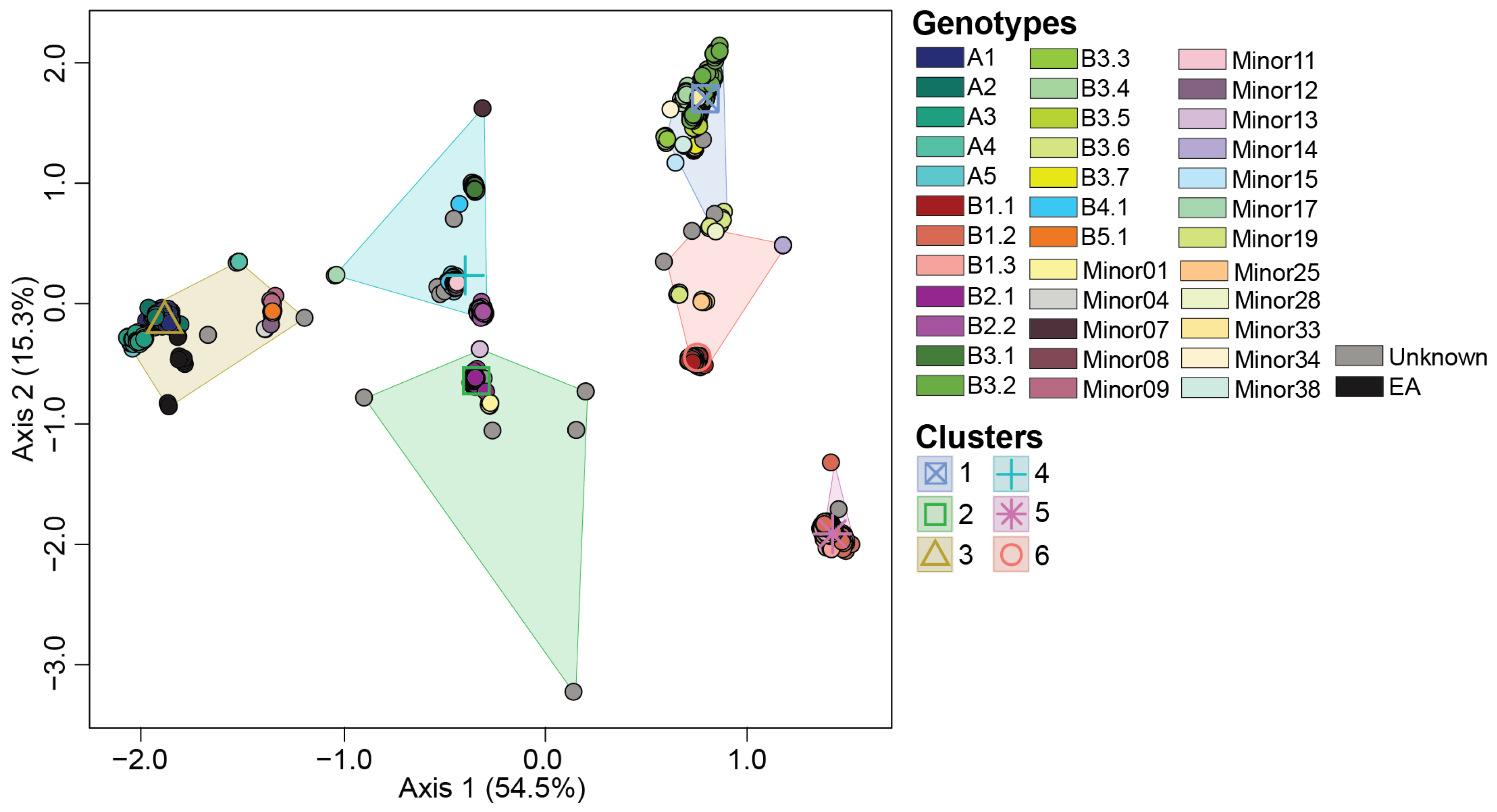
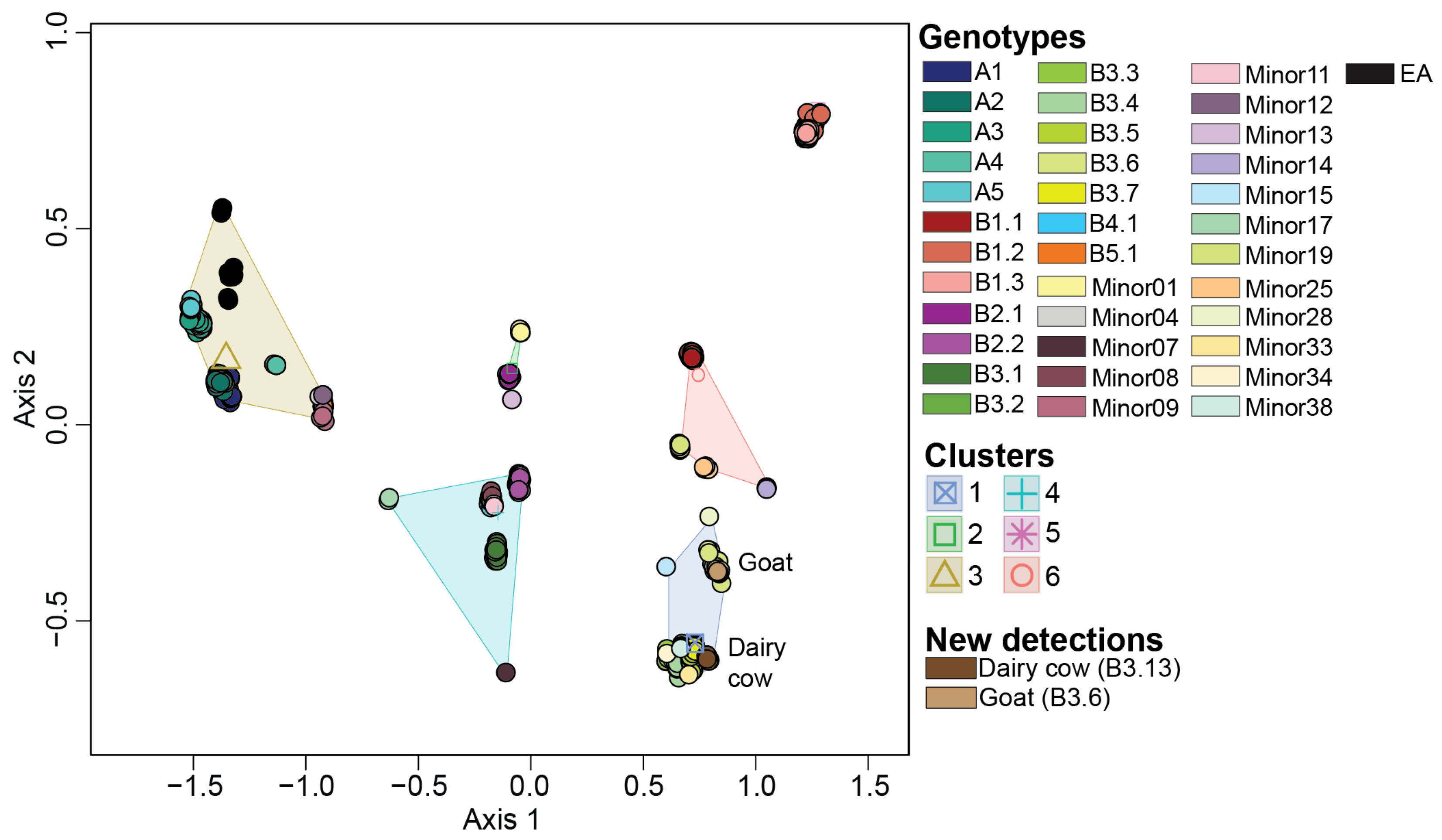
3.2. Ordination Analysis Recapitulates H5N1 Genotypic Diversity Observed by Phylogentic Analysis
3.3. Reassortant H5N1 Virus Genotypes
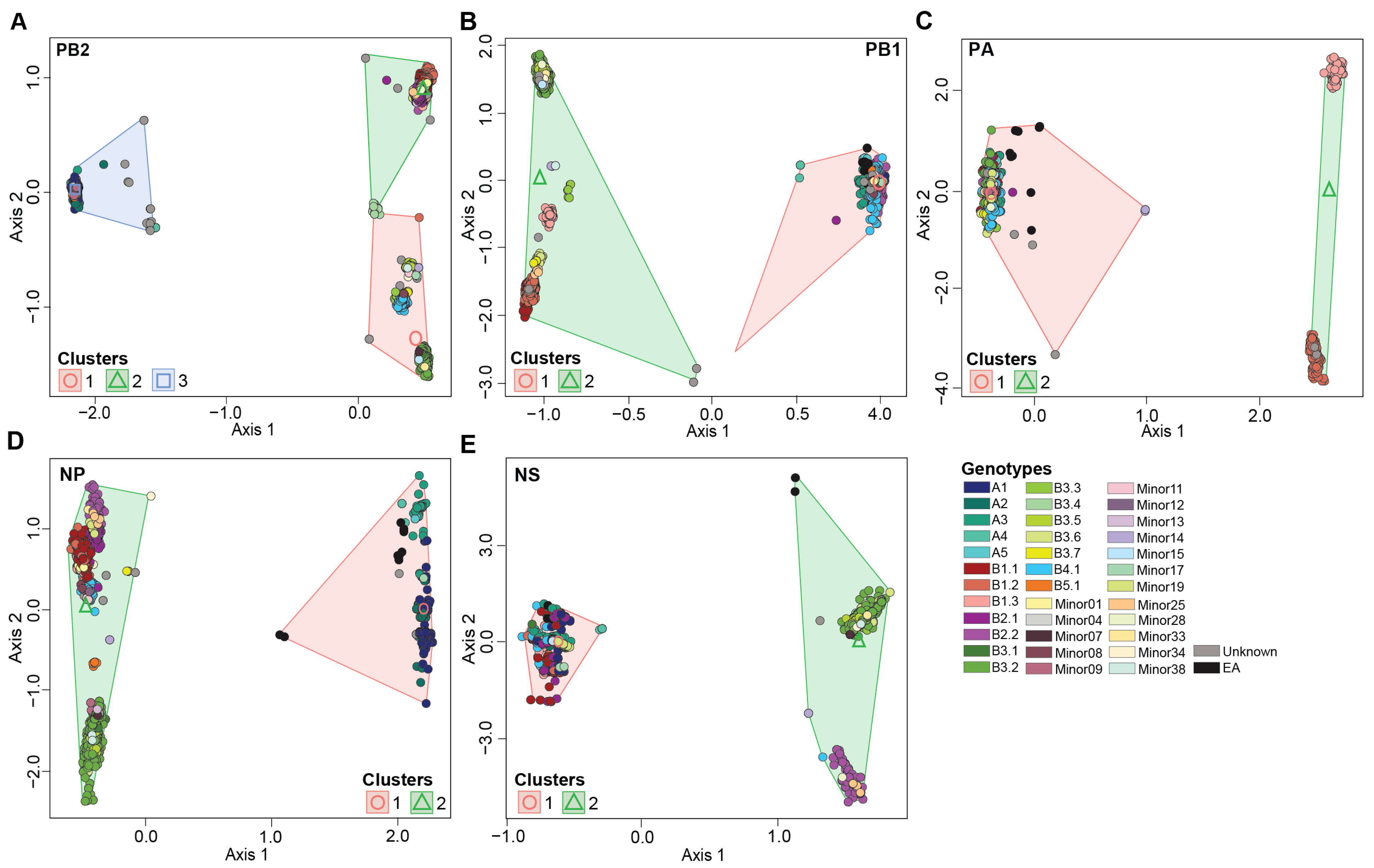
3.4. H5N1 Virus Reassortants Were Limited to Five Viral Gene Segments
3.4.1. PB2 Gene Segment
3.4.2. PB1 Gene Segment
3.4.3. PA Gene Segment
3.4.4. NP Gene Segment
3.4.5. NS Gene Segment
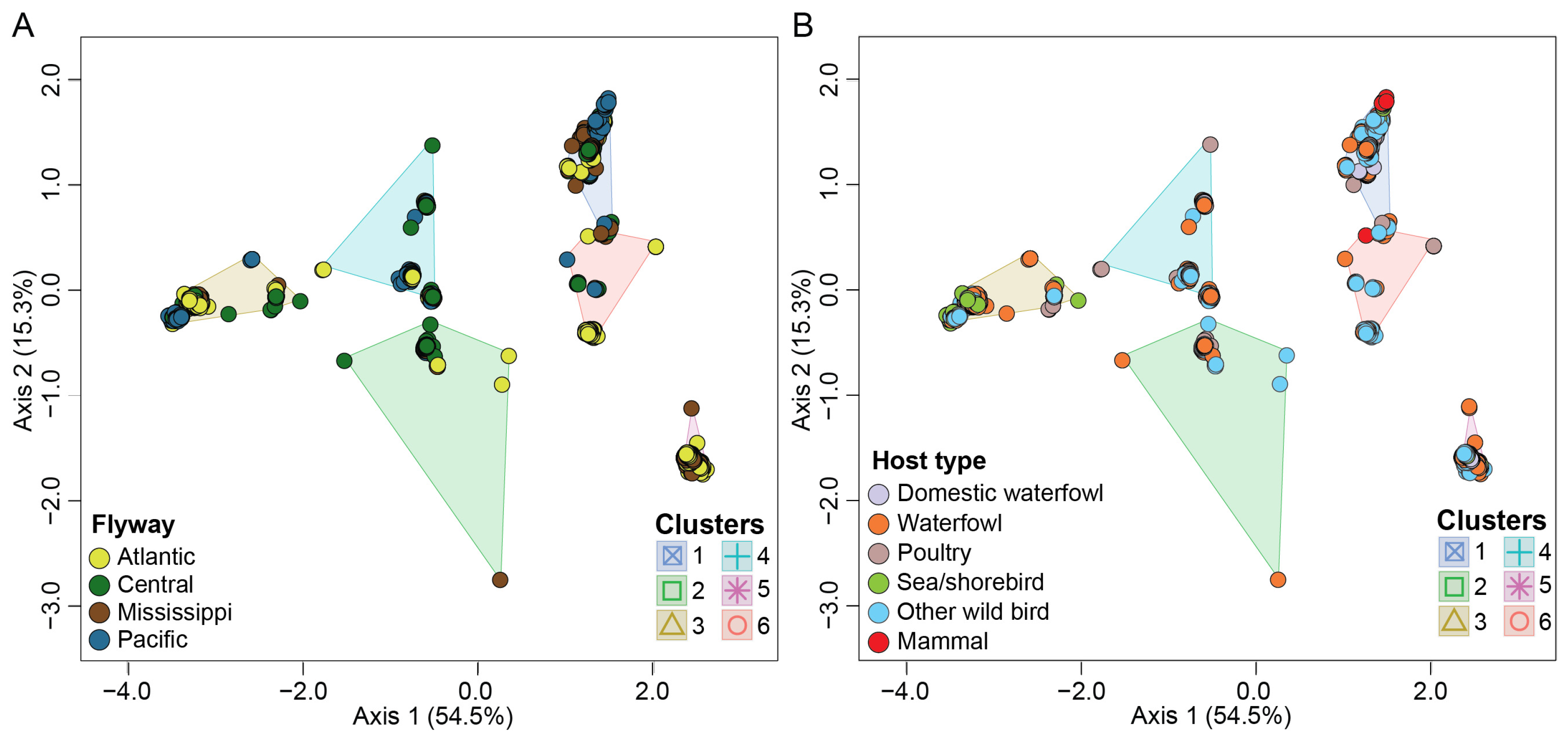
3.5. Migratory Flyway and Host Type May Impact Viral Genotypic Diversity
3.6. Reference Dataset Is Robust Enough to Be Used for Genotype Assignment of Newly Sequenced H5N1 Viruses Using Ordination Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Blagodatski, A.; Trutneva, K.; Glazova, O.; Mityaeva, O.; Shevkova, L.; Kegeles, E.; Onyanov, N.; Fede, K.; Maznina, A.; Khavina, E.; et al. Avian Influenza in Wild Birds and Poultry: Dissemination Pathways, Monitoring Methods, and Virus Ecology. Pathogens 2021, 10, 630. [Google Scholar] [CrossRef] [PubMed]
- Boulinier, T. Avian Influenza Spread and Seabird Movements between Colonies. TREE 2023, 38, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Sonnberg, S.; Webby, R.J.; Webster, R.G. Natural History of Highly Pathogenic Avian Influenza H5N1. Virus Res. 2013, 178, 63–77. [Google Scholar] [CrossRef]
- Lee, D.-H.; Bertran, K.; Kwon, J.-H.; Swayne, D.E. Evolution, Global Spread, and Pathogenicity of Highly Pathogenic Avian Influenza H5Nx Clade 2.3.4.4. J. Vet. Sci. 2017, 18, 269–280. [Google Scholar] [CrossRef]
- Verhagen, J.H.; Fouchier, R.A.M.; Lewis, N. Highly Pathogenic Avian Influenza Viruses at the Wild–Domestic Bird Interface in Europe: Future Directions for Research and Surveillance. Viruses 2021, 13, 212. [Google Scholar] [CrossRef]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert (Akberovna), S.; King, J.; Harder, T.; et al. Emergence and Spread of Novel H5N8, H5N5 and H5N1 Clade 2.3.4.4 Highly Pathogenic Avian Influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, A.; Zecchin, B.; Giussani, E.; Palumbo, E.; Agüero-García, M.; Bachofen, C.; Bálint, Á.; Banihashem, F.; Banyard, A.C.; Beerens, N.; et al. High Pathogenic Avian Influenza A(H5) Viruses of Clade 2.3.4.4b in Europe—Why Trends of Virus Evolution Are More Difficult to Predict. Virus Evol. 2024, 10, veae027. [Google Scholar] [CrossRef]
- Gu, W.; Shi, J.; Cui, P.; Yan, C.; Zhang, Y.; Wang, C.; Zhang, Y.; Xing, X.; Zeng, X.; Liu, L.; et al. Novel H5N6 Reassortants Bearing the Clade 2.3.4.4b HA Gene of H5N8 Virus Have Been Detected in Poultry and Caused Multiple Human Infections in China. Emerg. Microbes Infect. 2022, 11, 1174–1185. [Google Scholar] [CrossRef]
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic Spread of Highly Pathogenic Avian Influenza H5N1 by Wild Birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef]
- Bevins, S.N.; Shriner, S.A.; Cumbee, J.C.; Dilione, K.E.; Douglass, K.E.; Ellis, J.W.; Killian, M.L.; Torchetti, M.K.; Lenoch, J.B. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006–1011. [Google Scholar] [CrossRef]
- Günther, A.; Krone, O.; Svansson, V.; Pohlmann, A.; King, J.; Hallgrimsson, G.T.; Skarphéðinsson, K.H.; Sigurðardóttir, H.; Jónsson, S.R.; Beer, M.; et al. Iceland as Stepping Stone for Spread of Highly Pathogenic Avian Influenza Virus between Europe and North America. Emerg. Infect. Dis. 2022, 28, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Elsmo, E.; Wünschmann, A.; Beckmen, K.; Broughton-Neiswanger, L.; Buckles, E.; Ellis, J.; Fitzgerald, S.; Gerlach, R.; Hawkins, S.; Ip, H.; et al. Highly Pathogenic Avian Influenza A(H5N1) Virus Clade 2.3.4.4b Infections in Wild Terrestrial Mammals, United States, 2022. Emerg. Infect. Dis. 2023, 29, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Puryear, W.; Sawatzki, K.; Hill, N.; Foss, A.; Stone, J.J.; Doughty, L.; Walk, D.; Gilbert, K.; Murray, M.; Cox, E.; et al. Highly Pathogenic Avian Influenza A(H5N1) Virus Outbreak in New England Seals, United States. Emerg. Infect. Dis. 2023, 29, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Youk, S.; Torchetti, M.K.; Lantz, K.; Lenoch, J.B.; Killian, M.L.; Leyson, C.; Bevins, S.N.; Dilione, K.E.; Ip, H.S.; Stallknecht, D.E.; et al. H5N1 Highly Pathogenic Avian Influenza Clade 2.3.4.4b in Wild and Domestic Birds: Introductions into the United States and Reassortments, December 2021-April 2022. Virology 2023, 587, 109860. [Google Scholar] [CrossRef]
- Blackshields, G.; Sievers, F.; Shi, W.; Wilm, A.; Higgins, D.G. Sequence Embedding for Fast Construction of Guide Trees for Multiple Sequence Alignment. Algorithms Mol. Biol. 2010, 5, 21. [Google Scholar] [CrossRef]
- Higgins, D.G. Sequence Ordinations: A Muitivariate Analysis Approach to Analysing Large Sequence Data Sets. Bioinformatics 1992, 8, 15–22. [Google Scholar] [CrossRef]
- Pelé, J.; Bécu, J.-M.; Abdi, H.; Chabbert, M. Bios2mds: An R Package for Comparing Orthologous Protein Families by Metric Multidimensional Scaling. BMC Bioinform. 2012, 13, 133. [Google Scholar] [CrossRef]
- Shi, W.; Lei, F.; Zhu, C.; Sievers, F.; Higgins, D.G. A Complete Analysis of HA and NA Genes of Influenza A Viruses. PLoS ONE 2010, 5, e14454. [Google Scholar] [CrossRef]
- Thijssen, M.; Khamisipour, G.; Maleki, M.; Devos, T.; Li, G.; Van Ranst, M.; Matthijnssens, J.; Pourkarim, M.R. Characterization of the Human Blood Virome in Iranian Multiple Transfused Patients. Viruses 2023, 15, 1425. [Google Scholar] [CrossRef]
- Shang, Y.; Xiao, G.; Zheng, P.; Cen, K.; Zhan, S.; Wang, C. Divergent and Convergent Evolution of Fungal Pathogenicity. GBE 2016, 8, 1374–1387. [Google Scholar] [CrossRef]
- GISAID. Available online: https://www.gisaid.org (accessed on 11 June 2024).
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 11 June 2024).
- Tjeldnes, H.; Labun, K.; Torres Cleuren, Y.; Chyżyńska, K.; Świrski, M.; Valen, E. ORFik: A Comprehensive R Toolkit for the Analysis of Translation. BMC Bioinform. 2021, 22, 336. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.; Stuber, T.; Lantz, K.; Torchetti, M.; Robbe-Austerman, S. vSNP: A SNP Pipeline for the Generation of Transparent SNP Matrices and Phylogenetic Trees from Whole Genome Sequencing Data Sets. BMC Genom. 2024, 25, 545. [Google Scholar] [CrossRef] [PubMed]
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. Msa: An R Package for Multiple Sequence Alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic Analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Res. 2024, 52, gkae268. [Google Scholar] [CrossRef] [PubMed]
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Cui, P.; Shi, J.; Wang, C.; Zhang, Y.; Xing, X.; Kong, H.; Yan, C.; Zeng, X.; Liu, L.; Tian, G.; et al. Global Dissemination of H5N1 Influenza Viruses Bearing the Clade 2.3.4.4b HA Gene and Biologic Analysis of the Ones Detected in China. Emerg. Microbes Infect. 2022, 11, 1693–1704. [Google Scholar] [CrossRef]
- Telele, N. Fikrie Predicting Interspecies Transmission and Pandemic Risks of Coronaviruses. Masters Thesis, University of Skovde, Skövde, Sweden, 2020. [Google Scholar]
- Thijssen, M.; Tacke, F.; Van Espen, L.; Cassiman, D.; Naser Aldine, M.; Nevens, F.; Van Ranst, M.; Matthijnssens, J.; Pourkarim, M.R. Plasma Virome Dynamics in Chronic Hepatitis B Virus Infected Patients. Front. Microbiol. 2023, 14, 1172574. [Google Scholar] [CrossRef]
- Mohamed, W.M.A.; Moustafa, M.A.M.; Thu, M.J.; Kakisaka, K.; Chatanga, E.; Ogata, S.; Hayashi, N.; Taya, Y.; Ohari, Y.; Naguib, D.; et al. Comparative Mitogenomics Elucidates the Population Genetic Structure of Amblyomma Testudinarium in Japan and a Closely Related Amblyomma Species in Myanmar. Evol. Appl. 2022, 15, 1062–1078. [Google Scholar] [CrossRef]
- Isoda, N.; Onuma, M.; Hiono, T.; Sobolev, I.; Lim, H.; Nabeshima, K.; Honjyo, H.; Yokoyama, M.; Shestopalov, A.; Sakoda, Y. Detection of New H5N1 High Pathogenicity Avian Influenza Viruses in Winter 2021–2022 in the Far East, Which Are Genetically Close to Those in Europe. Viruses 2022, 14, 2168. [Google Scholar] [CrossRef]
- Gass, J.D.; Hill, N.J.; Damodaran, L.; Naumova, E.N.; Nutter, F.B.; Runstadler, J.A. Ecogeographic Drivers of the Spatial Spread of Highly Pathogenic Avian Influenza Outbreaks in Europe and the United States, 2016–Early 2022. Int. J. Environ. Res. Public Health 2023, 20, 6030. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.A.; Mullinax, J.M.; Runge, M.C.; Prosser, D.J. The Changing Dynamics of Highly Pathogenic Avian Influenza H5N1: Next Steps for Management & Science in North America. Biol. Conserv. 2023, 282, 110041. [Google Scholar] [CrossRef]
- Ramey, A.M.; Scott, L.C.; Ahlstrom, C.A.; Buck, E.J.; Williams, A.R.; Kim Torchetti, M.; Stallknecht, D.E.; Poulson, R.L. Molecular Detection and Characterization of Highly Pathogenic H5N1 Clade 2.3.4.4b Avian Influenza Viruses among Hunter-Harvested Wild Birds Provides Evidence for Three Independent Introductions into Alaska. Virology 2024, 589, 109938. [Google Scholar] [CrossRef]
- Erdelyan, C.N.G.; Kandeil, A.; Signore, A.V.; Jones, M.E.B.; Vogel, P.; Andreev, K.; Bøe, C.A.; Gjerset, B.; Alkie, T.N.; Yason, C.; et al. Multiple Transatlantic Incursions of Highly Pathogenic Avian Influenza Clade 2.3.4.4b A(H5N5) Virus into North America and Spillover to Mammals. Cell Rep. 2024, 43, 114479. [Google Scholar] [CrossRef]
- Fourment, M.; Darling, A.E.; Holmes, E.C. The Impact of Migratory Flyways on the Spread of Avian Influenza Virus in North America. BMC Evol. Biol. 2017, 17, 118. [Google Scholar] [CrossRef]
- Alkie, T.N.; Lopes, S.; Hisanaga, T.; Xu, W.; Suderman, M.; Koziuk, J.; Fisher, M.; Redford, T.; Lung, O.; Joseph, T.; et al. A Threat from Both Sides: Multiple Introductions of Genetically Distinct H5 HPAI Viruses into Canada via Both East Asia-Australasia/Pacific and Atlantic Flyways. Virus Evol. 2022, 8, veac077. [Google Scholar] [CrossRef]
- World Organisation of Animal Health (WAHIS). Available online: https://wahis.woah.org/#/in-review/5065 (accessed on 26 July 2023).
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Alexakis, L.; Fusaro, A.; Kuiken, T.; Mirinavičiūtė, G.; Ståhl, K.; Staubach, C.; Svartström, O.; et al. Avian Influenza Overview March–June 2024. EFSA J. 2024, 22, e8930. [Google Scholar] [CrossRef] [PubMed]
- Leguia, M.; Garcia-Glaessner, A.; Muñoz-Saavedra, B.; Juarez, D.; Barrera, P.; Calvo-Mac, C.; Jara, J.; Silva, W.; Ploog, K.; Amaro, L.; et al. Highly Pathogenic Avian Influenza A (H5N1) in Marine Mammals and Seabirds in Peru. Nat. Commun. 2023, 14, 5489. [Google Scholar] [CrossRef]
- Kandeil, A.; Patton, C.; Jones, J.C.; Jeevan, T.; Harrington, W.N.; Trifkovic, S.; Seiler, J.P.; Fabrizio, T.; Woodard, K.; Turner, J.C.; et al. Rapid Evolution of A(H5N1) Influenza Viruses after Intercontinental Spread to North America. Nat. Commun. 2023, 14, 3082. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef]
- Murawski, A.; Fabrizio, T.; Ossiboff, R.; Kackos, C.; Jeevan, T.; Jones, J.C.; Kandeil, A.; Walker, D.; Turner, J.C.M.; Patton, C.; et al. Highly Pathogenic Avian Influenza A(H5N1) Virus in a Common Bottlenose Dolphin (Tursiops Truncatus) in Florida. Commun. Biol. 2024, 7, 476. [Google Scholar] [CrossRef] [PubMed]
- Garg, S. Outbreak of Highly Pathogenic Avian Influenza A(H5N1) Viruses in U.S. Dairy Cattle and Detection of Two Human Cases—United States, 2024. MMWR Morb. Mortal. Wkly. Rep. 2024, 73, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-Q.; Hutter, C.; Markin, A.; Thomas, M.; Lantz, K.; Killian, M.L.; Janzen, G.M.; Vijendran, S.; Wagle, S.; Inderski, B.; et al. Emergence and Interstate Spread of Highly Pathogenic Avian Influenza A(H5N1) in Dairy Cattle 2024. bioRxiv 2024. [Google Scholar] [CrossRef]
- Hu, X.; Saxena, A.; Magstadt, D.R.; Gauger, P.C.; Burrough, E.; Zhang, J.; Siepker, C.; Mainenti, M.; Gorden, P.J.; Plummer, P.; et al. Genomic Characterization of Highly Pathogenic Avian Influenza A H5N1 Virus Newly Emerged in Dairy Cattle. Emerg. Microbes Infect. 2024, 13, 2380421. [Google Scholar] [CrossRef]
- Beerens, N.; Heutink, R.; Harders, F.; Bossers, A.; Koch, G.; Peeters, B. Emergence and Selection of a Highly Pathogenic Avian Influenza H7N3 Virus. J. Virol. 2020, 94, e01818-19. [Google Scholar] [CrossRef]
- Monne, I.; Fusaro, A.; Nelson, M.I.; Bonfanti, L.; Mulatti, P.; Hughes, J.; Murcia, P.R.; Schivo, A.; Valastro, V.; Moreno, A.; et al. Emergence of a Highly Pathogenic Avian Influenza Virus from a Low-Pathogenic Progenitor. J. Virol. 2014, 88, 4375–4388. [Google Scholar] [CrossRef]
- Engering, A.; Hogerwerf, L.; Slingenbergh, J. Pathogen–Host–Environment Interplay and Disease Emergence. Emerg. Microbes Infect. 2013, 2, 1–7. [Google Scholar] [CrossRef]
- Laughlin, A.J.; Hall, R.J.; Taylor, C.M. Ecological Determinants of Pathogen Transmission in Communally Roosting Species. Theor. Ecol. 2019, 12, 225–235. [Google Scholar] [CrossRef]
- Lam, T.T.-Y.; Ip, H.S.; Ghedin, E.; Wentworth, D.E.; Halpin, R.A.; Stockwell, T.B.; Spiro, D.J.; Dusek, R.J.; Bortner, J.B.; Hoskins, J.; et al. Migratory Flyway and Geographical Distance Are Barriers to the Gene Flow of Influenza Virus among North American Birds. Ecol. Lett. 2012, 15, 24–33. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Genotype | First Detected | Last Detected | Flyways (N Viruses) | Total Viruses | |||
|---|---|---|---|---|---|---|---|---|
| Atlantic | Central | Mississippi | Pacific | |||||
| 1 | B3.2 | 20 March 2022 | 22 December 2023 | 40 | 261 | 348 | 343 | 1111 |
| B3.3 | 8 October 2022 | 6 April 2023 | 3 | 2 | 2 | x | ||
| B3.4 | 23 November 2022 | 7 March 2023 | x | 2 | 15 | x | ||
| B3.5 | 30 October 2022 | 18 May 2023 | 25 | 17 | 4 | x | ||
| B3.6 | 22 November 2022 | 12 October 2023 | x | 13 | 6 | 12 | ||
| B3.7 | 11 September 2023 | 11 October 2023 | x | 1 | x | 12 | ||
| Minor15 | 29 April 2022 | x | x | 1 | x | |||
| Minor33 | 21 February 2023 | 1 | x | x | x | |||
| Minor34 | 17 September 2022 | x | x | 1 | x | |||
| Minor38 | 18 October 2022 | 2 | x | x | x | |||
| 2 | B2.1 | 1 March 2022 | 7 September 2023 | 5 | 334 | 396 | 99 | 843 |
| Minor01 | 18 February 2022 | 27 April 2022 | 5 | x | 3 | x | ||
| Minor13 | 15 March 2022 | x | 1 | x | x | |||
| 3 | A1 | 30 December 2021 | 28 November 2022 | 263 * | 74 | 162 | 1 | 824 |
| A2 | 16 February 2022 | 30 September 2023 | 186 * | x | x | x | ||
| A3 | 3 February 2022 | 19 September 2023 | x | 6 | 1 | 89 * | ||
| A4 | 14 October 2022 | x | x | x | 6 * | |||
| A5 | 31 October 2022 | 3 February 2023 | 8 * | x | x | x | ||
| B5.1 | 1 March 2022 | 7 April 2022 | x | 13 | 6 | x | ||
| Minor04 | 9 April 2022 | x | 2 | x | x | |||
| Minor09 | 1 February 2022 | 1 March 2022 | 4 | x | 1 | x | ||
| Minor12 | 1 April 2022 | x | 2 | x | x | |||
| 4 | B2.2 | 28 March 2022 | 20 April 2023 | 22 | 78 | 59 | 38 | 742 |
| B3.1 | 4 March 2022 | 1 November 2022 | 2 | 65 | 24 | 23 | ||
| B4.1 | 5 March 2022 | 11 April 2023 | 1 | 28 | 4 | 385 | ||
| Minor07 | 26 April 2022 | x | 1 | x | x | |||
| Minor08 | 1 February 2022 | 25 April 2022 | 8 | x | x | x | ||
| Minor11 | 1 April 2022 | 4 April 2022 | x | 2 | x | x | ||
| Minor17 | 1 November 2022 | 2 | x | x | x | |||
| 5 | B1.2 | 12 February 2022 | 6 March 2023 | 124 | 6 | 131 | x | 605 |
| B1.3 | 26 June 2022 | 28 April 2023 | 130 | 7 | 207 | x | ||
| 6 | B1.1 | 25 January 2022 | 6 April 2023 | 460 | 4 | 107 | 4 | 596 |
| Minor14 | 22 March 2022 | 25 March 2022 | 4 | x | x | x | ||
| Minor19 | 18 November 2022 | 9 January 2023 | x | 7 | 2 | x | ||
| Minor25 | 22 November 2022 | 13 December 2022 | x | 5 | 1 | 1 | ||
| Minor28 | 21 November 2022 | x | x | 1 | x | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tawidian, P.; Torchetti, M.K.; Killian, M.L.; Lantz, K.; Dilione, K.E.; Ringenberg, J.M.; Bevins, S.N.; Lenoch, J.B.; Ip, H.S. Genotypic Clustering of H5N1 Avian Influenza Viruses in North America Evaluated by Ordination Analysis. Viruses 2024, 16, 1818. https://doi.org/10.3390/v16121818
Tawidian P, Torchetti MK, Killian ML, Lantz K, Dilione KE, Ringenberg JM, Bevins SN, Lenoch JB, Ip HS. Genotypic Clustering of H5N1 Avian Influenza Viruses in North America Evaluated by Ordination Analysis. Viruses. 2024; 16(12):1818. https://doi.org/10.3390/v16121818
Chicago/Turabian StyleTawidian, Patil, Mia K. Torchetti, Mary L. Killian, Kristina Lantz, Krista E. Dilione, Jourdan M. Ringenberg, Sarah N. Bevins, Julianna B. Lenoch, and Hon S. Ip. 2024. "Genotypic Clustering of H5N1 Avian Influenza Viruses in North America Evaluated by Ordination Analysis" Viruses 16, no. 12: 1818. https://doi.org/10.3390/v16121818
APA StyleTawidian, P., Torchetti, M. K., Killian, M. L., Lantz, K., Dilione, K. E., Ringenberg, J. M., Bevins, S. N., Lenoch, J. B., & Ip, H. S. (2024). Genotypic Clustering of H5N1 Avian Influenza Viruses in North America Evaluated by Ordination Analysis. Viruses, 16(12), 1818. https://doi.org/10.3390/v16121818