
Host-Driven Ubiquitination Events in Vector-Transmitted RNA Virus Infections as Options for Broad-Spectrum Therapeutic Intervention Strategies
 , ,
, ,
Abstract
1. Introduction
2. Alphaviruses
2.1. Chikungunya (CHIKV)
2.2. New World Alphaviruses
3. Bunyaviruses
3.1. Crimean–Congo Hemorrhagic Fever (CCHF)
3.2. Rift Valley Fever Virus (RVFV)
4. Flaviviruses
4.1. Dengue (DENV)
4.2. Japanese Encephalitis Virus (JEV)
4.3. Yellow Fever Virus (YFV)
4.4. Zika Virus (ZIKV)
5. Expert Opinion
5.1. Small Molecules Targeting Vector Transmitted RNA Viruses Ubiquitin Pathways
5.2. Small Molecules Targeting Other Virus Ubiquitin Pathways
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Socha, W.; Kwasnik, M.; Larska, M.; Rola, J.; Rozek, W. Vector-Borne Viral Diseases as a Current Threat for Human and Animal Health-One Health Perspective. J. Clin. Med. 2022, 11, 3026. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of Viral Mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.; Sajid, M.S.; Saqib, M.; Anjum, F.R.; Tayyab, M.H.; Rizwan, H.M.; Rashid, M.I.; Rashid, I.; Iqbal, A.; Siddique, R.M.; et al. Potential Mechanisms of Transmission of Tick-Borne Viruses at the Virus-Tick Interface. Front. Microbiol. 2022, 13, 846884. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B. The Ubiquitin System: From Cell Signalling to Disease Biology and New Therapeutic Opportunities. Cell Death Differ. 2021, 28, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Kondoh, T.; Yoshimoto, M.; Fujieda, K.; Matsuura, N.; Matsuda, I.; Miike, T.; Yano, K.; Okuno, A.; Aoki, Y. Complex Glycerol Kinase Deficiency: Molecular-Genetic, Cytogenetic, and Clinical Studies of Five Japanese Patients. Am. J. Med. Genet. 1988, 31, 603–616. [Google Scholar] [CrossRef]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin Enzymes in the Regulation of Immune Responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 425–460. [Google Scholar] [CrossRef]
- George, A.J.; Hoffiz, Y.C.; Charles, A.J.; Zhu, Y.; Mabb, A.M. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in Neurological Disorders. Front. Genet. 2018, 9, 29. [Google Scholar] [CrossRef]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like Protein Activation by E1 Enzymes: The Apex for Downstream Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 Enzymes: More than Just Middle Men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 Ubiquitin Ligases: Styles, Structures and Functions. Mol. Biomed. 2021, 2, 23. [Google Scholar] [CrossRef]
- Timms, R.T.; Mena, E.L.; Leng, Y.; Li, M.Z.; Tchasovnikarova, I.A.; Koren, I.; Elledge, S.J. Defining E3 Ligase-Substrate Relationships through Multiplex CRISPR Screening. Nat. Cell Biol. 2023, 25, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Horwood, P.F.; Buchy, P. Chikungunya. Rev. Sci. Tech. 2015, 34, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sherman, M.B.; Chafets, D.; Dinglasan, N.; Lu, K.; Lee, T.-H.; Carlson, L.-A.; Muench, M.O.; Simmons, G. An Attenuated Replication-Competent Chikungunya Virus with a Fluorescently Tagged Envelope. PLoS Neglected Trop. Dis. 2018, 12, e0006693. [Google Scholar] [CrossRef] [PubMed]
- Althouse, B.M.; Guerbois, M.; Cummings, D.A.T.; Diop, O.M.; Faye, O.; Faye, A.; Diallo, D.; Sadio, B.D.; Sow, A.; Faye, O.; et al. Role of Monkeys in the Sylvatic Cycle of Chikungunya Virus in Senegal. Nat. Commun. 2018, 9, 1046. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, A. An Intercellular Bridge for Chikungunya Virus Transmission. Nat. Rev. Microbiol. 2023, 21, 702. [Google Scholar] [CrossRef] [PubMed]
- Mahto, S.K.; Gupta, P.K.; Singh, A.; Meena, R.C. Atypical Neurological Manifestations of Chikungunya Fever: Two Case Reports. Indian J. Crit. Care Med. 2018, 22, 306–308. [Google Scholar] [CrossRef]
- Goupil, B.A.; Mores, C.N. A Review of Chikungunya Virus-Induced Arthralgia: Clinical Manifestations, Therapeutics, and Pathogenesis. Open Rheumatol. J. 2016, 10, 129–140. [Google Scholar] [CrossRef]
- Lima Neto, A.S.; Sousa, G.S.; Nascimento, O.J.; Castro, M.C. Chikungunya-Attributable Deaths: A Neglected Outcome of a Neglected Disease. PLoS Neglected Trop. Dis. 2019, 13, e0007575. [Google Scholar] [CrossRef]
- Silva, J.V.J.; Ludwig-Begall, L.F.; de Oliveira-Filho, E.F.; Oliveira, R.A.S.; Durães-Carvalho, R.; Lopes, T.R.R.; Silva, D.E.A.; Gil, L.H.V.G. A Scoping Review of Chikungunya Virus Infection: Epidemiology, Clinical Characteristics, Viral Co-Circulation Complications, and Control. Acta Trop. 2018, 188, 213–224. [Google Scholar] [CrossRef]
- Ly, H. Ixchiq (VLA1553): The First FDA-Approved Vaccine to Prevent Disease Caused by Chikungunya Virus Infection. Virulence 2024, 15, 2301573. [Google Scholar] [CrossRef]
- Bhalla, N.; Sun, C.; Metthew Lam, L.K.; Gardner, C.L.; Ryman, K.D.; Klimstra, W.B. Host Translation Shutoff Mediated by Non-Structural Protein 2 Is a Critical Factor in the Antiviral State Resistance of Venezuelan Equine Encephalitis Virus. Virology 2016, 496, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the Innate Immune Response: The Old World Alphavirus nsP2 Protein Induces Rapid Degradation of Rpb1, a Catalytic Subunit of RNA Polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Jian, X.; Liu, Y.; Liu, Y.; Lv, L.; Cui, H.; Zhang, L. Elucidating Cellular Interactome of Chikungunya Virus Identifies Host Dependency Factors. Virol. Sin. 2023, 38, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Terán, C.; Calderón-Rangel, A.; Rodriguez-Morales, A.; Mattar, S. Venezuelan Equine Encephalitis Virus: The Problem Is Not over for Tropical America. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Stienlauf, S.; Eisenkraft, A.; Robenshtok, E.; Hourvitz, A. Viral encephalitis caused by biowarfare agents. Harefuah 2002, 141, 51–56. [Google Scholar]
- Stromberg, Z.R.; Fischer, W.; Bradfute, S.B.; Kubicek-Sutherland, J.Z.; Hraber, P. Vaccine Advances against Venezuelan, Eastern, and Western Equine Encephalitis Viruses. Vaccines 2020, 8, 273. [Google Scholar] [CrossRef]
- Simon, L.V.; Coffey, R.; Fischer, M.A. Western Equine Encephalitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Crosby, B.; Crespo, M.E. Venezuelan Equine Encephalitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Armstrong, P.M.; Andreadis, T.G. Ecology and Epidemiology of Eastern Equine Encephalitis Virus in the Northeastern United States: An Historical Perspective. J. Med. Entomol. 2022, 59, 1–13. [Google Scholar] [CrossRef]
- Omoga, D.C.A.; Tchouassi, D.P.; Venter, M.; Ogola, E.O.; Osalla, J.; Kopp, A.; Slothouwer, I.; Torto, B.; Junglen, S.; Sang, R. Transmission Dynamics of Crimean-Congo Haemorrhagic Fever Virus (CCHFV): Evidence of Circulation in Humans, Livestock, and Rodents in Diverse Ecologies in Kenya. Viruses 2023, 15, 1891. [Google Scholar] [CrossRef]
- Amaya, M.; Keck, F.; Lindquist, M.; Voss, K.; Scavone, L.; Kehn-Hall, K.; Roberts, B.; Bailey, C.; Schmaljohn, C.; Narayanan, A. The Ubiquitin Proteasome System Plays a Role in Venezuelan Equine Encephalitis Virus Infection. PLoS ONE 2015, 10, e0124792. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef]
- Palchevska, O.; Dominguez, F.; Frolova, E.I.; Frolov, I. Alphavirus-Induced Transcriptional and Translational Shutoffs Play Major Roles in Blocking the Formation of Stress Granules. J. Virol. 2023, 97, e0097923. [Google Scholar] [CrossRef]
- Kehn-Hall, K.; Bradfute, S.B. Understanding Host Responses to Equine Encephalitis Virus Infection: Implications for Therapeutic Development. Expert Rev. Anti Infect. Ther. 2022, 20, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Boghdeh, N.A.; McGraw, B.; Barrera, M.D.; Anderson, C.; Baha, H.; Risner, K.H.; Ogungbe, I.V.; Alem, F.; Narayanan, A. Inhibitors of the Ubiquitin-Mediated Signaling Pathway Exhibit Broad-Spectrum Antiviral Activities against New World Alphaviruses. Viruses 2023, 15, 655. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Taylor, A. Ubiquitin-Proteasome Pathway and Cellular Responses to Oxidative Stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef]
- Xie, Y.; Cao, J.; Gan, S.; Xu, L.; Zhang, D.; Qian, S.; Xu, F.; Ding, Q.; Schoggins, J.W.; Fan, W. TRIM32 Inhibits Venezuelan Equine Encephalitis Virus Infection by Targeting a Late Step in Viral Entry. BioRxiv 2024. [Google Scholar] [CrossRef]
- Hawman, D.W.; Feldmann, H. Crimean-Congo Haemorrhagic Fever Virus. Nat. Rev. Microbiol. 2023, 21, 463–477. [Google Scholar] [CrossRef]
- Appannanavar, S.B.; Mishra, B. An Update on Crimean Congo Hemorrhagic Fever. J. Glob. Infect. Dis. 2011, 3, 285–292. [Google Scholar] [CrossRef]
- Scholte, F.E.M.; Zivcec, M.; Dzimianski, J.V.; Deaton, M.K.; Spengler, J.R.; Welch, S.R.; Nichol, S.T.; Pegan, S.D.; Spiropoulou, C.F.; Bergeron, É. Crimean-Congo Hemorrhagic Fever Virus Suppresses Innate Immune Responses via a Ubiquitin and ISG15 Specific Protease. Cell Rep. 2017, 20, 2396–2407. [Google Scholar] [CrossRef]
- Scholte, F.E.M.; Hua, B.L.; Spengler, J.R.; Dzimianski, J.V.; Coleman-McCray, J.D.; Welch, S.R.; McMullan, L.K.; Nichol, S.T.; Pegan, S.D.; Spiropoulou, C.F.; et al. Stable Occupancy of the Crimean-Congo Hemorrhagic Fever Virus-Encoded Deubiquitinase Blocks Viral Infection. mBio 2019, 10, e01065-19. [Google Scholar] [CrossRef]
- Zivcec, M.; Scholte, F.E.M.; Spiropoulou, C.F.; Spengler, J.R.; Bergeron, É. Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses 2016, 8, 106. [Google Scholar] [CrossRef]
- Lapa, D.; Pauciullo, S.; Ricci, I.; Garbuglia, A.R.; Maggi, F.; Scicluna, M.T.; Tofani, S. Rift Valley Fever Virus: An Overview of the Current Status of Diagnostics. Biomedicines 2024, 12, 540. [Google Scholar] [CrossRef] [PubMed]
- Nair, N.; Osterhaus, A.D.M.E.; Rimmelzwaan, G.F.; Prajeeth, C.K. Rift Valley Fever Virus-Infection, Pathogenesis and Host Immune Responses. Pathogens 2023, 12, 1174. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue Simonet, P.N.; Alexandre Michel, N.-N.; Abel, W.; Albert, E.; Martin Hermann, G.; Franziska, S. Diversity and Abundance of Potential Vectors of Rift Valley Fever Virus in the North Region of Cameroon. Insects 2020, 11, 814. [Google Scholar] [CrossRef] [PubMed]
- Rostal, M.K.; Cleaveland, S.; Cordel, C.; van Staden, L.; Matthews, L.; Anyamba, A.; Karesh, W.B.; Paweska, J.T.; Haydon, D.T.; Ross, N. Farm-Level Risk Factors of Increased Abortion and Mortality in Domestic Ruminants during the 2010 Rift Valley Fever Outbreak in Central South Africa. Pathogens 2020, 9, 914. [Google Scholar] [CrossRef]
- Kwaśnik, M.; Rożek, W.; Rola, J. Rift Valley Fever—A Growing Threat to Humans and Animals. J. Vet. Res. 2021, 65, 7–14. [Google Scholar] [CrossRef]
- Javelle, E.; Lesueur, A.; de Santi, V.P.; de Laval, F.; Lefebvre, T.; Holweck, G.; Durand, G.A.; Leparc-Goffart, I.; Texier, G.; Simon, F. The Challenging Management of Rift Valley Fever in Humans: Literature Review of the Clinical Disease and Algorithm Proposal. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 4. [Google Scholar] [CrossRef]
- Matsiela, M.S.; Naicker, L.; Khoza, T.; Mokoena, N. Safety and Immunogenicity of Inactivated Rift Valley Fever Smithburn Viral Vaccine in Sheep. Virol. J. 2023, 20, 221. [Google Scholar] [CrossRef]
- Alkan, C.; Jurado-Cobena, E.; Ikegami, T. Advancements in Rift Valley Fever Vaccines: A Historical Overview and Prospects for next Generation Candidates. NPJ Vaccines 2023, 8, 171. [Google Scholar] [CrossRef]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Kota, K.P.; Whitehouse, C.A.; Bavari, S. Protein Kinase R Degradation Is Essential for Rift Valley Fever Virus Infection and Is Regulated by SKP1-CUL1-F-Box (SCF)FBXW11-NSs E3 Ligase. PLoS Pathog. 2016, 12, e1005437. [Google Scholar] [CrossRef]
- Kalveram, B.; Lihoradova, O.; Ikegami, T. NSs Protein of Rift Valley Fever Virus Promotes Posttranslational Downregulation of the TFIIH Subunit P62. J. Virol. 2011, 85, 6234–6243. [Google Scholar] [CrossRef]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence Factor NSs of Rift Valley Fever Virus Recruits the F-Box Protein FBXO3 to Degrade Subunit P62 of General Transcription Factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.J.; Ansari, A. Critical Involvement of TFIIB in Viral Pathogenesis. Front. Mol. Biosci. 2021, 8, 669044. [Google Scholar] [CrossRef]
- Bracci, N.; de la Fuente, C.; Saleem, S.; Pinkham, C.; Narayanan, A.; García-Sastre, A.; Balaraman, V.; Richt, J.A.; Wilson, W.; Kehn-Hall, K. Rift Valley Fever Virus Gn V5-Epitope Tagged Virus Enables Identification of UBR4 as a Gn Interacting Protein That Facilitates Rift Valley Fever Virus Production. Virology 2022, 567, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, S.S.; Schwarz, M.M.; McMillen, C.M.; Price, D.A.; Feng, A.X.; Albe, J.R.; Wang, W.; Miersch, S.; Orvedahl, A.; Cole, A.R.; et al. Lrp1 Is a Host Entry Factor for Rift Valley Fever Virus. Cell 2021, 184, 5163–5178.e24. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Jamdar, S.F.; Alalowi, M.; Al Ageel Al Beaiji, S.M. Dengue Virus: A Global Human Threat: Review of Literature. J. Int. Soc. Prev. Community Dent. 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; et al. Global Spread of Dengue Virus Types: Mapping the 70 Year History. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef]
- Ashraf, S.; Patwary, M.M.; Rodriguez-Morales, A.J. Demographic Disparities in Incidence and Mortality Rates of Current Dengue Outbreak in Bangladesh. New Microbes New Infect. 2024, 56, 101207. [Google Scholar] [CrossRef]
- Singh, R.K.; Tiwari, A.; Satone, P.D.; Priya, T.; Meshram, R.J. Updates in the Management of Dengue Shock Syndrome: A Comprehensive Review. Cureus 2023, 15, e46713. [Google Scholar] [CrossRef]
- Torres-Flores, J.M.; Reyes-Sandoval, A.; Salazar, M.I. Dengue Vaccines: An Update. BioDrugs 2022, 36, 325–336. [Google Scholar] [CrossRef]
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. mBio 2016, 7, e00804-16. [Google Scholar] [CrossRef]
- Cai, D.; Liu, L.; Tian, B.; Fu, X.; Yang, Q.; Chen, J.; Zhang, Y.; Fang, J.; Shen, L.; Wang, Y.; et al. Dual-Role Ubiquitination Regulation Shuttling the Entire Life Cycle of the Flaviviridae. Front. Microbiol. 2022, 13, 835344. [Google Scholar] [CrossRef]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.-Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef]
- Hao, J.; Li, J.; Zhang, Z.; Yang, Y.; Zhou, Q.; Wu, T.; Chen, T.; Wu, Z.; Zhang, P.; Cui, J.; et al. NLRC5 Restricts Dengue Virus Infection by Promoting the Autophagic Degradation of Viral NS3 through E3 Ligase CUL2 (Cullin 2). Autophagy 2023, 19, 1332–1347. [Google Scholar] [CrossRef]
- Murphy Schafer, A.R.; Smith, J.L.; Pryke, K.M.; DeFilippis, V.R.; Hirsch, A.J. The E3 Ubiquitin Ligase SIAH1 Targets MyD88 for Proteasomal Degradation During Dengue Virus Infection. Front. Microbiol. 2020, 11, 24. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, A.; Yadav, P.; Dubey, S.K.; Azhar, E.I.; Maitra, S.S.; Dwivedi, V.D. Molecular Pathogenesis of Japanese Encephalitis and Possible Therapeutic Strategies. Arch. Virol. 2022, 167, 1739–1762. [Google Scholar] [CrossRef]
- Nemeth, N.; Bosco-Lauth, A.; Oesterle, P.; Kohler, D.; Bowen, R. North American Birds as Potential Amplifying Hosts of Japanese Encephalitis Virus. Am. J. Trop. Med. Hyg. 2012, 87, 760–767. [Google Scholar] [CrossRef]
- Fagre, A.C.; Kading, R.C. Can Bats Serve as Reservoirs for Arboviruses? Viruses 2019, 11, 215. [Google Scholar] [CrossRef]
- Kuwata, R.; Torii, S.; Shimoda, H.; Supriyono, S.; Phichitraslip, T.; Prasertsincharoen, N.; Takemae, H.; Bautista, R.C.J.T.; Ebora, V.D.B.M.; Abella, J.A.C.; et al. Distribution of Japanese Encephalitis Virus, Japan and Southeast Asia, 2016–2018. Emerg. Infect. Dis. 2020, 26, 125–128. [Google Scholar] [CrossRef]
- Nemeth, N.M.; Bosco-Lauth, A.M.; Bowen, R.A. Cross-Protection between West Nile and Japanese Encephalitis Viruses in Red-Winged Blackbirds (Agelaius phoeniceus). Avian Dis. 2009, 53, 421–425. [Google Scholar] [CrossRef]
- Xia, Q.; Yang, Y.; Zhang, Y.; Zhou, L.; Ma, X.; Xiao, C.; Zhang, J.; Li, Z.; Liu, K.; Li, B.; et al. Shift in Dominant Genotypes of Japanese Encephalitis Virus and Its Impact on Current Vaccination Strategies. Front. Microbiol. 2023, 14, 1302101. [Google Scholar] [CrossRef]
- Srivastava, K.S.; Jeswani, V.; Pal, N.; Bohra, B.; Vishwakarma, V.; Bapat, A.A.; Patnaik, Y.P.; Khanna, N.; Shukla, R. Japanese Encephalitis Virus: An Update on the Potential Antivirals and Vaccines. Vaccines 2023, 11, 742. [Google Scholar] [CrossRef]
- Firbas, C.; Jilma, B. Product Review on the JE Vaccine IXIARO. Hum. Vaccines Immunother. 2015, 11, 411–420. [Google Scholar] [CrossRef]
- Xiong, W.; Lu, L.; Xiao, Y.; Li, J.; Zhou, D. Mortality and Disability Due to Japanese Encephalitis in Elderly Adults: Evidence From an Adult Tertiary Care Center in West China. Front. Neurol. 2019, 10, 918. [Google Scholar] [CrossRef]
- Wang, S.; Liu, H.; Zu, X.; Liu, Y.; Chen, L.; Zhu, X.; Zhang, L.; Zhou, Z.; Xiao, G.; Wang, W. The Ubiquitin-Proteasome System Is Essential for the Productive Entry of Japanese Encephalitis Virus. Virology 2016, 498, 116–127. [Google Scholar] [CrossRef]
- van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 2017, 5, 23. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, N.; Chen, S.; Zhao, P.; Ren, H.; Zhu, S.; Tang, H.; Zhu, Y.; Qi, Z. E3 Ubiquitin Ligase Nedd4 Promotes Japanese Encephalitis Virus Replication by Suppressing Autophagy in Human Neuroblastoma Cells. Sci. Rep. 2017, 7, 45375. [Google Scholar] [CrossRef]
- Aizaz, M.; Kiani, Y.S.; Nisar, M.; Shan, S.; Paracha, R.Z.; Yang, G. Genomic Analysis, Evolution and Characterization of E3 Ubiquitin Protein Ligase (TRIM) Gene Family in Common Carp (Cyprinus carpio). Genes 2023, 14, 667. [Google Scholar] [CrossRef]
- Huang, N.; Sun, X.; Li, P.; Liu, X.; Zhang, X.; Chen, Q.; Xin, H. TRIM Family Contribute to Tumorigenesis, Cancer Development, and Drug Resistance. Exp. Hematol. Oncol. 2022, 11, 75. [Google Scholar] [CrossRef]
- Fan, W.; Wu, M.; Qian, S.; Zhou, Y.; Chen, H.; Li, X.; Qian, P. TRIM52 Inhibits Japanese Encephalitis Virus Replication by Degrading the Viral NS2A. Sci. Rep. 2016, 6, 33698. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Cianchi, V.; Torelli, A.; Montomoli, E. Yellow Fever: Origin, Epidemiology, Preventive Strategies and Future Prospects. Vaccines 2022, 10, 372. [Google Scholar] [CrossRef]
- Tyagi, P.; Ganguly, M.; Manney, S.; Wadkar, K.; Ingle, N.; Gairola, S.; Dhere, R.; Koide, F.; Grimes, S. Neurovirulence, Viscerotropism and Immunogenicity of Live Attenuated Yellow Fever 17D Vaccine Virus in Non-Human Primates. Vaccine 2023, 41, 836–843. [Google Scholar] [CrossRef]
- Gardner, C.L.; Ryman, K.D. Yellow Fever: A Reemerging Threat. Clin. Lab. Med. 2010, 30, 237–260. [Google Scholar] [CrossRef]
- Damasceno-Caldeira, R.; Nunes-Neto, J.P.; Aragão, C.F.; Freitas, M.N.O.; Ferreira, M.S.; de Castro, P.H.G.; Dias, D.D.; da Silva Araújo, P.A.; Brandão, R.C.F.; Nunes, B.T.D.; et al. Vector Competence of Aedes Albopictus for Yellow Fever Virus: Risk of Reemergence of Urban Yellow Fever in Brazil. Viruses 2023, 15, 1019. [Google Scholar] [CrossRef]
- Rojas, A.; Hachey, W.; Kaur, G.; Korejwo, J.; Muhammad, R. Enhanced Safety Surveillance of STAMARIL® Yellow Fever Vaccine Provided under the Expanded Access Investigational New Drug Program in the USA. J. Travel Med. 2023, 30, taad037. [Google Scholar] [CrossRef]
- Ramanathan, H.N.; Zhang, S.; Douam, F.; Mar, K.B.; Chang, J.; Yang, P.L.; Schoggins, J.W.; Ploss, A.; Lindenbach, B.D. A Sensitive Yellow Fever Virus Entry Reporter Identifies Valosin-Containing Protein (VCP/P97) as an Essential Host Factor for Flavivirus Uncoating. mBio 2020, 11, e00467-20. [Google Scholar] [CrossRef]
- Das, P.; Dudley, J.P. How Viruses Use the VCP/P97 ATPase Molecular Machine. Viruses 2021, 13, 1881. [Google Scholar] [CrossRef]
- Shen, Z.; Wei, L.; Yu, Z.-B.; Yao, Z.-Y.; Cheng, J.; Wang, Y.-T.; Song, X.-T.; Li, M. The Roles of TRIMs in Antiviral Innate Immune Signaling. Front. Cell. Infect. Microbiol. 2021, 11, 628275. [Google Scholar] [CrossRef]
- Rawal, G.; Yadav, S.; Kumar, R. Zika Virus: An Overview. J. Fam. Med. Prim. Care 2016, 5, 523–527. [Google Scholar] [CrossRef]
- Li, P.; Wu, J.; Liu, S.; Lu, R.; Jiang, H.; Wang, N.; Luo, M.; Guo, L.; Xiao, J.; Bu, L.; et al. The RNA Polymerase of Cytoplasmically Replicating Zika Virus Binds with Chromatin DNA in Nuclei and Regulates Host Gene Transcription. Proc. Natl. Acad. Sci. USA 2022, 119, e2205013119. [Google Scholar] [CrossRef]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and Emergence of Zika Virus. J. Infect. Dis. 2017, 216, S860–S867. [Google Scholar] [CrossRef]
- Hu, T.; Li, J.; Carr, M.J.; Duchêne, S.; Shi, W. The Asian Lineage of Zika Virus: Transmission and Evolution in Asia and the Americas. Virol. Sin. 2019, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.J.; Oduyebo, T.; Brault, A.C.; Brooks, J.T.; Chung, K.-W.; Hills, S.; Kuehnert, M.J.; Mead, P.; Meaney-Delman, D.; Rabe, I.; et al. Modes of Transmission of Zika Virus. J. Infect. Dis. 2017, 216, S875–S883. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Bugallo, G.; Piedra, L.A.; Rodriguez, M.; Bisset, J.A.; Lourenço-de-Oliveira, R.; Weaver, S.C.; Vasilakis, N.; Vega-Rúa, A. Vector-Borne Transmission and Evolution of Zika Virus. Nat. Ecol. Evol. 2019, 3, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, L.S.; Parra, B.; Pardo, C.A. Neuroviruses Emerging in the Americas Study Neurological Implications of Zika Virus Infection in Adults. J. Infect. Dis. 2017, 216, S897–S905. [Google Scholar] [CrossRef]
- Pielnaa, P.; Al-Saadawe, M.; Saro, A.; Dama, M.F.; Zhou, M.; Huang, Y.; Huang, J.; Xia, Z. Zika Virus-Spread, Epidemiology, Genome, Transmission Cycle, Clinical Manifestation, Associated Challenges, Vaccine and Antiviral Drug Development. Virology 2020, 543, 34–42. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope Protein Ubiquitination Drives Entry and Pathogenesis of Zika Virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef]
- Kaur, G.; Pant, P.; Bhagat, R.; Seth, P. Zika Virus E Protein Modulates Functions of Human Brain Microvascular Endothelial Cells and Astrocytes: Implications on Blood-Brain Barrier Properties. Front. Cell. Neurosci. 2023, 17, 1173120. [Google Scholar] [CrossRef]
- Yang, D.; Li, N.L.; Wei, D.; Liu, B.; Guo, F.; Elbahesh, H.; Zhang, Y.; Zhou, Z.; Chen, G.-Y.; Li, K. The E3 Ligase TRIM56 Is a Host Restriction Factor of Zika Virus and Depends on Its RNA-Binding Activity but Not miRNA Regulation, for Antiviral Function. PLoS Neglected Trop. Dis. 2019, 13, e0007537. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Hu, D.; Gao, D.; Wang, W.; Wu, K.; Wu, J. USP38 Inhibits Zika Virus Infection by Removing Envelope Protein Ubiquitination. Viruses 2021, 13, 2029. [Google Scholar] [CrossRef]
- Luo, H. Interplay between the Virus and the Ubiquitin-Proteasome System: Molecular Mechanism of Viral Pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef]
- Schneider, S.M.; Lee, B.H.; Nicola, A.V. Viral Entry and the Ubiquitin-Proteasome System. Cell. Microbiol. 2021, 23, e13276. [Google Scholar] [CrossRef] [PubMed]
- Nag, D.K.; Finley, D. A Small-Molecule Inhibitor of Deubiquitinating Enzyme USP14 Inhibits Dengue Virus Replication. Virus Res. 2012, 165, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, Y.; Ding, S.; Li, J.; Song, N.; Ren, Y.; Hong, D.; Wu, C.; Li, B.; Wang, F.; et al. Small Molecule Inhibitors Reveal Allosteric Regulation of USP14 via Steric Blockade. Cell Res. 2018, 28, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, N.A.; Paparisto, E.; Barr, S.D.; Spratt, D.E. HERC5 and the ISGylation Pathway: Critical Modulators of the Antiviral Immune Response. Viruses 2021, 13, 1102. [Google Scholar] [CrossRef]
- Lopata, A.; Kniss, A.; Löhr, F.; Rogov, V.V.; Dötsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369. [Google Scholar] [CrossRef]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Płaszczyca, A.; Bartenschlager, R.; Götz, M.; Pichlmair, A. An Orthogonal Proteomic Survey Uncovers Novel Zika Virus Host Factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.-G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef]
- Mairiang, D.; Zhang, H.; Sodja, A.; Murali, T.; Suriyaphol, P.; Malasit, P.; Limjindaporn, T.; Finley, R.L. Identification of New Protein Interactions between Dengue Fever Virus and Its Hosts, Human and Mosquito. PLoS ONE 2013, 8, e53535. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA Interference Screen for Human Genes Associated with West Nile Virus Infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef]
- Ruan, J.; Liang, D.; Yan, W.; Zhong, Y.; Talley, D.C.; Rai, G.; Tao, D.; LeClair, C.A.; Simeonov, A.; Zhang, Y.; et al. A Small-Molecule Inhibitor and Degrader of the RNF5 Ubiquitin Ligase. Mol. Biol. Cell 2022, 33, ar120. [Google Scholar] [CrossRef]
- Doroudgar, S.; Völkers, M.; Thuerauf, D.J.; Khan, M.; Mohsin, S.; Respress, J.L.; Wang, W.; Gude, N.; Müller, O.J.; Wehrens, X.H.T.; et al. Hrd1 and ER-Associated Protein Degradation, ERAD, Are Critical Elements of the Adaptive ER Stress Response in Cardiac Myocytes. Circ. Res. 2015, 117, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Rothan, H.A.; Zhong, Y.; Yan, W.; Henderson, M.J.; Chen, F.; Fang, S. A Small Molecule Inhibitor of ER-to-Cytosol Protein Dislocation Exhibits Anti-Dengue and Anti-Zika Virus Activity. Sci. Rep. 2019, 9, 10901. [Google Scholar] [CrossRef] [PubMed]
- Ci, Y.; Yao, B.; Yue, K.; Yang, Y.; Xu, C.; Li, D.-F.; Qin, C.-F.; Shi, L. Bortezomib Inhibits ZIKV/DENV by Interfering with Viral Polyprotein Cleavage via the ERAD Pathway. Cell Chem. Biol. 2023, 30, 527–539.e5. [Google Scholar] [CrossRef] [PubMed]
- López, L.S.; Calvo, E.P.; Castellanos, J.E. Deubiquitinating Enzyme Inhibitors Block Chikungunya Virus Replication. Viruses 2023, 15, 481. [Google Scholar] [CrossRef]
- Feng, W.; Sun, X.; Shi, N.; Zhang, M.; Guan, Z.; Duan, M. Influenza a Virus NS1 Protein Induced A20 Contributes to Viral Replication by Suppressing Interferon-Induced Antiviral Response. Biochem. Biophys. Res. Commun. 2017, 482, 1107–1113. [Google Scholar] [CrossRef]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J.M.; et al. Human Papillomavirus (HPV) Upregulates the Cellular Deubiquitinase UCHL1 to Suppress the Keratinocyte’s Innate Immune Response. PLoS Pathog. 2013, 9, e1003384. [Google Scholar] [CrossRef]
- Nair, S.R.; Abraham, R.; Sundaram, S.; Sreekumar, E. Interferon Regulated Gene (IRG) Expression-Signature in a Mouse Model of Chikungunya Virus Neurovirulence. J. Neurovirol. 2017, 23, 886–902. [Google Scholar] [CrossRef]
- Keck, F.; Amaya, M.; Kehn-Hall, K.; Roberts, B.; Bailey, C.; Narayanan, A. Characterizing the Effect of Bortezomib on Rift Valley Fever Virus Multiplication. Antiviral Res. 2015, 120, 48–56. [Google Scholar] [CrossRef]
- Deschamps, T.; Waisner, H.; Dogrammatzis, C.; Roy, A.; Chacko, S.; Perera, C.; Prisinzano, T.E.; Kalamvoki, M. Discovery of Small-Molecule Inhibitors Targeting the E3 Ubiquitin Ligase Activity of the Herpes Simplex Virus 1 ICP0 Protein Using an In Vitro High-Throughput Screening Assay. J. Virol. 2019, 93, e00619-19. [Google Scholar] [CrossRef]
- Yuan, Y.; Miao, Y.; Zeng, C.; Liu, J.; Chen, X.; Qian, L.; Wang, X.; Qian, F.; Yu, Z.; Wang, J.; et al. Small-Molecule Inhibitors of Ubiquitin-Specific Protease 7 Enhance Type-I Interferon Antiviral Efficacy by Destabilizing SOCS1. Immunology 2020, 159, 309–321. [Google Scholar] [CrossRef]
- Perry, J.W.; Ahmed, M.; Chang, K.-O.; Donato, N.J.; Showalter, H.D.; Wobus, C.E. Antiviral Activity of a Small Molecule Deubiquitinase Inhibitor Occurs via Induction of the Unfolded Protein Response. PLoS Pathog. 2012, 8, e1002783. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.-F. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Akari, H.; Sakurai, A.; Yoshida, A.; Chiba, T.; Tanaka, K.; Strebel, K.; Adachi, A. Expression of HIV-1 Accessory Protein Vif Is Controlled Uniquely to Be Low and Optimal by Proteasome Degradation. Microbes Infect. 2004, 6, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Strebel, K.; Daugherty, D.; Clouse, K.; Cohen, D.; Folks, T.; Martin, M.A. The HIV “A” (Sor) Gene Product Is Essential for Virus Infectivity. Nature 1987, 328, 728–730. [Google Scholar] [CrossRef]
- Fisher, A.G.; Ensoli, B.; Ivanoff, L.; Chamberlain, M.; Petteway, S.; Ratner, L.; Gallo, R.C.; Wong-Staal, F. The Sor Gene of HIV-1 Is Required for Efficient Virus Transmission in Vitro. Science 1987, 237, 888–893. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Duan, S.; Wang, S.; Song, Y.; Gao, N.; Meng, L.; Gai, Y.; Zhang, Y.; Wang, S.; Wang, C.; Yu, B.; et al. A Novel HIV-1 Inhibitor That Blocks Viral Replication and Rescues APOBEC3s by Interrupting Vif/CBFβ Interaction. J. Biol. Chem. 2020, 295, 14592–14605. [Google Scholar] [CrossRef]
- Chen, C.; Meng, Y.; Wang, L.; Wang, H.-X.; Tian, C.; Pang, G.-D.; Li, H.-H.; Du, J. Ubiquitin-Activating Enzyme E1 Inhibitor PYR41 Attenuates Angiotensin II-Induced Activation of Dendritic Cells via the IκBa/NF-κB and MKP1/ERK/STAT1 Pathways. Immunology 2014, 142, 307–319. [Google Scholar] [CrossRef]
- Ortiz, L.; Geiger, G.; Ferreri, L.; Moran, D.; Mendez, D.; Gonzalez-Reiche, A.S.; Alvarez, D.; Motta, M.; Escobar, F.; Rajao, D.; et al. Blue-Winged Teals in Guatemala and Their Potential Role in the Ecology of H14 Subtype Influenza a Viruses. Viruses 2023, 15, 483. [Google Scholar] [CrossRef]
- El-Daly, M.M.; Al-Raddadi, R.; Alharbi, A.; Azhar, A.E.; Khallaf, A.M.; Hassan, A.M.; Alwafi, O.M.; Shabouni, O.I.; Alandijany, T.A.; Li, T.-C.; et al. Hepatitis E Virus (HEV) in Makkah, Saudi Arabia: A Population-Based Seroprevalence Study. Viruses 2023, 15, 484. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Voza, A.; Riccardi, A.; Vanni, S.; De Iaco, F. Study & Research Center of the Italian Society of Emergency Medicine (SIMEU) Unfavorable Outcome and Long-Term Sequelae in Cases with Severe COVID-19. Viruses 2023, 15, 485. [Google Scholar] [CrossRef]
- Yin, M.; Xu, W. Special Issue: “Evolution, Ecology and Diversity of Plant Virus”. Viruses 2023, 15, 487. [Google Scholar] [CrossRef] [PubMed]
- Shiryaev, S.A.; Farhy, C.; Pinto, A.; Huang, C.-T.; Simonetti, N.; Elong Ngono, A.; Dewing, A.; Shresta, S.; Pinkerton, A.B.; Cieplak, P.; et al. Characterization of the Zika Virus Two-Component NS2B-NS3 Protease and Structure-Assisted Identification of Allosteric Small-Molecule Antagonists. Antiviral Res. 2017, 143, 218–229. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kwon, E.; Hartley, P.D.; Crosby, D.C.; Mann, S.; Krogan, N.J.; Gross, J.D. CBFβ Stabilizes HIV Vif to Counteract APOBEC3 at the Expense of RUNX1 Target Gene Expression. Mol. Cell 2013, 49, 632–644. [Google Scholar] [CrossRef]
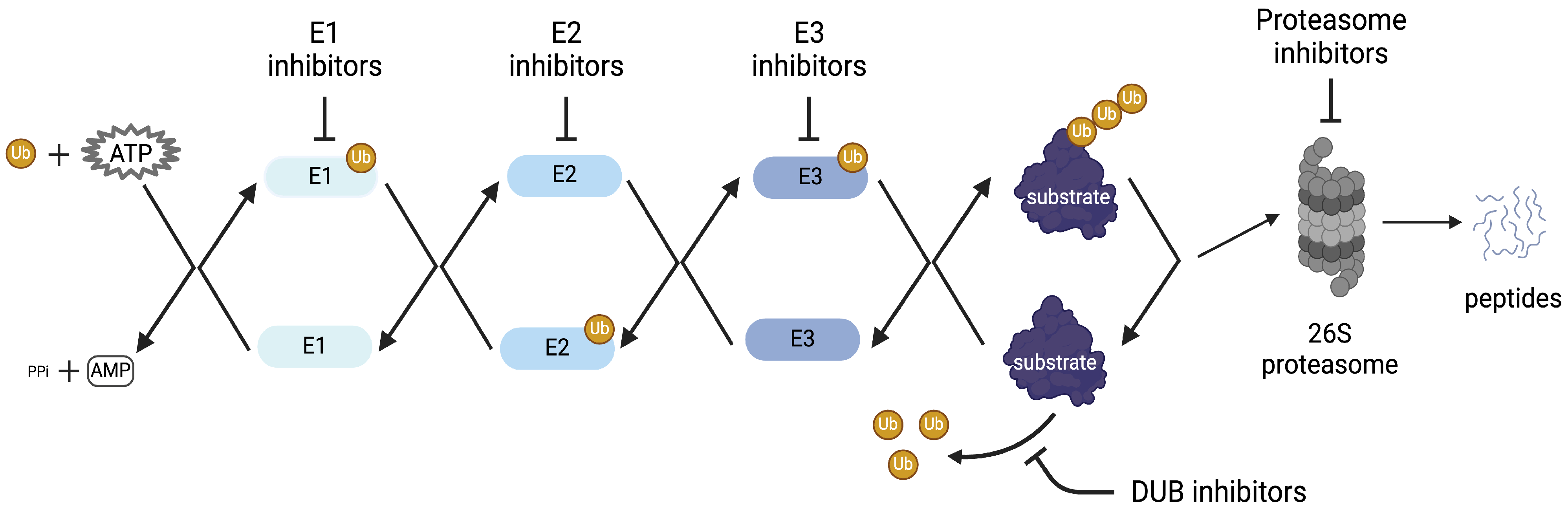
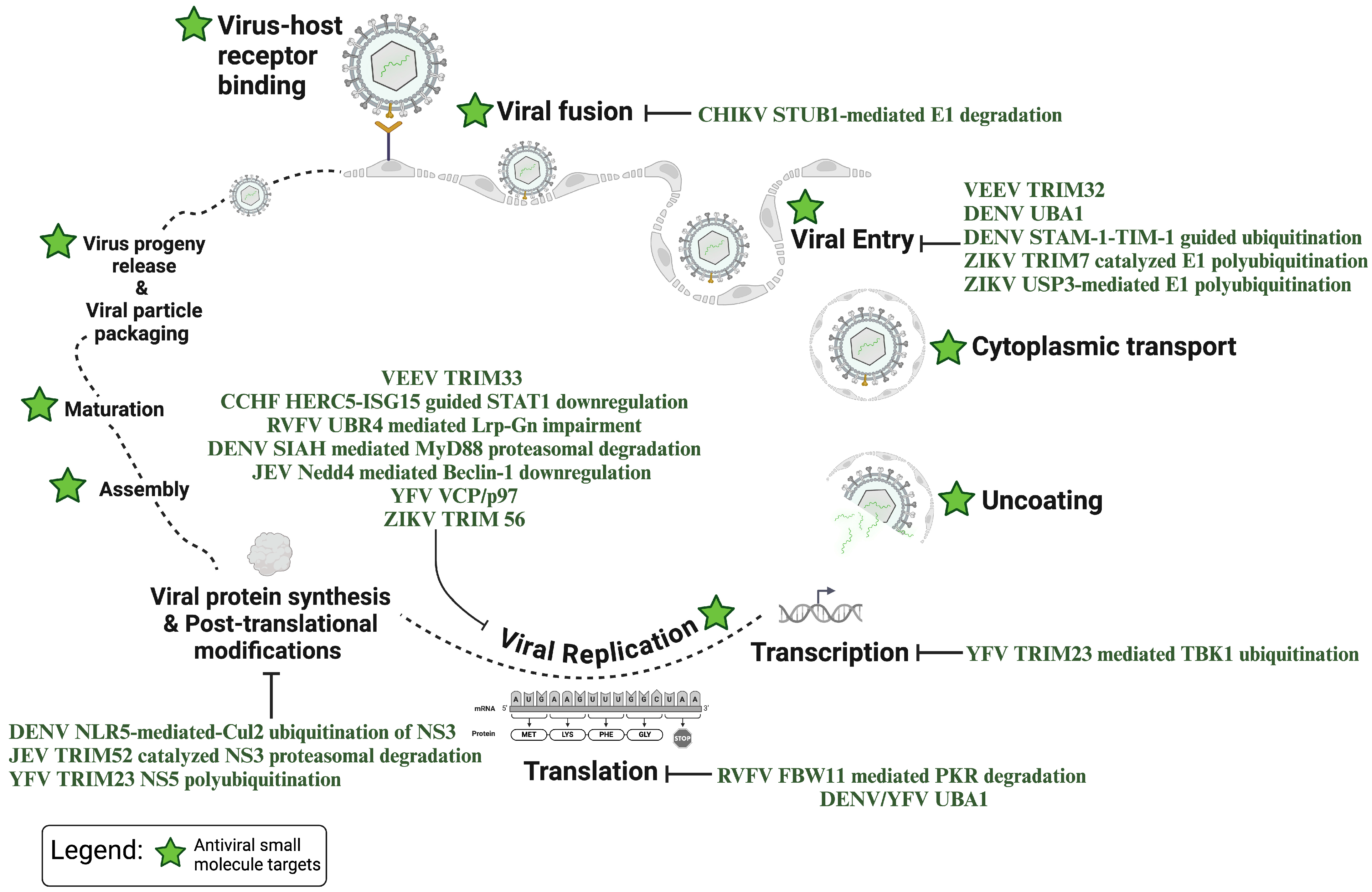

{kind=link}
{kind=link}
| Virus | Host or Viral Protein | Target Ub Pathway | Mechanism of Action | Proviral or Antiviral | Reference |
|---|---|---|---|---|---|
| CHIKV | Host/Viral | STUB1-mediated E1 degradation | Viral Fusion | Antiviral | [23] |
| VEEV | Host | TRIM33 | Viral Replication | Proviral | [37] |
| Host | TRIM32 | Late Viral Entry | Antiviral | [37] | |
| CCHF | Host | HERC5-ISG15-guided STAT1 downregulation | Viral Replication | Proviral | [106] |
| RVFV | Host | Skp1 and FBXO3-mediated p62 TFIIH degradation | Host Transcription | Proviral | [52,53] |
| Host | FBXW11-mediated PKR degradation | Viral Translation | Proviral | [55] | |
| Host | UBR4-mediated Lrp-Gn impairment | Viral Replication | Proviral | [55] | |
| DENV | Host | UBA1 | Viral Entry/Translation | Antiviral | [62] |
| Host | STAM-1-TIM-1-guided ubiquitination | Viral Entry | Proviral | [63,64] | |
| Host | NLRC5-mediated-Cul2 K48-linked polyubiquitination of NS3 | Viral Replication | Antiviral | [65] | |
| Host | SIAH-mediated MyD88 proteasomal degradation | Viral Replication | Proviral | [66] | |
| JEV | Host | TRIM52 catalyzed NS3 proteasomal degradation | Viral Replication | Antiviral | [77] |
| Host | Nedd4-mediated Beclin-1 downregulation | Viral Replication/NS3 Expression | Proviral | [78] | |
| YFV | Host | TRIM23-mediated K63-linked polyubiquitination of NS5 | Viral Replication | Proviral | [89] |
| Host | TRIM23-mediated TBK1 ubiquitination | Viral Replication/Transcription | Antiviral | [89] | |
| Host | VCP/p97 | Initial Viral Replication | Proviral | [88] | |
| Host | UBA1 | Initial Viral Translation | Proviral | [87] | |
| ZIKV | Host/Viral | TRIM7 catalyzed K63-linked E1 polyubiquitination | Viral Entry/Attachment | Proviral | [98] |
| Host | USP3-mediated impairment of K63-linked ZIKV E1 polyubiquitination | Viral Entry | Antiviral | [101] | |
| Host | TRIM56 | Viral Replication | Antiviral | [100] |
| Small Molecule | Function | Effect | Antiviral/ Proviral | Virus | Reference |
|---|---|---|---|---|---|
| IU1 | Inhibits DUB USP14 | Inhibits viral replication | Antiviral | DENV | [104] |
| CP26 | Inhibits Hrd1- mediated ERAD pathway | Inhibits ubiquitination and degradation of misfolded ER proteins | Antiviral | DENV/ZIKV | [113] |
| PYR-41 | Inhibits ubiquitin- activating enzyme E1 (UBA1) | Restricts viral translation | Antiviral | DENV | [131] |
| Bortezomib | Proteasomal inhibitor | Ubiquitin-mediated capsid degradation | Antiviral | VEEV | [31] |
| Inhibits viral replication | Antiviral | VEEV/EEEV/WEEV | [31] | ||
| NS3 ubiquitination | Antiviral | DENV/ZIKV | [115] | ||
| SAP30 and mSin3A ubiquitination | Antiviral | RVFV | [120] | ||
| PR619 | Broad-spectrum DUB inhibitor of USP and UCHL5 family. | Impairs viral RNA and protein synthesis | Antiviral | CHIKV | [116] |
| WP1130 | DUB inhibitor of USP5, USP9X, USP14, USP37, and UCHL5 | Impairs viral RNA and protein synthesis | Antiviral | CHIKV | [116] |
| WP1130 | Targets DUB USP14 | Binds to IRE1 | Antiviral | Human Norwalk/Murine noroviruses (MNV) | [123] |
| CV-3 | Rescues APOBEC3 activity and inhibits the interaction between Vif and CBFβ | Inhibits HIV-1 replication | Antiviral | HIV-1 | [130] |
| Virus | Protein | Viral/Host Protein | Proviral/ Antiviral | Mechanism of Action | Targeted Stage of Virus Life Cycle | Reference |
|---|---|---|---|---|---|---|
| CHIKV | NSP2 | Viral | Proviral | Inhibits host transcription and translation | NA | [22] |
| Rpb1 | Host | Proviral | Inhibits antiviral host genes | NA | [22] | |
| E1 | Viral | Proviral | Viral fusion | Viral fusion | [23] | |
| STUB1 | Host | Proviral | CHIKV host dependency factor | Degrades CHIKV E1 | [23] | |
| CYLD | Host | Antiviral | DUB | NA | [116] | |
| A20 | Host | Antiviral | DUB | NA | [116] | |
| UCHL1 | Host | Antiviral | DUB | NA | [132] | |
| STAMBP | Host | Antiviral | DUB | NA | [133] | |
| Otubain A20 | Host | Antiviral | DUB | NA | [134] | |
| USP10 | Host | Antiviral | DUB | NA | [135] | |
| VEEV | Capsid | Viral | Proviral | Inhibits host transcription | Increases vRNA | [31] |
| TRIM33 | Host | Proviral | Unknown | Unknown | [37] | |
| TRIM32 | Host | Antiviral | Capsid uncoating | Late viral entry | [37] | |
| CCHF | ISG15 | Host | Antiviral | Immune response regulator | NA | [106] |
| IFN | Host | Antiviral | Antiviral response | NA | [41] | |
| HERC5 | Host | Antiviral | Immune response regulator | NA | [106] | |
| NK-FB | Host | Antiviral | Immune response regulator | NA | [41,106] | |
| STAT1 | Host | Antiviral | IFN response mediator | NA | [41,106] | |
| RVFV | NSs | Viral | Proviral | RVFV virulence factor | Suppresses IFN signaling | [53] |
| Cullin-1 | Host | Proviral | Activates PKR | NA | [53] | |
| Skp1 | Host | Proviral | Degrade p62 TFIIH | NA | [53] | |
| FBXO3 | Host | Proviral | Degrade p62 TFIIH | NA | [53] | |
| FBXW11 | Host | Proviral | PKR degradation | Viral translation | [55] | |
| TFIIH | Host | Antiviral | IFN signaling | NA | [53] | |
| UBR4 | Host | Proviral | RVFV Gn interactor | Viral protein Transport | [55,56] | |
| Lrp | Host | Antiviral | RVFV host restriction factor | NA | [55,56] | |
| Gn | Host | Proviral | Binds to Lrp | NA | [55,56] | |
| PKR | Host | Antiviral | Inhibits viral translation | NA | [55] | |
| SAP30 | Host | Proviral | NSs interactors, inhibit IFN-β expression | Unknown inhibition | [120] | |
| mSin3a | Host | Proviral | NSs interactors, inhibit—β expression | Unknown inhibition | [120] | |
| DENV | TIM-1 | Host | Proviral | DENV entry receptor | Viral endocytosis | [63,64] |
| STAM-1 | Host | Proviral | Ubiquitinated cargo mediation | Viral entry and attachment | [63,64] | |
| NS3 | Viral | Proviral | RNA replication | RNA replication | [65] | |
| SIAH | Host | Proviral | Ubiquitinates, degrades MyD88 | NA | [66] | |
| MyD88 | Host | Antiviral | Mediates NF-KB signaling | Unknown inhibition | [66] | |
| Cullin-2 | Host | Antiviral | Polyubiquitinates NS3 | Unknown inhibition | [65] | |
| NLC5 | Host | Antiviral | Innate immunity | Unknown inhibition | [65] | |
| JEV | Nedd4 | Host | Proviral | Downregulates Beclin-1 | Increases NS3 viral protein | [77,78] |
| Beclin-1 | Host | Antiviral | Autophagy and ubiquitination regulator | Unknown inhibition | [77,78] | |
| TRIM21 | Host | Proviral | Downregulates IFN-β signaling | Viral replication | [77] | |
| TRIM52 | Host | Antiviral | Host restriction factor | Degrades viral NS2A | [81] | |
| NS2A | Viral | Proviral | Recruits vRNA | Viral replication | [81] | |
| NS3 | Viral | Proviral | Protease activity | Viral replication | [78] | |
| YFV | VCP/p97 | Host | Proviral | Protein homeostasis | Post-fusion and pre-translation | [87,88] |
| TRIM23 | Host | Proviral | Initiates TBK1 | Unknown inhibition | [89] | |
| TRIM23 | Host | Antiviral | Inhibit IFN signaling | Polyubiquitinates NS5 | [89] | |
| NS5 | Viral | Antiviral | Interferon signaling mediator | Unknown inhibition | [89] | |
| STAT2 | Host | Proviral | Inhibit type-1 IFN | Viral replication | [89,98] | |
| ZIKV | E | Viral | Proviral | Receptor binding | Viral entry | [98,99] |
| TRIM7 | Host | Proviral | Virus endosome membrane fusion | Viral entry | [89] | |
| TRIM56 | Host | Antiviral | RNA binding protein | Downregulates vRNA | [100] | |
| USP38 | Host | Antiviral | Binds to ZIKV E | Inhibits viral Replication | [101] | |
| ZIKV NS2A3 | Viral | Proviral | Cleaves ZIKV polyprotein precursor | Viral protein Generation | [136] | |
| RNF126 | Host | Antiviral | Ubiquitination of ZIKV NS3 | NA | [115] | |
| NS3 | Viral | Proviral | Promotes NS5-guided RNA synthesis | Viral replication | [115] | |
| DENV/ ZIKV | Hrd1 | Host | Antiviral | ERAD-mediated degradation of misfolded proteins | NA | [113] |
| Hrd1 | Host | Antiviral | Ubiquitination of ZIKV NS3 | NA | [115] | |
| Norovirus | USP14 | Host | Proviral | DUB binds to IRE1 | Viral replication | [104] |
| Measles Influenza HPV | UCHL1 | Host | Proviral | Downregulate immune response | Viral replication | [116] |
| A20 | Host | Proviral | TRAF6 ubiquitination modification | Viral replication | [116] | |
| HIV-1 | CBFB | Host | Proviral | Interacts with Vif-mediated cell cycle regulation | Unknown host inhibition | [137] |
| APOBEC3 | Host | Antiviral | Targeted for proteasomal degradation through CBFB | Unknown inhibition | [137] | |
| Vif | Viral | Proviral | HIV-1 accessory protein, degrades APOBEC3 | Viral replication | [137] | |
| HSV-1 | SOCS1 | Host | Proviral | Downregulation of IFN-1 expression | Viral replication | [122] |
| USP7 | Host | Proviral | Deubiquitinates and promotes SOCS1 stability | Viral immune evasion | [122] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sreepangi, S.; Baha, H.; Opoku, L.A.; Jones, N.X.; Konadu, M.; Alem, F.; Barrera, M.D.; Narayanan, A. Host-Driven Ubiquitination Events in Vector-Transmitted RNA Virus Infections as Options for Broad-Spectrum Therapeutic Intervention Strategies. Viruses 2024, 16, 1727. https://doi.org/10.3390/v16111727
Sreepangi S, Baha H, Opoku LA, Jones NX, Konadu M, Alem F, Barrera MD, Narayanan A. Host-Driven Ubiquitination Events in Vector-Transmitted RNA Virus Infections as Options for Broad-Spectrum Therapeutic Intervention Strategies. Viruses. 2024; 16(11):1727. https://doi.org/10.3390/v16111727
Chicago/Turabian StyleSreepangi, Sanskruthi, Haseebullah Baha, Lorreta Aboagyewa Opoku, Naomi X. Jones, Maame Konadu, Farhang Alem, Michael D. Barrera, and Aarthi Narayanan. 2024. "Host-Driven Ubiquitination Events in Vector-Transmitted RNA Virus Infections as Options for Broad-Spectrum Therapeutic Intervention Strategies" Viruses 16, no. 11: 1727. https://doi.org/10.3390/v16111727
APA StyleSreepangi, S., Baha, H., Opoku, L. A., Jones, N. X., Konadu, M., Alem, F., Barrera, M. D., & Narayanan, A. (2024). Host-Driven Ubiquitination Events in Vector-Transmitted RNA Virus Infections as Options for Broad-Spectrum Therapeutic Intervention Strategies. Viruses, 16(11), 1727. https://doi.org/10.3390/v16111727